


Abstract
The significance of the human multidrug resistance gene (MDR1) G1199A polymorphism, resulting in a Ser400Asn modification in P-glycoprotein (P-gp), remains unclear. We have developed stable recombinant LLC-PK1 epithelial cells expressing either MDR1wt or MDR11199 to evaluate functional consequences of G1199A [N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide]. P-gp activity observed in MDR1wt and MDR11199 cells was completely inhibited in the presence of the specific P-gp inhibitor GF120918. Comparable expression of mRNA and protein in the MDR1-expressed cells and correct localization of P-gp in the apical membrane of recombinant cells was verified. Mean intracellular rhodamine-123 (R123) accumulation, measured by flow cytometry, was approximately 4.75-fold higher in MDR11199 recombinant cells than MDR1wt cells. Cytotoxicity studies have shown that MDR1wt and MDR11199 cells exhibited similar resistance, as measured by EC50 values, to doxorubicin (155 ± 68 versus 120 ± 32 nM); however, MDR11199 cells were more resistant to vinblastine (1.41 ± 0.51 versus 15.7 ± 4.0 nM; p < 0.001) and vincristine (1.18 ± 0.56 versus 3.41 ± 1.47 nM; p < 0.05). The apparent transepithelial permeability ratios of R123 in MDR1wt and MDR11199 cells were 3.54 ± 0.94 and 2.02 ± 0.51 (p < 0.05), respectively. Therefore, the G1199A polymorphism alters the efflux and transepithelial permeability of a fluorescent substrate and sensitivity to select cytotoxic agents, which may influence drug disposition and therapeutic efficacy of some P-gp substrates.
The human multidrug resistance gene (MDR1) encodes a 170-kDa integral membrane protein that mediates ATP-dependent substrate efflux. The protein product, P-glycoprotein (P-gp), a member of the ATP-binding cassette superfamily of transporters, resides in the plasma membrane and functions as an efflux transporter of a variety of natural compounds and lipophilic xenobiotics (for review, see Lin, 2003). Although the contribution of P-gp in multidrug resistance for cancer chemotherapy is well documented, the role of P-gp in drug disposition is not fully understood and has continued to generate significant debate. P-gp mediates the energy-dependent efflux of a broad range of xenobiotics in epithelial tissues throughout the human body, including the intestinal mucosa, liver canalicular membrane, kidney proximal tubules, blood-brain barrier, and placenta (Schinkel, 1997). P-gp efflux may, therefore, act to decrease intestinal absorption, enhance biliary excretion and renal tubular secretion, and limit drug distribution to the fetus and brain. Because P-gp is found in tissues important in drug disposition, variation in expression and function of P-gp due to genetic polymorphisms of MDR1 may influence pharmacokinetics and, in turn, pharmacodynamics.
Recent progress has been made in identifying genetic polymorphisms in the MDR1 gene in normal human tissues. The first major screen of the MDR1 gene in 188 healthy Caucasian subjects, identified 15 single nucleotide polymorphisms; however, only one, a C → T transition at nucleotide position 3435 (C3435T), was shown to correlate with decreased intestinal P-gp expression and digoxin exposure in vivo (Hoffmeyer et al., 2000). Because the C3435T polymorphism in exon 26 is a synonymous polymorphism that does not modify the amino acid sequence of P-gp, several investigators have searched for clues to the significance of C3435T. Another study reported that C3435T is linked to a nonsynonymous G2677T polymorphism, resulting in an alanine-to-serine transition at amino acid 893, and another synonymous SNP, C1236T (Kim et al., 2001). Several other studies have attempted to link MDR1 polymorphisms, particularly C3435T, to changes in P-gp expression and function, and subsequent changes in drug disposition profiles (Hitzl et al., 2001; Sakaeda et al., 2001; Tanabe et al., 2001; von Ahsen et al., 2001; Calado et al., 2002; Drescher et al., 2002; Fellay et al., 2002; Gerloff et al., 2002; Goto et al., 2002; Horinouchi et al., 2002; Kurata et al., 2002; Min and Ellingrod, 2002; Moriya et al., 2002; Nakamura et al., 2002; Pauli-Magnus et al., 2002; Siegmund et al., 2002; Oselin et al., 2003; Verstuyft et al., 2003). However, much of the data has been contradictory and inconclusive as to the influence of MDR1 SNPs and haplotypes on P-gp expression and in vivo drug disposition. A systematic study, designed to address genetic variants at the cellular and molecular level, is needed to define the functional significance of and the linkage between genetic polymorphisms of MDR1 and their impact on clinical pharmacokinetics.
Therefore, we have developed a recombinant expression system in epithelial cells capable of expressing P-gp variants in a reproducible manner to systematically study the influence of genetic polymorphisms in MDR1. Thus far, in vitro studies in the literature to evaluate the influence of MDR1 polymorphisms on P-gp efflux have produced variable results. Discrepancies exist in the reported influence of G2677T and C3435T on P-gp activity in various vector and expression systems (Kim et al., 2001; Kimchi-Sarfaty et al., 2002; Morita et al., 2003). In addition, some transient expression systems used to evaluate changes in activity cannot be used to evaluate transepithelial transport, which is important in assessing drug uptake and permeability. Because linkage to C1236T and G2677T complicates the influence of C3435T, we have instead chosen to evaluate the functional role of the G1199A polymorphism as a model for our recombinant P-gp expression system. MDR1 G1199A, which results in a serineto-asparagine transition at amino acid 400 in a cytoplasmic domain of P-gp, has a reported allelic frequency of approximately 5.5% in Caucasians (Hoffmeyer et al., 2000; Cascorbi et al., 2001; Kim et al., 2001). Thus far, G1199A has not been shown to be linked to other SNPs. The goal of this study was to develop a recombinant MDR1 expression system to evaluate the functional significance of G1199A in epithelial cells. We found that the G1199A polymorphism alters efflux and transepithelial transport as well as altering the sensitivity profiles of cells to cytotoxic drugs.
Materials and Methods
Cell Culture. LLC-PK1 control and transfected cells were grown in complete media consisting of RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% (v/v) fetal calf serum and 1% (v/v) antibiotic-antimycotic and grown at 37°C in the presence of 5% CO2. For deconvolution immunofluorescent microscopy and transepithelial transport studies, cells were grown in Medium 199 (Invitrogen) supplemented with l-glutamine, 10% (v/v) fetal calf serum, and 1% (v/v) antibiotic-antimycotic and grown at 37°C in the presence of 5% CO2.
Generation of MDR1wt and MDR11199 Plasmids. Total RNA was extracted from the MDR1-overexpressing cell line, MES-SADX5, and full-length MDR1 cDNA was generated using the high-fidelity protocol developed previously (Yang et al., 2002). The isolated MDR1 cDNA was cloned into a linearized pCR3.1 TA vector (Invitrogen) containing cytomegalovirus and T7 promoters capable of transcription, also described previously (Yang et al., 2002). The plasmid stock was designated as MDR1wild-type (MDR1wt).
The plasmid containing the G1199A polymorphism was generated by subcloning a fragment containing the polymorphism into the MDR1wt plasmid using the restriction enzyme HindIII. This strategy employed two PCR steps to insert the 1199A variant and is further detailed in Fig. 1. Two pairs of primers were designed with an online primer design software program (Primer3) based on an MDR1 sequence available in the GenBank database (accession no. M14758). The first pair overlapped the G1199A region with the 1199A variation in the middle of the oligonucleotide: 5′ CAGAAATGTTCACTTCAATTACCCATCTCGAAAAG 3′ (residues 1182-1216; forward primer) and 5′ TTTCGAGATGGGTAATTGAAGTGAACATTTCTG 3′ (residues 1182-1214; reverse primer). The other set of primers overlapped the restriction enzyme sites for HindIII, one recognition site in the pCR3.1 vector, 5′ GTTTAACTTAAGCTTGGTACC 3′ (residues -697-674; forward primer), and one site in MDR1 cDNA, 5′ GGTACTAAGCTTTCTGTCTTGG 3′ (residues 2031-2052; reverse primer). The first PCR step generated two fragments containing 1199A at one end of the fragment and the HindIII site at the other end. The two fragments were annealed together and amplified using the HindIII primers. The amplified PCR fragment and the pCR3.1-hMDR1 plasmid were digested with HindIII and annealed together. Clones were screened by restriction enzyme mapping with PstI and EcoRI and the sequence was verified using Big-Dye 3.0 chemistry (Applied Biosystems, Foster City, CA) and an ABI Prism 377 DNA Sequencer (Applied Biosystems). The variant plasmid stock was designated MDR11199.

Schematic representation of the generation of the MDR11199 plasmid. The MDR11199 plasmid was generated by introducing a 1199A variant into the original MDR1wt plasmid. The first step in generating the MDR11199 plasmid used the HindIII and 1199A primer sets to generate two fragments each containing 1199A at one end and a HindIII site at the other from the original MDR1wt plasmid. Next, the fragments were annealed and amplified using the HindIII primer set. Finally, the fragment containing 1199A and the pCR3.1-hMDR1 vector were digested with HindIII and religated. The MDR11199 plasmid was validated by restriction enzyme and sequencing analyses. MDR1wt and MDR11199 plasmids contain T7 and cytomegalovirus promoters and ampicillin and neomycin resistance genes.
Isolation of Stable Recombinant Cells Expressing MDR1wt or MDR11199. The MDR1wt and MDR11199 plasmids were transfected into the porcine kidney epithelial cell line LLC-PK1. Ten million cells were resuspended in complete media and transferred to a 0.4-cm electroporation cuvette (Bio-Rad, Hercules, CA) with 10 μg of plasmid DNA (either MDR1wt or MDR11199). Electroporation was performed at 230 V and 975 μF in a Gene Pulser II (Bio-Rad). Cells were placed on ice for 10 min and cultured in complete media for selection. Transfected LLC-PK1 cells with the highest level of P-gp expression were selected to generate stable cell lines expressing MDR1wt or MDR11199. Cells were treated for 3 days with 300 μg/ml G418, a neomycin derivative (Calbiochem, San Diego, CA). A fraction of the cells were briefly exposed for another 3 days to 5 μM doxorubicin (VHA PLUS; VHA Inc., Irving, TX), a cytotoxic P-gp substrate, and continued selection by G418 pressure. The cells expressing high levels of P-gp were further expanded and verified to have stable expression, allowing for functional evaluation.
Characterization of P-gp Expression. Absolute quantitation of MDR1 mRNA transcripts in cellular samples was performed using the ABI Prism 7900HT sequence detection system (Applied Biosystems). An MDR1 RNA standard was generated by the method described previously, and concentration was measured by absorbance at 260 nm and converted to the number of copies of MDR1 RNA by the molecular weight (Yang et al., 2002). A dilution series of the MDR1 RNA standard was used to generate a standard curve of the number of copies of MDR1 mRNA versus CT value. After RNA isolation, 100 ng of total RNA of each cellular sample was analyzed in triplicate to obtain a CT value and estimation of the number of copies of MDR1 mRNA.
Immunoblot analysis was used to detect protein expression. Briefly, 2 × 106 cells were pelleted and lysed in a lysis buffer containing SDS and β-mercaptoethanol. Protein concentration was measured by a microplate assay protocol (Bio-Rad). The P-gp-positive control is derived from the MES-SA-DX5 cell line known to highly express P-gp. Electrophoresis and transfer was run according to instructions for Mini-PROTEAN II Electrophoresis Cell and Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad). Nitrocellulose blots were blocked with 5% evaporated milk in 0.1% Tween 20, 20 mM Tris-HCl, 0.9% NaCl, pH 7.6, buffer. Immunoblotting was performed with the anti-P-gp monoclonal antibody F4 (Sigma-Aldrich, St. Louis, MO) followed by a secondary horseradish peroxidaseconjugated goat anti-mouse IgG. Enhanced chemiluminescence reagents were used as a substrate and blots were exposed to X-ray film for visualization of the protein bands.
Cell surface expression of P-gp was analyzed by flow cytometry. Briefly, 5 × 105 cells were plated overnight on six-well plates (Corning Glassworks, Corning, NY). Cells were washed with phosphate-buffered saline with 1% bovine serum albumin (PBS/BSA) and detached after 10-min incubation with 2 mM EDTA in PBS. Cells were incubated with either anti-P-gp monoclonal antibody F4 or a matched mouse isotype control for 30 min on ice, followed by three washes with cold Hanks' balanced salt solution supplemented with 0.5% BSA and 0.1% NaN3 (HBSS-BSA-NaN3). Cells were then incubated with phycoerythrin (PE)-conjugated goat anti-mouse IgG (Calbiochem) for 30 min on ice in the dark, again followed by three washes with HBSS-BSA-NaN3. Cells were resuspended in HBSSBSA-NaN3, and the fluorescence intensity of PE-conjugated antibody was analyzed with a FACScan flow cytometer (BD Biosciences, Franklin Lakes, NJ) using CellQuest software (BD Biosciences) to evaluate cellular surface expression of P-gp. Analysis was made comparing anti-P-gp monoclonal antibody F4 to the isotype control to account for nonspecific binding.
For deconvolution immunofluorescent microscopy, 4 × 105 cells were plated on glass chamber slides (eight-well; Lab-Tek; Nalge Nunc International, Rochester, NY) and grown for 4 days. Cells were fixed with 2% paraformaldehyde in PBS (pH 7.4) and permeabilized with 0.2% Triton X-100. Blocking was performed with 2% goat serum in PBS/BSA. Slides were immunostained with anti-P-gp monoclonal antibodies F4 (Sigma-Aldrich) and C219 (Calbiochem) followed by goat anti-mouse secondary antibody, Alexa-594 (Molecular Probes, Eugene, OR). Slides were mounted with Fluoromount-G (Southern Biotechnology Associates, Birmingham, AL), and images were collected with an Axiovert 200 MAT deconvolution microscope (Carl Zeiss, Oberkochen, Baden-Wuerttemberg, Germany) with a 63× oil immersion objective lens, and analysis was performed with Slide-Book 3.0 software (Intelligent Imaging Innovation, Denver, CO).
Intracellular Accumulation Assay. One million cells per well were plated overnight on six-well plates (Corning Glassworks). For rhodamine-123 (R123) accumulation, cells were washed with PBS and incubated for 90 min in 5 μM R123 (Sigma-Aldrich) in serum-free RPMI 1640 medium at 37°C. Cells were again washed with PBS and then allowed to efflux for 60 min in complete media. After efflux, cells were washed in ice-cold PBS, trypsinized, and resuspended in 1% paraformaldehyde. Cells were analyzed with a FACScan flow cytometer (BD Biosciences) using CellQuest software (BD Biosciences). Ten thousand cells from each sample were analyzed for forward scatter, side scatter, and R123 accumulation.
Inhibition analyses were performed with the specific inhibitor GF120918 [N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide], kindly provided by Kenneth Brouwer (GlaxoSmithKline, Research Triangle Park, NC). To determine concentration necessary for 50% inhibition (IC50) values of GF120918, cells were exposed to a range of GF120918 concentrations from 2 nM to 2 μM in quadruplicate. The ability of cells to efflux R123 in the presence of GF120918 was used to estimate IC50 values based on a sigmoid Emax model on WinNonlin software (Pharsight, Mountain View, CA). Complete inhibition studies were performed at 1 μM GF120918.
Doxorubicin accumulation was performed in quadruplicate by incubating cells with 10 μM doxorubicin in PBS with 0, 20, 100, or 1000 nM GF120918 for 1 h at 37°C. Cells were washed with ice-cold PBS, trypsinized, and lysed in 0.5% deoxycholic acid. To precipitate the proteins, tricholoracetic acid was added to 5%, and the cell lysates were incubated for 30 min at 4°C and centrifuged at 20,000g. The supernatant was removed and fluorescence intensity of doxorubicin was measured with a fluorescence spectrophotometer F-4500 (Hitachi Instruments, San Jose, CA); excitation was set at 488 nm and emission at 560 nm.
Cytotoxic Drug Sensitivity Assay. LLC-PK1 control and MDR1 recombinant cells were plated at a density of 1 × 104 cells/well in a 96-well plate (Corning Glassworks) in complete media and allowed to attach for 4 h at 37°C. Varying concentrations of doxorubicin, vinblastine (Bedford Laboratories, Bedford, OH), and vincristine (Faulding, Paramus, NJ) were added to the cells in quadruplicate. Cells were incubated with the cytotoxic drugs for 3 days at 37°C. On the second day of the incubation, 1 μCi of [3H]thymidine (specific activity 81.1 Ci/mmol) was added to each well and incubated overnight. Cells were harvested on day 3 with a PHD cell harvester (Cambridge Technology Inc., Watertown, MA), and 3H radioactivity was measured with a 1600 TR liquid scintillation analyzer (Can-berra Industries, Zellik, Belgium). Cytotoxicity was measured as the effective concentration necessary for 50% cell death (EC50) for each drug; EC50 values were estimated using a sigmoid Emax model on WinNonlin software (Pharsight).
Transepithelial Transport Assay. LLC-PK1 control and recombinant MDR1 cells were plated at a density of 2 × 106 cells/24-mm well on permeable supports (Transwell; 3.0-μm membrane pore size; Corning) and grown for 4 days; media were refreshed after 2 days in culture. Fresh media were added to the cells 1 h before the initiation of the experiment, and transepithelial electrical resistance values were measured with a Millicell-ERS (Millipore, Billerica, MA). For transport of R123 across the epithelial cells, 1 μM R123 in Opti-MEM (Invitrogen) was added to either the apical or basolateral compartments with fresh Opti-MEM medium added to the opposite side. Inhibition was performed at 1 μM GF120918. Aliquots of 50 μl were taken from apical and basal compartments at 1, 2, 3, and 4 h. Fluorescence intensity of R123 was measured with a Gemini XS microplate spectrofluorometer (Molecular Devices, Sunnyvale, CA) with SoftMax Pro software (Molecular Devices); excitation was set at 488 nm and emission at 525 nm. Apparent permeability (Papp) was calculated in the apical-to-basolateral direction (PappA→ B) and in the basolateral-to-apical direction (PappB→A) as described previously (Polli et al., 2001). Briefly, Papp = (1/(A × C0)) × (dQ/dt), where A is the surface area of permeable support, C0 is the initial concentration in the donor compartment, and dQ/dt is the rate of transfer of compound into the acceptor compartment. The ratio of PappB→A/PappA→B was also estimated to evaluate P-gp-mediated directional efflux.
Statistical Analysis. Student's two-sided t test was used to evaluate differences between two sets of data. P values <0.05 were considered statistically significant.
Results
Generation of Cells Expressing Recombinant MDR1wt or MDR11199. We have successfully constructed MDR1 vectors for mammalian cell expression containing either the wild-type or variant allele at nucleotide position 1199. The final plasmid constructs were verified by directly sequencing the MDR1 insert (data not shown). The introduction of the 1199A polymorphism from a wild-type plasmid was based on a restriction enzyme approach and is described in Fig. 1 (additional details are described under Materials and Methods). These plasmids, referred to as MDR1wt and MDR11199, were used for generating stable recombinant cells for functional studies.
We chose LLC-PK1 cells as the host to isolate stable epithelial cells for continuous expression of MDR1. These cells differentiate to form polarized monolayers for transepithelial efflux studies. As mammalian cells, LLC-PK1 should provide proper post-translational modification, which has been demonstrated to be important in targeting of P-gp to the plasma membrane and P-gp function (Loo and Clarke, 1993a,b, 1994a,b). To further reduce the baseline levels of P-gp-like activity in these cells, we have selected and cloned a line of LLC-PK1 cells that were P-gp negative based on their retention of R123 and sensitivity to the cytotoxic P-gp substrate doxorubicin. P-gp-deficient LLC-PK1 cells were transfected with respective plasmids by electroporation, and systematically selected for high levels of P-gp expression. The recombinant cells expressing MDR1wt or MDR11199 in a stable and consistent manner were cloned and expanded for functional studies. Direct sequencing was performed to verify expression of the correct allele at nucleotide position 1199 (Fig. 2). P-gp activity was maintained for more than 100 days in culture with greater than 95% of MDR1wt and MDR11199 recombinant cell populations excluding R123, as measured by flow cytometry. These cells also retained P-gp activity after being frozen in liquid nitrogen and thawed.

Sequence verification of LLC-PK1 cells expressing MDR1wt or MDR11199. Recombinant MDR1-expressing LLC-PK1 cells were sequenced at nucleotide position 1199; wild-type allele G and variant allele A. Electropherograms of MDR1wt (A) and MDR11199 (B) are shown; arrows indicate nucleotide position 1199.
Characterization of P-gp Expression in Recombinant LLC-PK1 Cells. After transfection and selection of control and variant 1199 plasmids, it was important to verify MDR1 mRNA and protein expression. The level of MDR1 mRNA transcripts in MDR1 recombinant cells was assessed using an MDR1 standard that was generated with known copy number. MDR1 copy number values were similar in MDR1wt and MDR11199 recombinant cells with estimations of 2.90 × 106 ± 0.30 and 3.51 × 106 ± 0.30 copies of MDR1 mRNA/100 ng total RNA, respectively. Immunoblot analyses of LLC-PK1 cells, normalized by protein concentration, showed approximately equal expression of 150- and 170-kDa bands of P-gp in MDR1wt and MDR11199 cells (Fig. 3A, lanes 3 and 4). In addition, cell surface expression of P-gp, analyzed by flow cytometry with an anti-P-gp monoclonal antibody, revealed that the amount of P-gp on the surface of MDR1wt and MDR11199 cells was similar (Fig. 3B). Image deconvolution also confirmed that MDR1wt and MDR11199 cells express similar amounts of P-gp and that expression is predominantly on the apical membrane (Fig. 4).

P-gp expression in LLC-PK1 control and recombinant MDR1 cells. Immunoblot analysis of cellular P-gp expression, normalized by protein concentration, for the visualization of 150- and 170-kDa P-gp bands (A): lane 1, P-gp-positive control isolated from the P-gp overexpressed cell line MES-SA-DX5; lane 2, LLC-PK1 control cells; lane 3, recombinant MDR1wt cells; and lane 4, recombinant MDR11199 cells. Cell surface expression evaluated by detection of a PE-conjugated secondary antibody by flow cytometry (B); MDR1wt cells (solid line) and MDR11199 cells (bold line).

Confirmation of apical membrane expression of P-gp by deconvolution immunofluorescent microscopy in recombinant MDR1wt and MDR11199 cells. LLC-PK1 control cells (A), MDR1wt cells (B), and MDR11199 cells (C) plated on glass slides. The cells are orientated such that the apical membrane is on top and the basolateral membrane is on bottom; magnification at 63×.
P-gp-Mediated R123 Efflux. Flow cytometry was used to evaluate efflux transport activity of MDR1wt and MDR11199 recombinant cells by measuring intracellular accumulation of R123, a fluorescent P-gp substrate. Mean intracellular R123 fluorescence (given in arbitrary units) for MDR1wt and MDR11199 cells were 3.91 ± 0.11 and 18.56 ± 0.46 (p < 0.001), respectively, an approximate 4.75-fold higher accumulation of R123 in MDR11199 cells (Fig. 5A). To verify that the R123 efflux observed was due to P-gp activity, we next used a specific and potent P-gp inhibitor, GF120918, for R123 efflux assessment. Although there was differential efflux observed between cells expressing MDR1wt or MDR11199, R123 efflux activity was abolished in both types of cells, reversing cellular R123 accumulation similar to that of control LLC-PK1 cells (Fig. 5, B-D). Dose-dependent analysis of the ability of GF120918 to inhibit P-gp-mediated R123 efflux in MDR1wt and MDR11199 cells was completed, and IC50 values were estimated. Similar IC50 values for inhibition by GF120918 in MDR1wt and MDR11199 were recorded as 20.3 ± 10.31 and 28.6 ± 2.1 nM, respectively. These results suggest that recombinant cells expressing MDR1wt or MDR11199 proteins are both capable of mediating P-gp-specific R123 transport activity, but cells expressing MDR11199 are less efficient than those expressing MDR1wt.
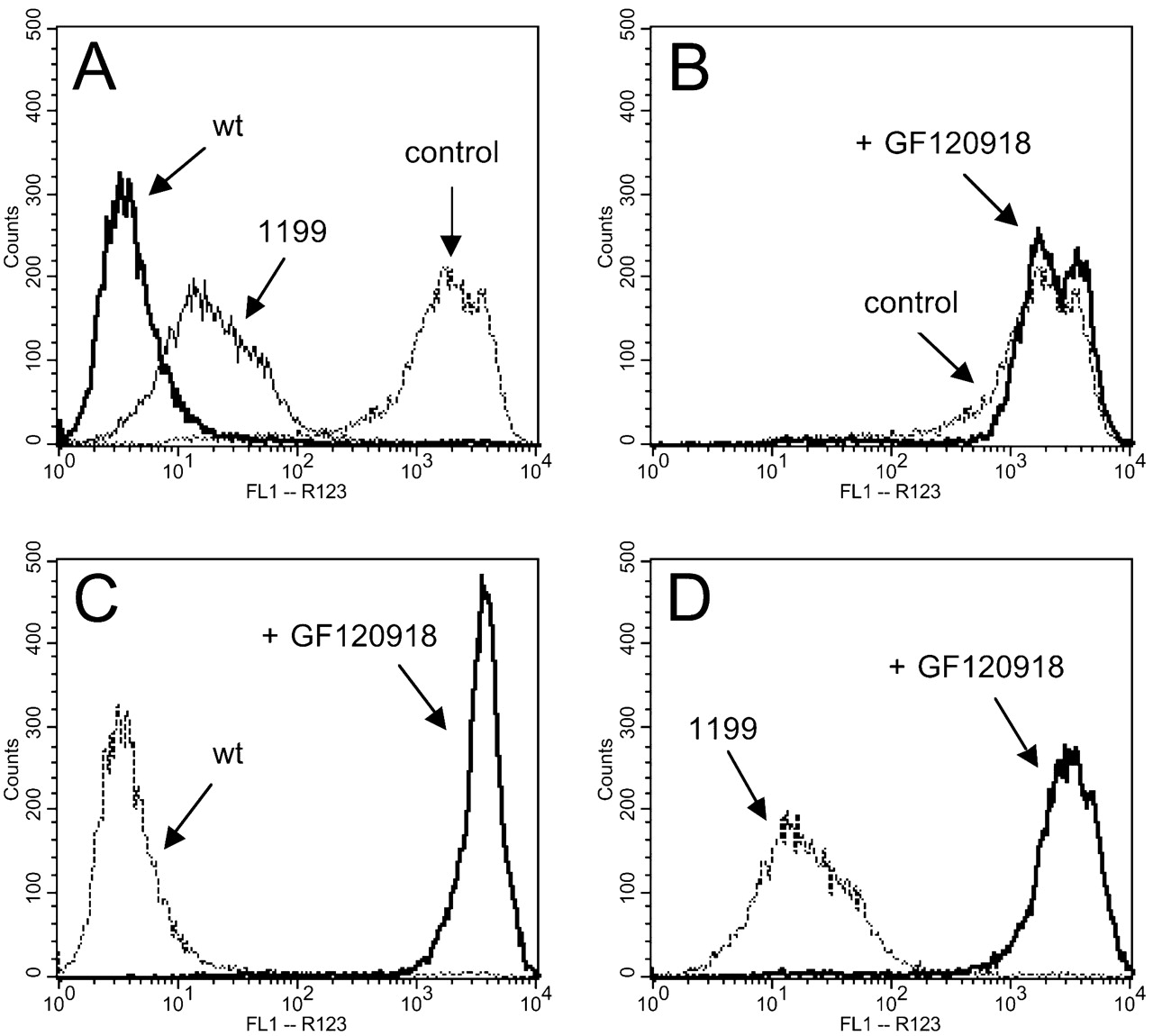
Intracellular accumulation of R123 in LLC-PK1 control and MDR1 recombinant cells. The intensity of intracellular R123 was evaluated by flow cytometry in LLC-PK1 control and MDR1 cells (A): control (dashed line), MDR1wt cells (bold line), and MDR11199 cells (solid line). The specificity of R123 efflux was evaluated in the absence (dashed line) and presence (solid line) of 1 μM GF120918: LLC-PK1 control cells (B), recombinant MDR1wt cells (C), and recombinant MDR11199 cells (D).
Sensitivity to Cytotoxic Agents. To evaluate the impact of differential efficiency in efflux activity of cells expressing MDR1wt or MDR11199, we used three cytotoxic chemotherapeutic agents that have been shown to exhibit P-gp-dependent drug resistance. Dose-response analysis was performed with P-gp substrates doxorubicin, vincristine, and vinblastine; these compounds were chosen because they have been shown to exhibit differential sensitivities across various MDR1 mutant cells generated by site-directed mutagenesis (Loo and Clarke, 1993a,b, 1994a,b). Drug sensitivities were estimated by EC50 values for each cytotoxic drug in LLC-PK1 control and MDR1wt and MDR11199 cells and are presented in Table 1. Recombinant cells expressing MDR1wt or MDR11199 exhibit significantly enhanced resistance to doxorubicin compared with the control cells, but their sensitivity to doxorubicin seems to be similar. A significant degree of resistance toward vinblastine and vincristine was also observed for both MDR1wt and MDR11199 cells; however, we found that cells expressing MDR11199 were more resistant to vinblastine and vincristine (about 11- and 2.9-fold, respectively; Table 1) than those expressing MDR1wt. Collectively, these data suggest that the P-gp product derived from MDR11199 exhibits differential sensitivity to vincristine and vinblastine compared with MDR1wt without significantly altering sensitivity to doxorubicin.
Sensitivities of LLC-PK1 control and recombinant MDR1 cells to cytotoxic P-gp substrates measured by EC50 values
Intracellular levels of doxorubicin were also investigated to ensure that the observed drug resistance in recombinant MDR1 cells was due to efflux transport of doxorubicin mediated by P-gp. Accumulation of intracellular doxorubicin was measured in the presence of 0, 20, 100, and 1000 nM GF120918 in MDR1wt and MDR11199 cells (Fig. 6). The presence of GF120918 had no effect on the accumulation of doxorubicin in LLC-PK1 parental cells used as a negative control. P-gp-specific inhibition by GF120918 abolished doxorubicin efflux in both MDR1wt and MDR11199 cells to a similar degree across inhibitor concentrations. Together, these data suggest that expression of both MDR1wt and MDR1199 in LLC-PK1 cells greatly reduces the intracellular concentration of the P-gp substrate doxorubicin, activity that is completely blocked by a specific P-gp inhibitor. Therefore, it is likely that drug resistance in these cells is due to P-gp expression.
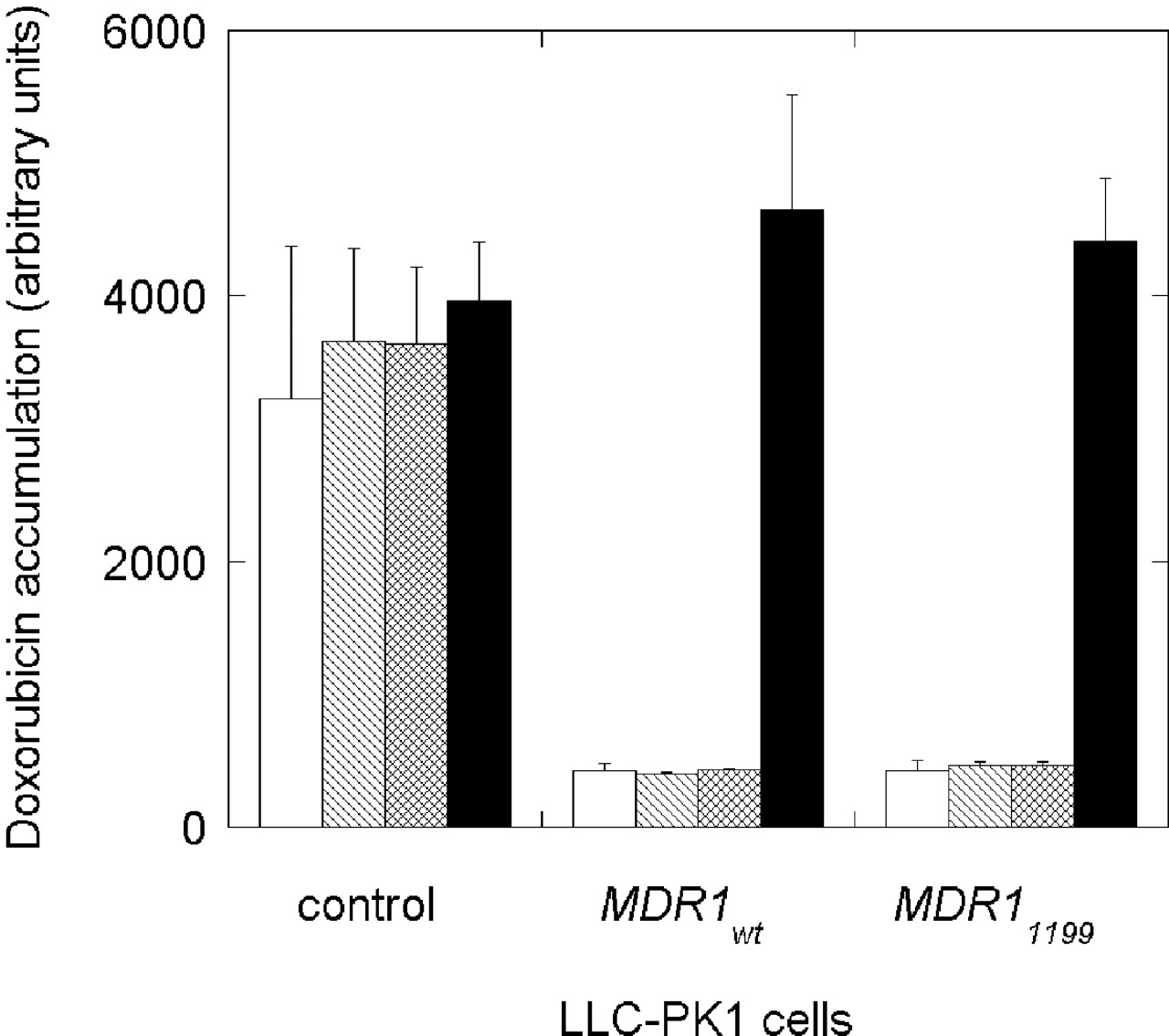
Intracellular accumulation of doxorubicin in LLC-PK1 control and MDR1 recombinant cells. The intracellular doxorubicin fluorescence was evaluated in the absence of GF120918 (open column) and presence of 20 nM (hatched column), 100 nM (crossed column), and 1000 nM (black bar) GF120918, in quadruplicate.
Transepithelial Transport. Differences in transcellular directional permeability in epithelial cells expressing MDR1wt and MDR11199 were evaluated by growing cells on a filter supports, providing for the establishment of polarized monolayers and tight junctions. P-gp is a directional transporter expressed on the apical membrane of epithelial cells and effluxes substrates in a basolateral-to-apical direction. transepithelial electrical resistance values, verifying the integrity of the monolayers and formation of tight junctions before an experiment, were 358 ± 47.8 Ω · cm2. The directional efflux transport was evaluated with a fluorescent substrate R123. The permeability ratios (PappB→A/PappA→B) were calculated for comparison. If cells display directional efflux mediated by P-gp, a ratio greater than 1.0 is expected, and in the presence of a P-gp-specific inhibitor, GF120918, the ratio should be reduced to near 1.0. Both MDR1wt and MDR11199 cells show increased basolateral-to-apical efflux and permeability ratios compared with control cells (Fig. 7; Table 2). However, cells expressing MDR11199 seem to have decreased directional efflux and permeability ratios of R123 compared with MDR1wt. In the presence of GF120918, a specific P-gp inhibitor, directional transport is eliminated, suggesting that the directional efflux is P-gp-mediated (Fig. 7; Table 2).

Transepithelial transport across LLC-PK1 epithelial cells. Transepithelial permeability was evaluated as the amount of R123 transported across an epithelial monolayer from the donor to recipient compartment over a 4-h time period: LLC-PK1 control cells (A), recombinant MDR1wt cells (B), and recombinant MDR11199 cells (C). R123 transport in the basolateral-to-apical direction without inhibitor (•) and with 1 μM GF120918 (○) and in the apical-to-basolateral direction without inhibitor (▴) and with 1 μM GF120918 (▵).
Apparent permeability ratios for rhodamine-123 transport in LLC-PK1 control and recombinant MDR1 cells
Discussion
Due to the broad substrate specificity and localization in tissues, the importance of P-gp-mediated efflux transport in drug delivery and disposition has become increasingly appreciated. Significant progress has been made in the discovery of MDR1 polymorphisms and the assessment of allelic frequencies. The search for key genetic determinants, including MDR1 genetic polymorphisms, which alter the disposition of drugs that are substrates or inhibitors of P-gp in individuals, has just begun. Toward this end, we have developed stable recombinant LLC-PK1 cells expressing either MDR1wt or MDR11199 and evaluated the significance of the G1199A polymorphism.
Alterations in the efflux transport and chemoresistance of P-gp due to the G1199A transition have been observed in our recombinant expression system. Variations in efflux were demonstrated with the fluorescence substrate R123; cells expressing MDR11199 displayed decreased efflux of R123 compared with MDR1wt. Regardless of the efficiency of efflux, cells expressing either MDR1wt or MDR11199 were both inhibited to the same degree by a P-gp-specific inhibitor, GF120918. This indicates that the observed differences were due to the influence of an MDR1 polymorphism on P-gp function. Variation in chemoresistance due to G1199A polymorphism was also observed (Table 1). Cells expressing MDR11199 seem to be more resistant to vinblastine and vincristine than cells expressing MDR1wt; however, MDR1wt and MDR11199 recombinant cells displayed similar resistance to doxorubicin. Site-directed mutagenesis studies have also shown that modifications in the nucleotide sequence of MDR1 alter substrate efflux of some anticancer agents but do not cause a change in efflux of others (Loo and Clarke, 1993a,b, 1994a,b). Multiple binding domains in the drug binding pocket of P-gp have been proposed and may account for the drug-specific alteration in P-gp efflux due to genetic polymorphisms (Martin et al., 2000; Loo et al., 2003). The observed differential sensitivity to cytotoxic agents due to G1199A may be important in modulating the efficacy of chemotherapy. The reported results could be used to provide alternate choices of drugs to overcome chemoresistance in some cancer patients. The use of LLC-PK1 epithelial cells as host cells allows for assessment of the role of MDR1 genetic variation in P-gp-dependent directional transcellular efflux. Permeability and directional efflux transport studies with the epithelial cells demonstrated that MDR11199 recombinant cells exhibited a lower PappB→A/PappA→B ratio for R123 (Table 2). Changes in drug permeability may impact absorption, bioavailability, and distribution into target tissues, including the central nervous system.
The variations in P-gp activity due to G1199A we found in our recombinant expression system are not comparable with those reported on MDR11199 expressed in HeLa cervical adenocarcinoma cells using a vaccinia virus-based transient expression system (Kimchi-Sarfaty et al., 2002). However, the HeLa cell system cannot evaluate transcellular permeability, which is an especially important component of P-gp activity, particularly in intestinal absorption and blood-brain barrier penetration. In this transient expression system, HeLa cells expressing MDR11199 did not exhibit significant differences in efflux of bodipy-FL-verapamil, bodipy-FL-vinblastine, calcein-AM, bodipy-FL-prazosin, bisantrene, and bodipy-FL-forskolin, and daunorubicin (Kimchi-Sarfaty et al., 2002). However, the fluorescent bodipy modification on the P-gp substrates in this study may influence the ability to detect functional variability due to MDR1 polymorphisms. It is also possible that expression of some vaccinia viral elements and cytolytic viral proteins in these transient expressed HeLa cells contribute to the discordance. Our system allows for long-term expression of P-gp variants without the complication of viral elements. Whether coexpression of viral components influences P-gp function is not known and may require further investigation.
A considerable amount of contradictory reports exist in the literature as to the influence of MDR1 polymorphisms (primarily G2677T and C3435T) on P-gp expression and function at the cellular level. In vitro retroviral vectors containing MDR1 variants at 2677, designed to express Ala893 or Ser893 in NIH-3T3 mouse embryonic cells, suggested that Ser893-expressing cells exhibited decreased digoxin accumulation compared with Ala893 cells, indicating enhanced P-gp efflux (Kim et al., 2001). On the other hand, a vaccinia virus-based transient expression system of several MDR1 polymorphisms (A61G, T307C, G1199A, G2677T, and G2995A) in HeLa cells showed no difference in P-gp expression and efflux of various drugs (Kimchi-Sarfaty et al., 2002). Similarly, expression of different combinations of variations at 2677 and 3435 in LLC-PK1 cells (2677G/3435C, 2677G/3435T, 2677A/3435C, 2677A/3435T, 2677T/3435C, and 2677T/3435T) were shown to have no apparent difference in transcellular transport and accumulation of verapamil, digoxin, vinblastine, and cyclosporine (Morita et al., 2003). At present, the data in the literature provide no consistent information on the influence of MDR1 polymorphisms in vitro. There is great potential for an in vitro transport system, based on recombinant MDR1 variants stably expressed in epithelial cells, that will allow for studying the impact of MDR1 SNPs and attempt to sort out some of the controversy in the literature.
There are several reasons that can account for the discrepancies observed between the in vitro observations of various groups of researchers. As described above, multiple binding domains may exist in P-gp, and variations in the amino acid sequence of the protein may alter binding and efflux of some drugs but not others. Therefore, the selection of substrates may be important in assessing changes in P-gp activity and could explain some observed differences. Also, some vectors carry unique membrane components that may influence the activity of P-gp. The cells used to express the variant MDR1 genes can also have variable intrinsic transporters, many of which contribute to the efflux of P-gp substrates. The recombinant expression system we have developed to generate cell lines that continuously express MDR1 variants will be useful for study in detailing the complex interactions of MDR1 polymorphisms.
We now have a validated expression system consisting of MDR1 mammalian expression vectors and epithelial host cells capable of reproducibly expressing P-gp variants and differentiating into polarized monolayers. This strategy can be readily adapted to investigate other SNPs and haplotypes of MDR1 and provide insight into the functional significance on the more than 30 SNPs reported in the literature. For example, the influence of C3435T may be evaluated to determine whether this synonymous polymorphism changes expression or activity of P-gp, with which it has been reported to be associated. In addition, the effect of MDR1 haplotypes can be evaluated in this system because multiple SNPs can be expressed at once in the same plasmid. Multiple SNPs found on the same chromosome are assigned to a specific haplotype, and some attempts have been made to determine the role of MDR1 haplotypes in P-gp variability, but the data are still inconclusive (Kim et al., 2001; Johne et al., 2002; Kroetz et al., 2003). Our expression system may be useful in sorting out the role of MDR1 haplotypes.
Mammalian epithelial host cells similar to LLC-PK1, including MDCKII and Caco-2, could potentially be used for evaluating the significance of MDR1 polymorphisms (Zhang and Benet, 1998; Polli et al., 2001; Stormer et al., 2001; Williams et al., 2003). In theory, the same MDR1 and variant constructs can be used to generate recombinant expression systems with these cells. Nevertheless, the described stable recombinant system expressed in LLC-PK1 cells has provided a unique method for evaluating MDR1 genetic polymorphisms. Because LLC-PK1 recombinant cells can be grown continuously in culture for several passages, they may serve as an in vitro tool to detect potential drug-drug interactions in drug development. However, our system will not readily allow for the elucidation of the functional significance of sequence variation in the promoter and intronic sequences of MDR1 that may also influence expression of P-gp. By design, this continuous system focuses on genetic modifications in the coding region of MDR1 that alter the structure of P-gp. Other strategies specifically designed to evaluate the effects of promoter and intronic SNPs may be needed to address regulation of MDR1 expression.
In summary, we have developed a stable expression system for MDR1 in epithelial cells and demonstrated that G1199A causes functional variance in efflux transport, drug resistance, and transepithelial transport and permeability. Deconvolution microscopy studies have shown that stable recombinant cells express P-gp in the correct cellular location and orientation, which has been confirmed by transepithelial analyses for MDR1wt and MDR11199 recombinant cells. Elucidating the significance of the G1199A polymorphism is the first step. Our system may have substantial impact in providing a method to consistently express other MDR1 SNPs and haplotypes to characterize the functional significance of MDR1 polymorphisms.
Acknowledgments
We thank Tami Daley for editorial assistance and the Center for DNA Sequencing and Gene Analysis (Department of Pharmaceutics, University of Washington).
Footnotes
-
This study was supported in part by National Institutes of Health Grants GM62883, AI52663, NS39178, AI31854, ES07033, and HL56548. E.L.W. is a recipient of the National Institutes of Health Pharmaceutical Sciences Training Grant (GM07750) and the William E. Bradley Fellowship in Pharmaceutics.
-
doi:10.1124/jpet.104.065383.
-
ABBREVIATIONS:MDR1, multidrug resistance gene; P-gp, P-glycoprotein; SNP, single nucleotide polymorphism; PCR, polymerase chain reaction; PBS, phosphate-buffered saline; BSA, bovine serum albumin; HBSS, Hanks' balanced salt solution; PE, phycoerythrin; R123, rhodamine-123.
- Received January 13, 2004.
- Accepted April 20, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}