


Abstract
Several of the hepatic microsomal cytochromes P-450 (CYP) including CYP3A are inducible by phenobarbital (PB). However, the intracellular pathways involved in the action of PB on CYP3A remain poorly known. With the aim to unravel some of the main aspects of PB signaling, we first devised a simple model of mouse cultured primary hepatocytes in which CYP3A mRNA and protein were strongly induced by PB in the absence of dexamethasone and were at maximum levels after a 48-h treatment with a 2-mM dose of PB. Under these culture conditions, we studied the effects of inhibitors and activators of different protein kinases or phosphatases on CYP3A mRNA and protein induction by PB. CYP3A-induced expression was inhibited by activators of cyclic AMP-dependent protein kinase (PKA) (dibutyryl-cyclic AMP and forskolin) whereas inhibition of PKA by PKA inhibitor enhanced induction. 8-br-cGMP produced effects similar to the activators of PKA, and so did the specific inhibitor of cGMP-dependent protein kinase, β-phenyl-1,N2-etheno-8-bromoguanosine-3,5′-cyclic monophosphorothioate, Rp-isomer (Rp-8-Br-PET-cGMPS). Inhibition of Ca2+/calmodulin-dependent protein kinase by KN-62 or the intracellular Ca2+ chelator BAPTA-AM produced an inhibition of CYP3A induction by PB. Specific inhibitors of protein kinase C, mitogen-activated protein kinase kinase, phosphatidylinositol-3-kinase, or serine/threonine phosphatase did not produce any effect. Taken together, our results suggest that CYP3A induction by PB is regulated positively by calmodulin-dependent protein kinase and cGMP-dependent protein kinase, and negatively by PKA in mouse hepatocytes in primary culture.
Cytochromes P-450 (CYPs) are hemoproteins encoded by a gene superfamily, and catalyze mainly monooxygenation reactions of a variety of substrates, including both endogenous compounds such as steroid hormones, fatty acids, biogenic amines, and prostaglandins, as well as xenobiotics (Porter and Coon, 1991; Nelson et al., 1993). Several major tissues are involved in CYP-mediated metabolic activation and detoxication, and include the liver, intestine, lung, skin, the central nervous system, and the adrenals among others (Leighton and Kemper, 1984). The expression of these enzymes is regulated by complex mechanisms, in which many different factors like sex, age, hormones, diet composition, or genetic background are involved (Shimada et al., 1995; Ganem and Jefcoate, 1998). Drugs and other foreign compounds can also participate in the regulatory mechanisms of CYP gene expression by either inducing or repressing CYP levels (Levine et al., 1998).
Phenobarbital (PB) is a typical inducer of many genes for which it displays a very strong liver selectivity (Ramsden et al., 1993). This molecule, and its structurally unrelated agonists, induce at the transcriptional level CYP members of subfamilies 1A, 2A, 2B, 2C, and 3A in different species, as well as other drug-metabolizing enzymes such as aldehyde dehydrogenase, epoxide hydrolase, NADPH:P-450 reductase, UDP-glucuronyltransferase and glutathioneS-transferase (Waxman and Azaroff, 1992). Despite the growing body of accumulating data on the regulatory pathways mobilized in response to the barbiturate, much remains to be uncovered concerning the role of key enzymes controlling the integrative response within the isolated liver cell. However, we have recently reported the ability of PB to stimulate phosphorylation of rat liver and hepatocyte nuclear proteins (Baffet and Corcos, 1995). It is well established that many signal transduction pathways are involved in gene regulation in numerous model cell systems, including those operating in the transcriptional control of CYP gene expression. With respect to PB, it is important to stress that different protein kinases, e.g., cyclic AMP (cAMP)-dependent protein kinase (PKA) and Ca2+/calmodulin-dependent protein kinase (CaM PK), and phosphatases have been reported to play a determinant role in the mechanism of induction of different CYPs (Sidhu and Omiecinski, 1995a; Brown et al., 1997; Bani et al., 1998; Honkakoski and Negishi, 1998).
CYP3As catalyze a remarkable number of oxidation reactions of clinically important drugs, including cyclosporin A, midazolam, nifedipine, verapamil, Taxol, and antibiotics like erythromycin or rifampicin (Cresteil et al., 1994; Takano et al., 1998). CYP3A mRNA and protein expression are highly inducible upon treatment with several therapeutic molecules, such as PB or glucocorticoids (Schuetz et al., 1993). Three different members of the CYP3A subfamily have been isolated from the mouse. CYP3A11 mRNA is predominantly expressed in mouse liver, in the adult animals, and it is more extensively induced by PB than CYP3A13, the other isoform of mouse CYP3A expressed in this tissue (Yanagimoto et al., 1997). CYP3A16 is the third mouse isoform, expressed only in fetal liver (Itoh et al., 1994). To our knowledge, no data have been reported yet about the PB-mediated regulation of expression of those genes in mouse primary hepatocyte cultures.
In this study, we have looked into the involvement of different regulatory pathways in CYP3A induction by PB in mouse cultured hepatocytes using different protein kinase/phosphatase activators and inhibitors. For this purpose, we have worked out the conditions under which primary mouse hepatocytes retained CYP3A inducibility in culture. Our data suggest both a positive effect of calcium, likely via CaM PK, and cGMP-dependent protein kinase (PKG) as well as a negative effect of the cyclic nucleotide cAMP through the PKA transduction pathway, on PB induction of CYP3A at the mRNA level. By contrast, tyrosine kinases, protein kinase C (PKC), and other serine/threonine kinases did not seem to play any significant role in the inductive response.
Materials and Methods
Chemicals.
Collagenase D was obtained from Boehringer Mannheim (Mannheim, Germany). Cell culture medium was purchased from Eurobio (Les Ulis, France) and fetal calf serum from Dominique Dutscher SA (Brumath, France). PB was obtained from Coopération Pharmaceutique Française (Melun, France). Dexamethasone, pyruvate, ITS (5 mg/l insulin, 2.25 mg/l transferrin, 2.5 mg/l sodium selenite),N6,O2-dibutyryl-cAMP (db-cAMP), and 8-bromo-cyclic guanosine monophosphate (8-br-cGMP) were purchased from Sigma (St. Louis, MO). Genistein, okadaic acid, [1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetra (acetoxymethyl) ester] (BAPTA-AM), {1-[N,O-bis-(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine} (KN-62), [1-(5-isoquinolinesulfonyl)-2-methylpiperazine, HCl] (H7), [N-(2-guanidinoethyl)-5-isoquinolinesulfonamide, HCl] (HA-1004), {N-[2-((p-bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide, HCl} (H89), {3-[1-[3-(amidinothio)propyl-1H-indol-3-yl]-3-(1-methyl-1H-indol-3-yl)maleimide methane sulfonate} (Ro-31–8220), [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one)] (LY-294002), 2′-amino-3′-methoxyflavone (PD-98059), forskolin, and protein kinase A inhibitor (PKAI) were purchased from Calbiochem (La Jolla, CA); β-phenyl-1,N2-etheno-8-bromoguanosine-3′,5′-cyclic monophosphorothioate, Rp-isomer (Rp-8-Br-PET-cGMPS) was purchased from Biolog Life Science Institute (Bremen, Germany).
Hepatocyte Cultures.
Primary hepatocytes were isolated from 8- to 10-week-old C57BL/6J male mice (CERJ, Le Genest, St.-Isle, France) by a two-step collagenase perfusion. Cell viability was estimated by the trypan blue exclusion test and was found to range between 80 and 90%. The isolated hepatocytes were suspended in Williams’ E medium containing ITS, penicillin G/streptomycin (100 I.U./ml), 30 mM pyruvate, and 7% (v/v) fetal calf serum (FCS) and were plated onto 60-mm diameter culture dishes (Nunc, Brand Products, Roskilde, Denmark) at a density of 2 × 106cells. The cultures were maintained at 37°C in a humidified incubator under an atmosphere of 5% CO2/95% air. After a 1-h incubation, nonattached cells were discarded by aspiration. Cells were washed with PBS, followed by changing medium to Williams’ E without FCS. The chemicals to be tested, dissolved in dimethyl sulfoxide (final concentration in culture of 0.1%; no apparent toxicity due to this vehicle alone was observed in hepatocytes), were added to the serum-free medium and were incubated at 37°C for 1 h before the addition to the cultures of PB or its vehicle. The concentrations tested were between 5 and 10 times above theKi of the molecules, and no toxicity was observed for any of them. Culture medium changes were performed thereafter on a daily basis. All experiments were performed at least three times using different cell culture preparations.
Analysis of Cellular mRNA.
Cells were washed with 0.1 M PBS. Total RNA was extracted according to the method of Chomczynski and Sacchi (1987). For Northern blotting, 7 μg of total RNA was separated by electrophoresis on a denaturing 16% (v/v) formaldehyde, 1.25% (w/v) agarose gel, and transferred onto Appligene nylon filters (Illkirch, France). RNAs were fixed by UV cross-linking. Integrity and relative amounts of RNA were ascertained by methylene blue staining of the filters. The filters were prehybridized in 3X standard sodium citrate (SSC), 0.1% SDS, 10% (w/v) dextran sulfate, and 10X Denhardt’s solution (0.1% BSA, 0.1% ficoll, 0.1% polyvinyl pyrolidone) and 100 μg/ml of herring sperm DNA at 65°C, and then hybridized with cDNA probes labeled with [α32P]dCTP by random priming using a DNA-labeling kit from Amersham Pharmacia Biotech (Amersham, UK). The CYP3A probe was a 1.6-kb full-length coding rabbit cDNA fragment (P-450LM3c; Dalet et al., 1986) that displays at least 70% identity with both CYP3A11 and CYP3A13 mRNAs. The albumin probe was a human cDNA. Filters were washed with 3X SSC, 0.1% SDS at 65°C twice for 10 min and with 1X SSC, 0.1% SDS at 65°C for 10 min. Filters were exposed to X-ray films at −80°C. Relative mRNA amounts were determined by densitometry (Densylab, Microvision instruments, Evry, France). Basal level of CYP3A mRNA was never detected, but fold induction was estimated by comparing the faintest signal the imaging software could detect to the signal observed after PB induction.
Microsomes.
Cells were scraped and homogenized for 20 s with an ultraturax (Janke and Kunkle GmbH & Co., IKA-labortechnik, Staufen, Germany) in 50 mM Tris-HCl buffer (pH 7.4) containing 0.25 M sucrose and 1 mM EDTA. After removing nuclear and mitochondrial fractions by centrifugation at 3000g for 10 min, and at 9000g for 20 min, microsomes were isolated from the supernatant by centrifugation at 100,000g for 1 h. They were resuspended in 10% glycerine, 0.1 M phosphate buffer (pH 7.4), and stored at −80°C until use. Protein concentration was determined according to the method of Bradford (1976)using BSA as a standard.
Western Blot.
Microsomal proteins were diluted in 10% SDS, 1% 2-mercaptoethanol, 10 mM Tris-HCl pH 6.8, and 20% glycerol. Ten-microgram proteins were separated by 10% polyacrylamide gel electrophoresis and transferred electrophoretically onto nitrocellulose membranes (Hybond-ECL, Amersham Life Science, Amersham, UK). Relative amounts of proteins were ascertained by staining membranes with Ponceau S. After blocking the filters in 3% BSA-Tris-buffered saline (TBS), they were incubated with an antihuman CYP3A4 polyclonal antibody (Valbiotech, Paris, France) diluted at 1/2000 in 3% BSA-TBS-0.3% NP40. The filters were then washed three times for 10 min in TBS and incubated with peroxidase-conjugated goat antirabbit IgG. All incubations were performed at room temperature for 2 h. Antibody binding was detected by an ECL system (Amersham Pharmacia Biotech, Amersham, UK).
Data Analysis.
Relative CYP mRNA levels were deduced from normalization of hybridization signals by densitometry analysis after reprobing of the filters with an albumin cDNA probe (Figs. 2, 3, 4A, and 5A) or reporting to the respective blue methylene staining 18S RNA (Figs. 4B and 5B). Results were expressed as percentages of the values obtained from PB alone. These values are given as the mean percentage ± S.E. from data obtained from three independent hepatocyte preparations (n = 3).
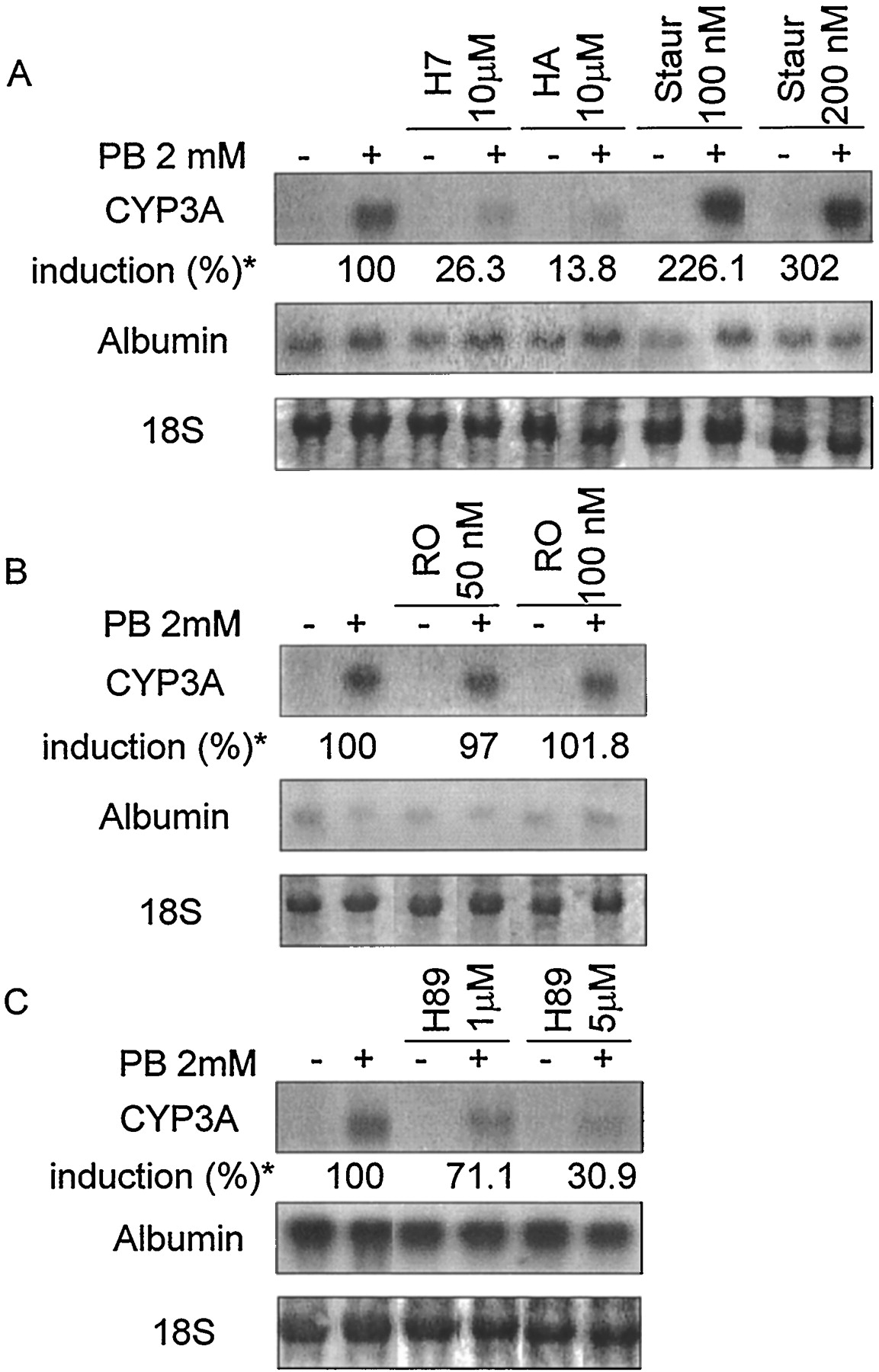
Effects of inhibitors of different transduction pathways on the induction by PB of CYP3A mRNA expression in cultured mouse hepatocytes. Uniform RNA loading was demonstrated by staining of the 18S ribosomal band. Albumin hybridization levels were used as a control. A, broad spectrum serine/threonine kinase inhibitors: H7, HA-1004 (HA), and staurosporine (Staur). B, specific PKC inhibitor: Ro-31–8220 (RO). C, specific inhibitor of PKA/PKG: H89. * % CYP3A induction is noted under each inhibitor as the mean of three different hepatocyte preparations; the respective S.E.s are: H7 (0.8); HA (1.2); Staur 100 nM (35.4); Staur 200 nM (43.4); RO 50 nM (8.8); RO 100 nM (10.6); H89–1 μM (7.4); H89–5 μM (7.3).

Effects of activators of PKA/PKG on the induction by PB of CYP3A mRNA expression in cultured mouse hepatocytes. Uniform RNA loading was demonstrated by staining of the 18S ribosomal band. Albumin hybridization levels were used as a control. A, PKA activators: db-cAMP and forskolin. B, PKG activator: 8-bromo-cGMP (br-cGMP). * % CYP3A induction is noted under each activator as the mean of three different hepatocyte preparations, except for br-cGMP (two different hepatocyte preparations); the respective S.E.s are: cAMP 10 μM (4.4); cAMP 100 μM (8.6); forskolin 5 μM (10.5); forskolin 50 μM (5.1); br-cGMP 10 μM (6.9); br-cGMP 100 μM (0.3).

Effects of specific inhibitors of PKA/PKG on the induction by PB of CYP3A mRNA expression in cultured mouse hepatocytes. Uniform RNA loading was demonstrated by staining of the 18S ribosomal band. Albumin hybridization levels were used as the control for PKA Inhibitor. 18S ribosomal bands were used as the control for Rp-8-Br-PET-cGMPS. A, PKAI. B, PKG inhibitor: Rp-8-Br-PET-cGMPS (Rp-GMPS). * % CYP3A induction is noted under each inhibitor as the mean of three different hepatocyte preparations; the respective S.E.s are: PKAI 25 nM (24.1); PKAI 50 nM (25.5); Rp-GMPS 300 nM (12.4); Rp-GMPS 600 nM (5.0).

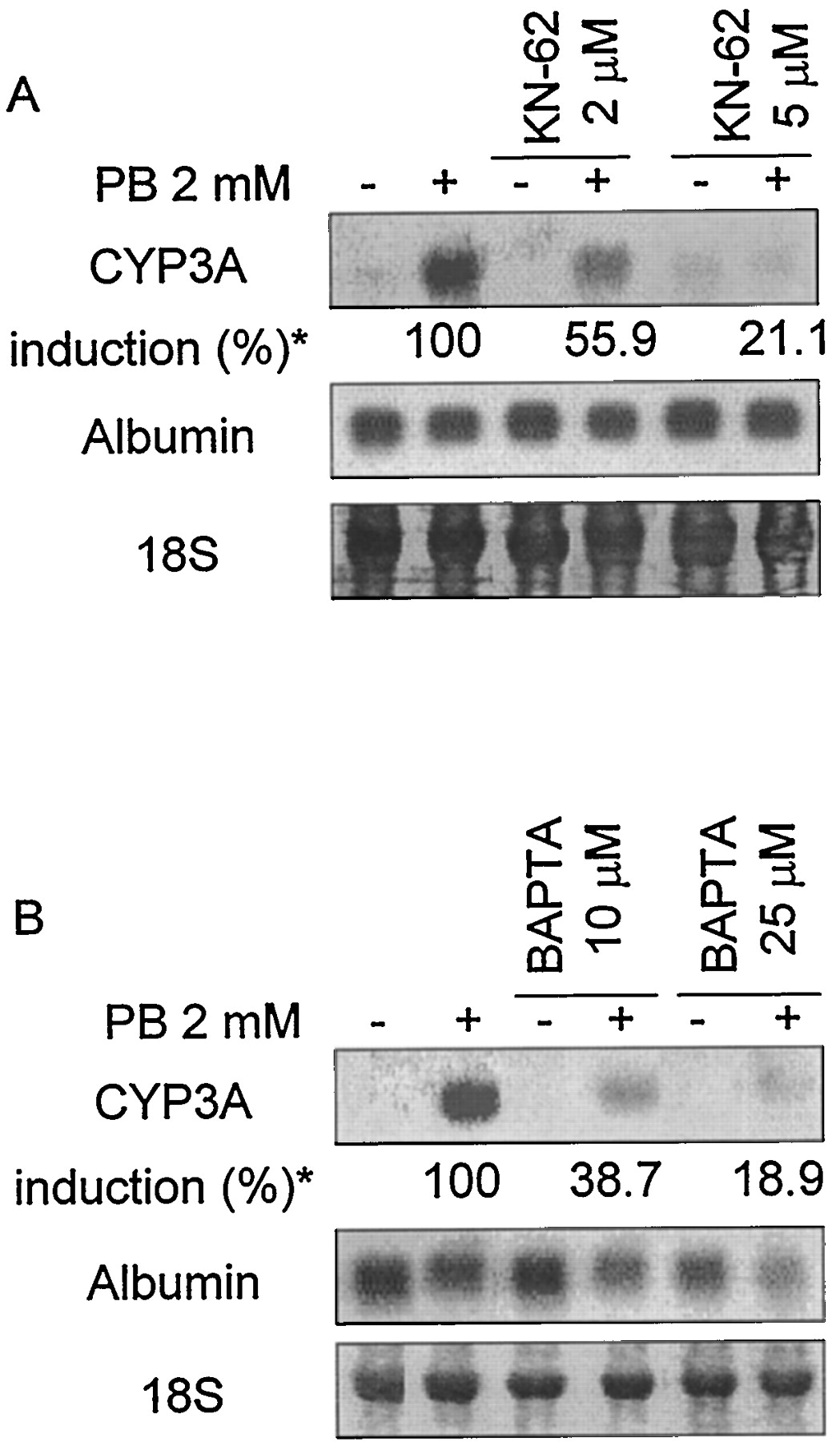
A, effect of a specific inhibitor of CaM PK, KN-62 on the induction by PB of CYP3A mRNA expression in cultured mouse hepatocytes. B, effect of the intracellular Ca2+ chelator BAPTA-AM on the induction by PB of CYP3A mRNA expression in cultured mouse hepatocytes. Uniform RNA loading was demonstrated by staining of the 18S ribosomal band. Albumin hybridization levels were used as the control for KN-62. 18S ribosomal bands were used as the control for BAPTA-AM. * % CYP3A induction is noted under each molecule as the mean of three different hepatocyte preparations; the respective S.E.s are: KN-62 2 μM (5.1); KN-62 5 μM (11.9); BAPTA-AM 10 μM (6.8); BAPTA-AM 25 μM (12.8).
Results
Expression of CYP3A mRNA in Primary Cultures of Mouse Hepatocytes.
To establish the optimal conditions for CYP3A induction in primary mouse hepatocytes, a series of experiments was performed. Different concentrations of ITS supplement were tested, as well as various attachment periods from 1 to 4 h. PB was added to the cultured cells immediately after those attachment periods, at three different concentrations, i.e., 0.1, 0.5, and 2 mM, for 24, 48, or 72 h (Fig. 1A). No basal CYP3A mRNA expression was detected in any of the conditions tested. The dose- and time-response studies revealed that PB induction of CYP3A mRNA expression was maximum at 2 mM PB and 48-h exposure to this molecule, to amount to at least 15-fold (Fig. 1A). No differences were found depending on the attachment periods, or at different concentrations of ITS supplement (data not shown). Therefore, we decided to choose the dose of 2 mM PB for 48 h for subsequent experiments. We also studied the influence of 5 nM dexamethasone on the basal and PB-induced expression of CYP3A mRNA (Fig. 1B). The presence of dexamethasone did not detectably affect CYP3A basal expression although it had a slightly positive effect on the PB induction of CYP3A at all PB concentrations tested, and after 24 and 72 h of treatment (data not shown). However, no effect was detected when compared with the cells cultured in the absence of dexamethasone and exposed to 2 mM PB for 48 h, a condition that gave the strongest induction (Fig. 1A). This result, as well as the fact that no morphological changes were observed between cells treated or not with dexamethasone, led us to avoid the use of this molecule in subsequent experiments.

A. Effects of PB on CYP3A mRNA expression. Dose and time response of CYP3A mRNA induction by PB in cultured mouse hepatocytes. Hepatocytes were cultured as described in Materials and Methods. After a 1-h attachment period, they were treated with different concentrations of PB or its vehicle. Treatments were maintained for 24, 48, or 72 h, changing culture media every day. B, absence of effect of dexamethasone (Dex) 5 nM on CYP3A mRNA in mouse cultured hepatocytes treated with different concentrations of PB for 48 h. Uniform RNA loading is demonstrated by staining of the 18S ribosomal band.
Effect of Inhibitors and Activators of Protein Kinases/Phosphatases on CYP3A mRNA Levels.
Various agents known to affect different regulatory pathways were screened for their ability to influence the PB-mediated CYP3A induction. Genistein, a tyrosine kinase inhibitor, had no detectable effect on either the basal or PB-induced expression of CYP3A mRNA at the concentrations of 10 and 50 μM (data not shown). Staurosporine (100 and 200 nM), a broad spectrum serine/threonine kinase inhibitor, had a positive dose-dependent effect on induced CYP3A expression (increased to 226.1 ± 35.4 and 302.0 ± 43.4%, respectively) whereas other broad spectrum kinase inhibitors, H7 and HA-1004 (10 μM), inhibited this induction (decreased to 26.3 ± 0.8 and 13.8 ± 1.2%, respectively; Fig.2A). At the concentration of 10 μM, H7 is known to inhibit PKA, PKC, and PKG, whereas HA-1004 only inhibits PKA and PKG. These latter results thus suggested the implication of PKA and/or PKG in PB induction. The effect of staurosporine could have been consistent with the involvement of a PKC-dependent pathway but this possibility was ruled out by testing a specific PKC inhibitor, Ro-31–8220 (50 and 100 nM), which had no effect on either basal or induced CYP3A expression (Fig. 2B). Similarly, other more specific serine/threonine protein kinase inhibitors such as PD-98059 (10 and 50 μM), a mitogen-activated protein kinase kinase inhibitor, or LY-294002 (5 and 10 μM), a potent and specific phosphatidylinositol-3-kinase inhibitor had no effect either (data not shown). None of these molecules had any detectable influence on basal expression of CYP3A mRNA. The same result was observed for okadaic acid: this serine/threonine phosphatase inhibitor showed no effect on basal or PB-induced CYP3A mRNA levels at the concentrations tested (10 and 100 nM; data not shown). In all these cases, albumin mRNA level remained unaffected (Fig. 2, A and B).
In view of these results, we decided to more specifically analyze the involvement of PKA/PKG pathways in controlling the effects of PB on CYP3A mRNA expression. A specific PKA/PKG inhibitor, H89, produced a significant dose-dependent inhibition of induced CYP3A expression at the concentrations of 1 and 5 μM (decreased to 71.1 ± 7.4 and 30.9 ± 7.3%, respectively), but no effect on albumin mRNA level (Fig. 2C). This observation, together with the as yet unexplained enhancement of CYP3A induction by PB upon staurosporine treatment, led us to verify what effect was elicited by specific activators and inhibitors of PKA or PKG.
db-cAMP, an analog of cAMP that activates the PKA pathway, produced an important inhibition of CYP3A expression at the concentration of 10 μM (decreased to 18.5 ± 4.4%), although this inhibition was less marked (decreased to 74.1 ± 8.6%) when the concentration of this molecule was increased to 100 μM (Fig.3A). Likewise, the PKA activator forskolin, an adenylate cyclase activator, inhibited CYP3A induction at the concentration of 50 μM (decreased to 26.8 ± 10.5%). The inhibitory effect of forskolin when tested at 5 μM was lower (decreased to 62.8 ± 5.1%; Fig. 3A). With respect to the PKG pathway, the analog of cGMP assayed, 8-br-cGMP, produced an effect similar to that of db-cAMP, namely an important drop of PB-induced CYP3A expression at the dose of 10 μM (decreased to 57 ± 6.9%) and a barely detectable effect (decreased to 80.3 ± 0.3%) when tested at 100 μM (Fig. 3B). The effect of 8-br-cGMP was not, however, reproducible, as it was observed in only two of the three experiments carried out, and should, therefore, be taken as indicative. None of PKA or PKG activators showed any effect on albumin mRNA expression (Fig. 3, A and B). Treatment of cells with the PKA-specific inhibitor PKAI (25 and 50 nM) resulted in an enhancement of the PB-induced CYP3A mRNA expression (increased to 161.3 ± 24.1 and 133.2 ± 25.5%, respectively; Fig. 4A), which was in agreement with the inhibition of this expression upon PKA activation by the analog of cAMP; this observation was in line with the stimulatory effect of staurosporine over PB-induced CYP3A mRNA level. By contrast, the specific and potent inhibitor of PKG, Rp-8-Br-PET-cGMPS (Butt et al., 1995), produced an inhibition at the highest concentration tested (600 nM; decreased to 44.6 ± 5.0%) together with a reduction of albumin mRNA level (Fig. 4B). The specific inhibitor of PKA showed no effect on albumin mRNA expression (Fig. 4A).
In other culture models, Ca2+ has been suggested to be involved in PB signaling (Sadar et al., 1996). We also looked into the role of Ca2+ by using a specific inhibitor of CaM PK, KN-62 (2 and 5 μM). This molecule inhibited PB-induced CYP3A expression in a dose-dependent manner (decreased to 55.9 ± 5.1 and 21.1 ± 11.9%, respectively), although it had a marginal positive effect on basal mRNA expression (Fig.5A). We also studied the effect of an intracellular calcium chelator, BAPTA-AM (10 and 25 μM), which produced, like KN-62, an inhibition of PB-induced CYP3A mRNA (decreased to 38.7 ± 6.8 and 18.9 ± 12.8%, respectively; Fig. 5B). Taken together, these results pointed to a positive effect of Ca2+ in CYP3A induction by PB. Albumin mRNA levels remained unaffected after treatment with KN-62 but were reduced by BAPTA-AM (Fig. 5, A and B).
Effect of Inhibitors and Activators of Protein Kinases on CYP3A Protein Expression.
For some of the molecules showing an effect on PB-induced CYP3A mRNA expression, we also studied CYP3A protein levels in mouse primary hepatocytes under the same culture conditions by Western blot analysis. For this purpose, microsomal preparations were pooled from two independent hepatocyte cultures and, therefore, these results should be taken only as indicative. db-cAMP (10 μM), as it did at the mRNA level, produced an inhibition of CYP3A-induced expression (roughly 50% inhibition) (Fig.6A), and 8-br-cGMP (10 μM) produced a similar effect (Fig. 6B). However, this was not the case of forskolin, for which no differences were observed compared to PB-treated hepatocytes (Fig. 6A). Nevertheless, it was noteworthy that forskolin (50 μM) produced an important induction of basal CYP3A protein expression, to a much larger extent than for the mRNA, which basal level was barely detectable. With respect to PKA and PKG inhibitors, PKAI (25 nM) was found to marginally decrease PB-induced CYP3A protein levels, whereas Rp-8-Br-PET-cGMPS produced a slight (roughly 30%) inhibition of the induced protein expression only at 600 nM (Fig. 6B). Hepatocytes treated with KN-62 (5 μM) exhibited enhanced basal CYP3A protein expression but no modification of the PB-induced expression was observed (Fig. 6C).

Effects of different activators and inhibitors of different transduction pathways on the induction by PB of CYP3A protein expression in cultured mouse hepatocytes. A, PKA activators db-cAMP and forskolin. B, PKG activator 8-bromo-cGMP (br-cGMP); PKG inhibitor: Rp-8-Br-PET-cGMPS (Rp-GMPS) PKAI. C, inhibitor of CaM PK, KN-62.
Discussion
In the present study, we describe a model of mouse primary hepatocyte cultures in which CYP3A mRNA was induced by PB in the absence of additional extracellular matrix, or compounds like dexamethasone, a well known inducer of different CYPs, reported by others as essential to detect PB induction at the mRNA level in cultured rat or mouse hepatocytes (Salonpää et al., 1994;Sidhu and Omiecinski, 1995b; Honkakoski and Negishi, 1998). Sidhu and Omiecinski (1995b) described an additive effect on CYP3A induction when using both PB and dexamethasone compared with the individual action of each agent. These effects were already evident for dexamethasone concentrations of 1 nM, and PB concentrations of 1 and 2 mM, and were dramatically enhanced with increasing concentrations of dexamethasone. The present results showed that under our conditions of maximum induction by PB, dexamethasone had no additive effect. This observation, together with numerous lines of evidence showing that glucocorticoids can regulate various transduction pathways (Vintermyr et al., 1993; Matsunaga et al., 1994) and thus may interfere with PB signaling, led us to avoid the use of this molecule.
To get focused insights into transduction of the PB signal, we tested several inhibitors or activators of different kinases and phosphatases. Tyrosine kinase and PKC inhibitors showed no effect on the PB-induced expression of CYP3A. Activation of PKA has been reported to play a positive role in PB induction of the CYP2a5 gene in mouse hepatocytes (Salonpää et al., 1994). This pathway has also been involved in CYP2B and CYP3A induction by PB in rat cultured hepatocytes, but contradictory data have been reported. Sidhu and Omiecinski (1995a) found an inhibition of this induction by activators of PKA whereas Brown et al. (1997) described the opposite effect for this type of molecule. The in vitro induction of CYP3A31 by PB in Syrian hamster hepatocytes is also regulated by a cAMP-dependent pathway (Bani et al., 1998). Some authors suggested that the PKA pathway may not be involved predominantly in the regulation of CYP2B mRNA expression induced by PB in cultured mouse primary hepatocytes (Honkakoski and Negishi, 1998), in contradistinction with our results with PKA activators and inhibitors. Although the effects of 8-br-cGMP were not clear, the selective and potent PKG inhibitor Rp-8-Br-PET-cGMPS markedly suppressed CYP3A induction, thus suggesting that PKG might also take part in the regulatory mechanism of PB induction of CYP3A gene expression.
In our study, low concentrations of db-cAMP produced a more repressive effect on the PB-induced CYP3A expression than when tested at higher concentrations. The observation that high doses seem to be ineffective could be explained by a negative feed-back regulatory mechanism on the PKA, elicited either by inhibition of the expression of the PKA catalytic subunit, a process which takes place during long-term elevation of cAMP in hepatocytes (Houge et al., 1990), or by the regulation of other enzymes dependent on cyclic nucleotides such as phosphodiesterases or cyclases. Forskolin has been reported previously to induce basal CYP3A mRNA expression in rat and hamster cultured hepatocytes (Sidhu and Omiecinski, 1996; Bani et al., 1998). In our model, induction of this expression was barely detectable. In the present case, and unlike the effects of db-cAMP, inhibition of PB-induced CYP3A was more efficient at a higher (50 μM) than at a lower concentration (5 μM). This result agrees with the fact that at lower concentrations, activation of adenylate cyclase by forskolin produces less cAMP, leading to a weaker activation of PKA.
PKG is responsible for phosphorylation reactions in different cell types, particularly smooth muscle cells and platelets (Butt et al., 1995) as well as hepatocytes, where it mediates enhancement of intracellular Ca2+ concentration (Rooney et al., 1996). Its activation can be produced by either autophosphorylation or cGMP binding, through an apparently similar conformational change in the enzyme (Chu et al., 1998). The levels of cAMP and cGMP control transduction pathways involving PKA and PKG. cAMP is thought to participate directly in mechanisms of gene transcription, via PKA, by phosphorylation of the cAMP response element-binding protein (CREB;Gonzalez and Montminy, 1989). In the case of CYP3A, Bani et al. (1998)have recently reported that increasing amounts of transfected CREB or CREB-binding proteins in hamster hepatocytes reduced the PB induction of CYP3A31 mRNA expression, suggesting regulation by a cAMP-dependent pathway. The involvement of such a protein in the regulation of CYP3A expression has yet to be tested in mouse hepatocytes. CREB has also been described to be activated by other second messengers such as Ca2+, and it appears that CREB can respond to both PKA and CaM PK pathways; the latter kinase could thus mediate the Ca2+ response of CREB (Sheng et al., 1991; Dash et al., 1991). In the present work, inhibition of CaM PK, or cell treatment with an intracellular calcium chelator (BAPTA-AM), resulted in an important inhibition of PB induction of the CYP3A gene, suggesting the involvement of Ca2+ in the mechanism.
The modulation of Ca2+ transport and intracellular concentration by cyclic nucleotide-dependent cross-activation of protein kinases has been thoroughly investigated in vascular smooth muscle cells (Ruiz-Velasco et al., 1998). In these studies, it has been reported that cAMP elevations might lead to profound changes in Ca2+ metabolism through activation of both PKA and PKG. It has also been suggested that PKGs would mediate the reduction in Ca2+ levels elicited upon elevation of intracellular cGMP (Cornwell and Lincoln, 1989). However, in cultured rat hepatocytes, it has been recently demonstrated that cGMP analogs, i.e., 8-br-cGMP and dibutyryl-cGMP (db-cGMP), produce oscillatory increases in intracellular Ca2+ concentration, according to a mechanism in which only PKG would be involved (Rooney et al., 1996). In agreement with this hypothesis, our results might therefore point to a complex and antagonistic involvement of all of these second messengers and transduction pathways in the mechanism of PB-induction of CYP3A mRNA expression (Fig. 7). Thus, specific inhibition of PKA led to the enhancement of induced CYP3A expression, whereas the use of a specific inhibitor of CaM PK, KN-62, inhibited PB induction of this gene, an effect also produced by the specific inhibition of PKG. In this respect, the negative effect of Rp-8-Br-PET-cGMPS could be related to a reduction of intracellular Ca2+ levels, opposite conditions to those under which PB seemed to exert its induction on CYP3A mRNA expression. The contradictory fact that an activator and an inhibitor of PKG produced the same effect on the PB-induced CYP3A mRNA expression could be explained by the inhibition that continuous exposure to analogs of cyclic nucleotides cGMP and cAMP produces on type I PKG expression (Soff et al., 1997), together with the cross-activation of PKA and PKG signal pathways.

Schematic representation of the different regulatory pathways possibly involved in the induction by PB of CYP3A mRNA expression in mouse cultured hepatocytes. PKG and CaM PK regulate this induction in a positive manner. Both pathways might be related as PKG could be directly involved in the enhancement of intracellular Ca2+ needed to activate CaM PK. By contrast, activation of PKA exerts a negative regulation on PB induction of CYP3A, which would be either direct or mediated by down-regulation of intracellular Ca2+ levels.
Interestingly, other candidate regulatory proteins could play a role in the control of PB-induced CYP3A gene expression, such as the recently described nuclear orphan CAR receptor, atrans-activator shown to be important for the PB induction of mouse CYP2b10 (Honkakoski et al., 1998) as well as humanCYP2B6 and CYP3A4 (Sueyoshi et al., 1999) genes. This novel DNA-binding protein could very well be a major target of one or more of the pathways analyzed in the present study. In addition, PKA and cAMP activators have been recently shown to regulate activity of the hepatocyte nuclear factor 4 orphan receptor, which might result in the transcriptional inhibition of several CYP genes (Viollet et al., 1997).
CYP3A protein expression in our culture model was induced by PB. The regulation of this inductive response could be mediated by cyclic nucleotides and PKA/PKG, which produced negative effects similar to those described for mRNA expression. In this study, the adenylate cyclase activator, forskolin, had a different effect, as it enhanced basal expression of CYP3A protein. This effect could be predominant over the inhibitory one such that activation of PKA produces no effect on induction of this protein by PB, thus underlying the lack of inhibition when treating cells simultaneously with both forskolin and PB. Similarly, KN62 showed no effect on PB-induced CYP3A protein expression in cultured mouse hepatocytes, contrary to the effect on the mRNA. It is, however, possible that protein induction was blocked by KN-62, the suppressive effect being masked by the increase in protein level upon treatment by KN-62 alone.
In conclusion, our results suggest that, in mouse hepatocytes, CYP3A mRNA induction by PB observed under our culture conditions is regulated by a complex mechanism in which different regulatory pathways have counteracting effects. PKG and CaM PK modulate induction positively whereas PKA mediates a negative regulation. The possible involvement of PKG in the regulation of CYP gene induction by PB is suggested here for the first time.
Footnotes
-
Send reprint requests to: Dr. D. Lagadic-Gossmann, INSERM U456, Détoxication et Réparation Tissulaire, Facultédes Sciences Pharmaceutiques et Biologiques, Université de Rennes I, 2, Avenue du Professeur Léon Bernard, 35043 Rennes cedex, France. E-mail: Dominique.Lagadic{at}rennes.inserm.fr
-
↵1 This work was supported by the Institut National de la Sante et de la Recherche Medicale and European Economic Community contract EUROCYP: BMH4-CT96–0254. M.G. is a recipient of a postdoctoral fellowship from the “Universidad de Granada” (Spain). N.M. is a recipient of a fellowship from the “Ministère de la Recherche et de l’Enseignement Supérieur” (France).
-
↵2 Present address: Institut National de la Sante et de la Recherche Medicale U517, Faculté de Médecine, 7 Boulevard Jeanne d’Arc, 21033 Dijon Cedex, France.
- Abbreviations:
- CYP
- cytochrome P-450
- PB
- phenobarbital
- cAMP
- cyclic AMP
- PKA
- cAMP-dependent protein kinase
- PKG
- cGMP-dependent protein kinase
- CaM PK
- Ca2+/calmodulin-dependent protein kinase
- PKC
- protein kinase C
- MAP
- mitogen-activated protein
- Rp-8-Br-PET-cGMPS
- β-phenyl-1,N2-etheno-8-bromoguanosine-3′,5′-cyclic monophosphorothioate, Rp-isomer
- db-cAMP
- N6,O2-dibutyryl-cAMP
- PKAI
- protein kinase A inhibitor
- ITS
- 5 mg/l insulin, 2.25 mg/l transferrin, 2.5 mg/l sodium selenite
- SSC
- standard sodium citrate
- TBS
- Tris-buffered saline
- CREB
- cAMP response element-binding protein
- Received March 31, 1999.
- Accepted May 18, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}