Article Text

Abstract
Objective: Ursodeoxycholic acid (UDCA) exerts anticholestatic effects in part by protein kinase C (PKC)-dependent mechanisms. Its taurine conjugate, TUDCA, is a cPKCα agonist. We tested whether protein kinase A (PKA) might contribute to the anticholestatic action of TUDCA via cooperative cPKCα-/PKA-dependent mechanisms in taurolithocholic acid (TLCA)-induced cholestasis.
Methods: In perfused rat liver, bile flow was determined gravimetrically, organic anion secretion spectrophotometrically, lactate dehydrogenase (LDH) release enzymatically, cAMP response-element binding protein (CREB) phosphorylation by immunoblotting, and cAMP by immunoassay. PKC/PKA inhibitors were tested radiochemically. In vitro phosphorylation of the conjugate export pump, Mrp2/Abcc2, was studied in rat hepatocytes and human Hep-G2 hepatoma cells.
Results: In livers treated with TLCA (10 μmol/l)+TUDCA (25 μmol/l), combined inhibition of cPKC by the cPKC-selective inhibitor Gö6976 (100 nmol/l) or the non-selective PKC inhibitor staurosporine (10 nmol/l) and of PKA by H89 (100 nmol/l) reduced bile flow by 36% (p<0.05) and 48% (p<0.01), and secretion of the Mrp2/Abcc2 substrate, 2,4-dinitrophenyl-S-glutathione, by 31% (p<0.05) and 41% (p<0.01), respectively; bile flow was unaffected in control livers or livers treated with TUDCA only or TLCA+taurocholic acid. Inhibition of cPKC or PKA alone did not affect the anticholestatic action of TUDCA. Hepatic cAMP levels and CREB phosphorylation as readout of PKA activity were unaffected by the bile acids tested, suggesting a permissive effect of PKA for the anticholestatic action of TUDCA. Rat and human hepatocellular Mrp2 were phosphorylated by phorbol ester pretreatment and recombinant cPKCα, nPKCϵ, and PKA, respectively, in a staurosporine-sensitive manner.
Conclusion: UDCA conjugates exert their anticholestatic action in bile acid-induced cholestasis in part via cooperative post-translational cPKCα-/PKA-dependent mechanisms. Hepatocellular Mrp2 may be one target of bile acid-induced kinase activation.
Statistics from Altmetric.com
Ursodeoxycholic acid (UDCA) has been used for the treatment of jaundice in Chinese traditional medicine since the Tang dynasty (618–907 ad) in the form of dried black bear’s bile. Today, UDCA represents the only drug approved by the US Food and Drug Administration for the treatment of primary biliary cirrhosis (PBC), a model cholestatic liver disease.1 UDCA improves biliary secretion in PBC and a number of other cholestatic disorders, such as primary sclerosing cholangitis (PSC) or intrahepatic cholestasis of pregnancy (ICP).2 In early-stage PBC, UDCA delays progression to cirrhosis3 4 as well as development of complications,5 and normalises life expectancy.6–8 Several mechanisms of action of UDCA have been discussed, and stimulation of impaired hepatobiliary secretion, detoxification of bile, and anti-apoptotic effects are assumed to contribute to the beneficial effect of UDCA in cholestatic disorders.9
UDCA conjugates, such as tauroursodeoxycholic acid (TUDCA), are potent signalling molecules both in hepatocytes and cholangiocytes.9 10 In hepatocytes, TUDCA has been shown to induce increases of cytosolic free calcium [Ca2+]i and Ca2+ influx,11–13 to selectively activate Ca2+-dependent conventional protein kinase Cα (cPKCα),14 15 to stimulate an integrin-dependent dual signalling pathway leading to activation of mitogen-activated protein kinases (MAPKs), Erk1/2 and p38MAPK,16–19 and to induce targeting and insertion of key apical transporters like the bile salt export pump, Bsep/Abcb11, and the conjugate export pump, Mrp2/Abcc2, into canalicular membranes of hepatocytes.17 20 21 In normal hepatocytes, MAPK-dependent mechanisms mediate, in part, the choleretic effect of TUDCA,16 17 whereas in experimental cholestasis, PKC-dependent mechanisms appear to contribute to the anticholestatic action of TUDCA.20 22
Taurolithocholic acid (TLCA) is the most potent cholestatic agent among the major human bile acids23 and has recently been shown to exert its cholestatic action at the hepatocyte level by phosphatidylinositol-3-kinase, and putatively nPKCϵ-dependent mechanisms in isolated perfused rat livers (IPRLs) and isolated rat hepatocyte couplets.24 Like TUDCA, TLCA is a potent signalling molecule which elevates hepatocellular [Ca2+]i without stimulation of Ca2+ influx,25 selectively translocates nPKCϵ to canalicular membranes and activates membrane-bound PKC,26 and induces retrieval of key apical transporters such as the bile salt export pump, Bsep/Abcb11, from canalicular membranes of hepatocytes.21
Direct effects of bile acids such as TLCA or TUDCA on PKA activity in hepatocytes have not been disclosed although glucagon-induced cAMP formation was impaired by TUDCA in a staurosporine-sensitive fashion in hamster hepatocytes.27 PKC agonists at moderate concentrations and PKA are known to stimulate liver cell secretion.10 A cooperative PKC-/PKA-dependent mechanism has recently been described to potentiate chloride secretion via the Xenopus cystic fibrosis transmembrane conductance regulator, XCFTR,28 in Xenopus oocytes, and concomitant activation of cPKCα and PKA led to marked stimulation of insulin secretion in pancreatic beta cells by a convergent mechanism.29 Therefore, we tested the hypothesis that TUDCA may exert anticholestatic effects in the well-established model of TLCA-induced cholestasis by a cooperative cPKCα-/PKA-dependent mechanism.
MATERIALS AND METHODS
Materials
Bile acids and dimethylsulfoxide (DMSO) were purchased from Sigma (St. Louis, Missouri, USA). 1-Chloro-2,4-dinitrobenzene (CDNB) was from ICN Biomedicals (Aurora, Ohio, USA). The nonselective PKC inhibitor stauroporine (ST), the cPKC inhibitor Gö6976, the PKA inhibitor H89, and the recombinant catalytic subunits of cPKCα, nPKCϵ and PKA were from Calbiochem–Novabiochem (Nottingham, UK). The anti-MRP2/ABCC2 antibody was from Alexis (Lausen, Switzerland). A rabbit anti-pCREB antibody and a monoclonal CREB antibody were from Cell Signalling (Danvers, Massachusetts, USA), and a monoclonal mouse anti-GAPDH antibody was from Abcam (Cambridge, UK). A goat-anti-rabbit-IgG-HRP-conjugate-antibody was from Bio-Rad Lab (Munich, Germany) and a goat-anti-mouse-IgG-HRP antibody was from Santa Cruz Biotechnology (Santa Cruz, California, USA). Complete protease inhibitor cocktail was from Roche Diagnostics (Mannheim, Germany). Marker molecular weight standard was from Santa Cruz Biotechnology. The Renaissance western blot chemiluminescence reagent was from NEN (Boston, Massachusetts, USA). Hyperfilm ECL was from Amersham (Little Chalfont, UK). Polyvinylidene difluoride membranes were from Millipore (Bedford, Massachusetts, USA). A cAMP enzyme immunoassay kit was from Amersham Biosciences (Freiburg, Germany). FosCholin-12 was from Anatrace (Maumee, Ohio, USA). Protein A-Sepharose was from Zymed (San Francisco, California, USA). All other chemicals were of the highest purity commercially available.
Protein kinase inhibitors
The effect of inhibitors on activity of cPKCα, nPKCϵ, and PKA was tested in vitro at levels at least 5- to 10-fold above their IC50 for cPKCα, as indicated in the literature, using myelin basic protein (MBP) as substrate to apply concentrations in the IPRLs high enough to be effective, but not too high to cause cholestatic effects. The results showed that cPKCα was more effectively blocked by the nonselective PKC inhibitor staurosporine (10 nmol/l) than by the cPKC-specific inhibitor Gö6976 (10–100 nmol/l) (table 1). nPKCϵ was inhibited by staurosporine (10 nmol/l), but not by Gö6976 (10–100 nmol/l). In addition, the selective PKA inhibitor, H89 (100 nmol/l), and staurosporine (10 nmol/l), but not Gö6976 blocked PKA effectively (table 1).
Animals
Male Sprague–Dawley rats (208 (SD 22) g) were obtained from Charles River (Sulzfeld, Germany) and were subjected to a 12 h day–night rhythm with free access to rodent chow and water.
Isolated rat liver perfusions were performed as described in detail previously.20 24 Bile flow was determined gravimetrically in pretared tubes.
Perfusion protocol
Rat livers were perfused for a total of 115 min in a non-recirculating fashion with Krebs–Ringer bicarbonate solution (pH 7.4, 37°C). Twenty-five minutes after starting the perfusion, the nonselective PKC inhibitor staurosporine (final concentration in the portal vein, 10 nmol/l), the cPKC inhibitor Gö6976 (100 nmol/l), and/or the PKA inhibitor H89 (100 nmol/l) were infused continuously into the perfusion medium. Forty-five minutes after start of the perfusion, bile acids (TUDCA, TLCA, TCA, TUDCA+TLCA, TCA+TLCA), dibutyryl cAMP (dbcAMP) or the carrier DMSO only (control, 0.1%, v/v) were added continuously to the buffer by using an infusion pump to reach final portal venous concentrations of 25 μmol/l (TUDCA, TCA) or 10 μmol/l (TLCA, dbcAMP). Sixty-five minutes after start of the perfusion, 1-chloro-2,4-dinitrobenzene (CDNB; 30 μmol/l), the precursor of the model Mrp2/Abcc2 substrate, 2,4-dinitrophenyl-S-glutathione (GS-DNP), was infused for 10 min. At this concentration, saturation of the biliary GS-DNP secretion was observed in the perfused rat liver.20
Biliary secretion of 2,4-dinitrophenyl-S-glutathione
The model Mrp2/Abcc2 substrate, GS-DNP, was determined in bile spectrophotometrically as described previously.20
Hepatovenous efflux of lactate dehydrogenase
Lactate dehydrogenase (LDH) was determined as an indicator of liver cell damage in the hepatovenous effluate by use of a standard enzymatic test.20
cAMP in liver tissue
cAMP was extracted from liver tissue with a liquid phase extraction method conforming to the supplier’s protocol and was determined by an enzyme immunoassay.
pCREB in liver tissue
An aliquot of shock-frozen liver tissue (−80°C) was homogenised, and proteins were separated by western blotting. pCREB and GAPDH were identified by use of specific antibodies. pCREB and GAPDH were semiquantified by densitometry and pCREB was expressed as a ratio pCREB/GAPDH.
Rat hepatocyte isolation and in vitro phosphorylation of Mrp2/Abcc2
Rat hepatocytes were isolated and cultured on collagen-coated wells as described previously.30 A sample of 5×106cells/well was incubated for 2 h after plating for 4 h with 32P-orthophosphoric acid (74 MBq) and were then treated for 30 min with DMSO (0.1%, v/v), 100 nmol/l phorbol 12-myristate 13-acetate (PMA), 100 nmol/l PMA + 10 nmol/l staurosporine, 25 μmol/l TUDCA, or 10 μmol/l TLCA. Cells were then washed three times with phosphate-buffered saline (PBS).
Immunoprecipitation of rat Mrp2/Abcc2 or human MRP2/ABCC2
Rat Mrp2/Abcc2 and human MRP2/ABCC2 were immunoprecipitated from rat hepatocytes and human HepG2 hepatoma cells, respectively, as published previously.31 Efficacy of Mrp2/Abcc2 immunoprecipitation was tested by western blotting.
Phosphorylation of human MRP2/ABCC2 in vitro
Phosphorylation was peformed using a phosphorylation buffer containing 10 μmol/l ATP and per sample 370 kBq [γ32]ATP, and 400 U activated PKA or 90 ng activated cPKCα or/and 110 ng activated nPKCϵ. MBP was used as a phosphorylation control. Samples were immunoblotted and autoradiography of the gels was performed for 18 h. The bands corresponding to 32P-MRP2/ABCC2 were excised and radioactivity was counted in a scintillation counter.
Statistics
Data are expressed as mean (SD). Results were compared between different groups using ANOVA post hoc test (Tukey). Comparison of two groups only was performed using an unpaired two-tailed Student t test. A value of p<0.05 was considered statistically significant.
RESULTS
Bile flow
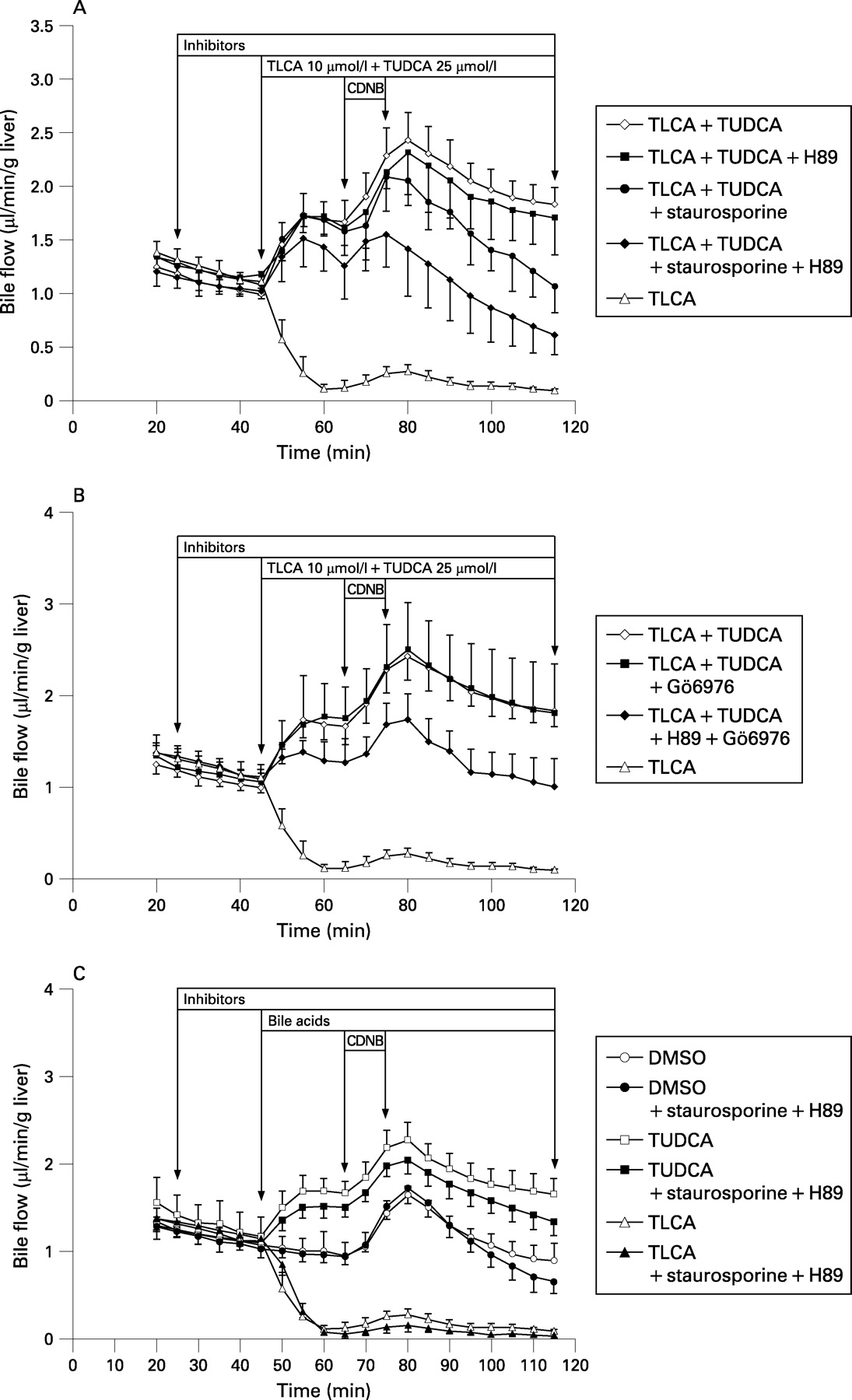
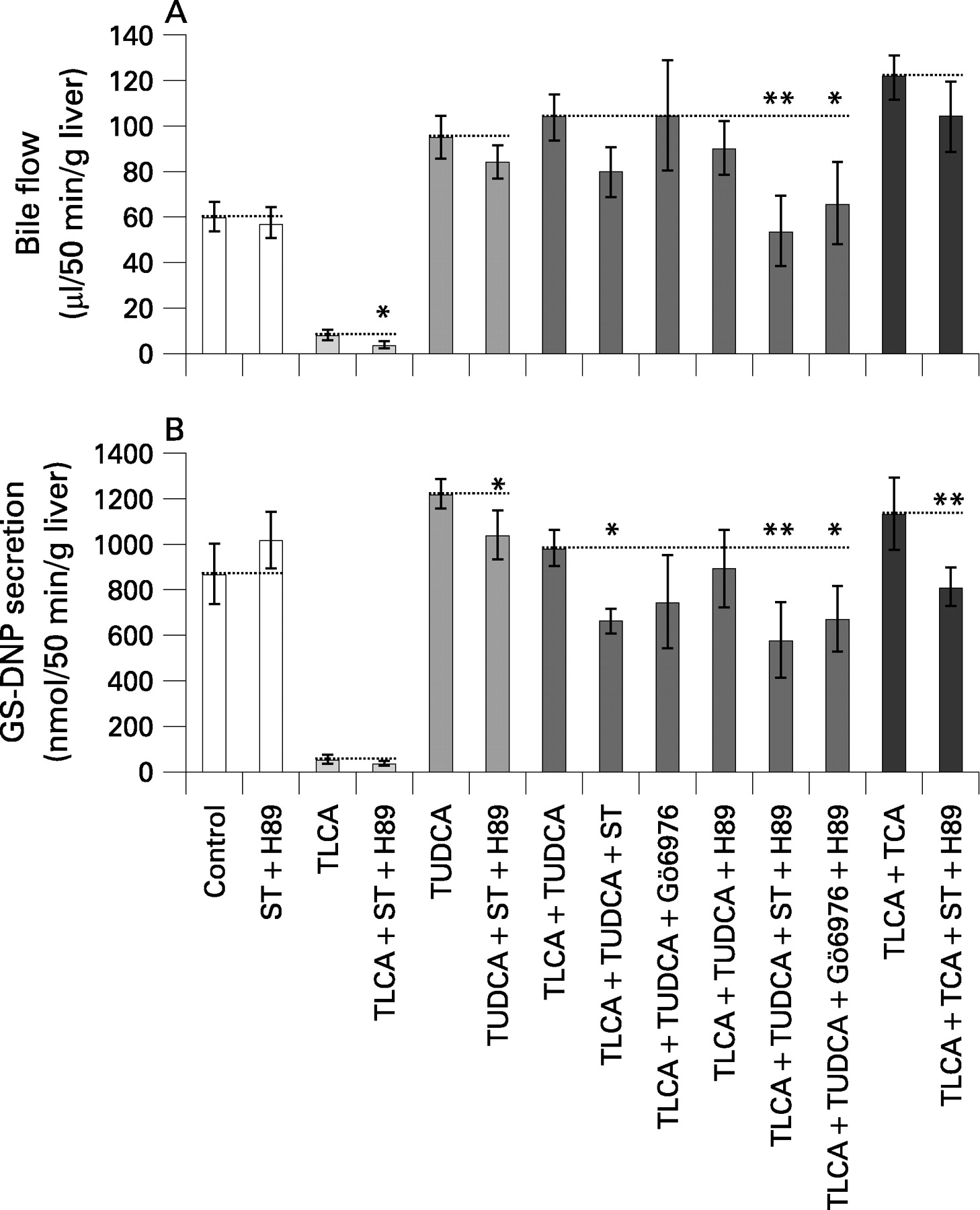
TLCA (10 μmol/l) reduced bile flow in isolated perfused rat livers (IPRLs) to 14% of controls (figs 1A,B and 2A). TUDCA (25 μmol/l) reversed TLCA-induced inhibition of bile flow to 173% of controls. The nonselective PKC inhibitor staurosporine (10 nmol/l), the selective cPKC inhibitor Gö6976 (100 nmol/l), and the selective PKA inhibitor H89 (100 nmol/l) did not significantly affect bile flow in either control livers or in livers treated with TLCA+TUDCA (figs 1A,B and 2A). In contrast, when administered concomitantly, staurosporine+H89 as well as Gö6976+H89 induced a significant reduction of bile flow in livers treated with TLCA+TUDCA by 48% (p<0.01) and 36% (p<0.05), respectively, but again, did not affect bile flow in controls or livers treated with TUDCA only (figs 1A–C and 2A).
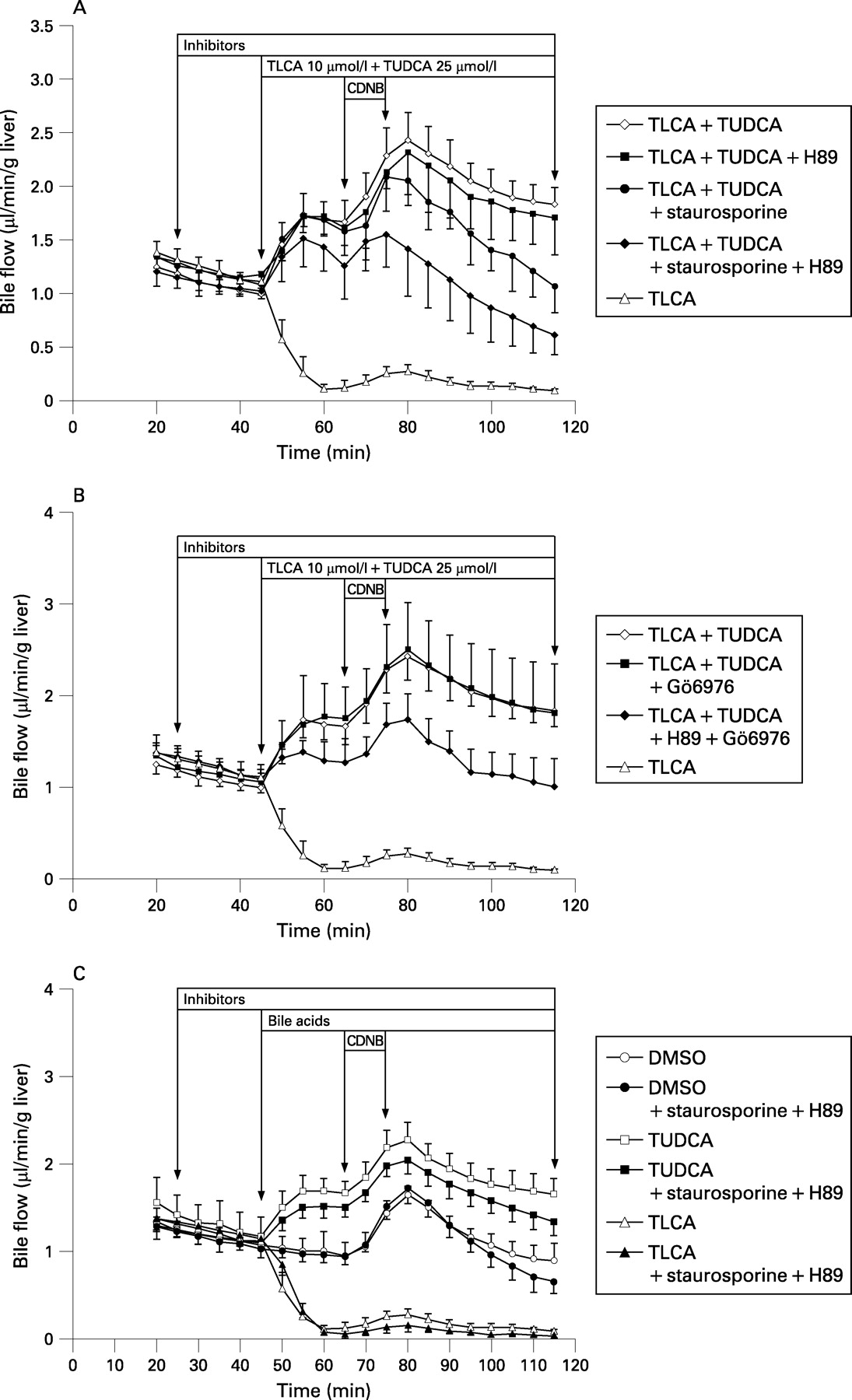

The cAMP analogue, dibutyryl cAMP (dbcAMP, 10 μmol/l), stimulated bile flow in controls by 55% (p<0.01), but did not significantly enhance bile flow in livers treated with TLCA (+81%, NS) or TLCA+TUDCA (+16%, NS) indicating that stimulation of PKA only does not reverse TLCA-induced cholestasis in IPRL.
The taurine conjugate of the major human trihydroxy bile acid, cholic acid (TCA, 25 μmol/l) also reverses TLCA-induced inhibition of bile flow in rat liver.32 Unlike TUDCA, TCA does not affect hepatocellular [Ca2+]i and PKC isoform distribution at physiological concentrations.11 14 Indeed, reversal of TLCA-induced inhibition of bile flow by TCA was not affected by staurosporine+H89 (fig 2A) suggesting different molecular mechanisms mediating the anticholestatic action of TUDCA and TCA in TLCA-induced cholestasis.
Together, these data suggest that recovery of bile flow by TUDCA in TLCA-induced cholestasis – but not stimulation of bile flow under non-cholestatic conditions – is mediated by a cooperative cPKC-/PKA-dependent mechanism.
Secretion of 2,4-dinitrophenyl-S-glutathione
TLCA reduced biliary secretion of the Mrp2/Abcc2 substrate, 2,4-dinitrophenyl-S-glutathione (GS-DNP), to 7% of controls. TUDCA reversed TLCA-induced inhibition of biliary GS-DNP secretion to 113% of controls (fig 2B). The nonselective PKC inhibitor staurosporine impaired biliary GS-DNP secretion by 32% (p<0.05) and the cPKC inhibitor Gö6976 tended to impair biliary GS-DNP secretion by 24% in livers treated with TLCA+TUDCA (fig 2B), but not in control livers. The PKA inhibitor H89 did not affect biliary GS-DNP secretion in livers treated with TLCA+TUDCA (fig 2B) when given alone. When administered concomitantly, staurosporine+H89 as well as Gö6976+H89 reduced biliary GS-DNP secretion by 41% (p<0.01) and 31% (p<0.05), respectively, in livers treated with TLCA + TUDCA. In contrast, biliary GS-DNP secretion in controls or livers treated with TUDCA only was barely affected by PKC/PKA inhibitors (fig 2B). Biliary GS-DNP secretion was not further enhanced by the cAMP analogue, dbcAMP, in controls, livers treated with TLCA or livers treated with TLCA+TUDCA (−5.7%, +1.6%, and −49%, respectively). Together, these data indicate that TUDCA-induced recovery of organic anion secretion via Mrp2/Abcc2 is mediated by a cooperative cPKC-/PKA-dependent mechanism in TLCA-induced cholestasis.
Hepatic cAMP levels and PKA activity
Hepatic cAMP levels were unaffected by any of the bile acids and inhibitors of PKC and PKA tested (table 2). In addition, CREB phosphorylation as a readout of PKA activity in liver tissue was unaffected by the bile acids used (table 2) suggesting that PKA activity, in line with previous findings,10 27 is rather permissive for the anticholestatic action of TUDCA.
Hepatovenous LDH release
The cholestatic effect of TLCA is associated with a serious cytotoxic action as demonstrated by a 32-fold increase of hepatovenous LDH release as a readout of liver cell damage. TLCA-induced LDH release was attenuated by addition of TUDCA, but markedly increased after combined inhibition of PKC and PKA in livers treated with TLCA+TUDCA – but not after inhibition of either PKC or PKA alone – in parallel with the inhibition of the anticholestatic action of TUDCA (table 3). In contrast, LDH release was unaffected by combined inhibition of PKC and PKA in controls and livers treated with TUDCA or TLCA alone (table 3). These data suggest that TUDCA alleviates TLCA-induced liver cell damage in part by a cooperative cPKCα-/PKA-dependent mechanism in association with its anticholestatic action.
Phosphorylation of rat Mrp2 by the nonselective PKC agonist, phorbol 12-myristate 13-acetate
Rat Mrp2 was immunoprecipitated from freshly isolated rat hepatocytes in short-term culture after incubation of hepatocytes with 32P-orthophosphoric acid for 4 h and with DMSO (0.1%), PMA (100 nmol/l), PMA+staurosporine (10 nmol/l), TUDCA (25 μmol/l), and TLCA (10 μmol/l) for 30 min. The results show that Mrp2 was phosphorylated under control conditions. The PMA-induced increase in phosphorylation was reversed by staurosporine. Bile acids at low micromolar concentrations did not affect total phosphorylation of Mrp2 (fig 3A).

{kind=link}
{kind=link}
{kind=link}
Phosphorylation of human MRP2 by cPKCα, nPKCϵ and PKA
Human MRP233 was immunoprecipitated from HepG2 hepatoma cells and MRP2 phosphorylation was studied in vitro using recombinant activated PKCα, PKCϵ or PKA. The results show that the MRP2 complex after immunoprecipitation was phosphorylated by cPKCα, nPKCϵ and PKA (fig 3B). Combined incubation with cPKCα and nPKCϵ tended to induce a more pronounced phosphorylation than incubation with cPKCα or nPKCϵ alone in line with the assumption that different PKC isoforms induce phosphorylation of the MRP2 complex at different sites.
DISCUSSION
The present study demonstrates that the short-term anticholestatic effect of TUDCA in the established experimental model of TLCA-induced cholestasis is mediated mainly by a cooperative cPKCα-/PKA-dependent mechanism.
We and others have previously observed that TUDCA at low micromolar concentrations stimulates Ca2+ entry into hepatocytes independent of inositol-1,4,5-trisphosphate,11–13 selectively translocates Ca2+-dependent cPKCα to hepatocyte membranes,14 20 34 stimulates formation of s,n-1,2-diacylglycerol (DAG),14 activates membrane-bound PKC14 34 and stimulates impaired biliary secretion of organic anions and bile acids in TLCA-induced cholestasis by PKC-dependent mechanisms in isolated perfused rat liver (IPRL)20 and isolated rat hepatocyte couplets,22 respectively. Similar effects of TUDCA on [Ca2+]i and Ca2+-dependent cPKCα have been observed in cholangiocytes.35 The present study not only confirms a role of PKC, but, by use of the cPKC-specific inhibitor Gö6976 (figs 1B and 2A,B), specifically shows that a Ca2+-dependent PKC isoform mediates the anticholestatic effect of TUDCA. Among the known cPKC isoforms, cPKCα, cPKCβ-I, cPKCβ-II and cPKCγ, only cPKCα and cPKCβ-II have been detected in hepatocytes. We were not able to disclose translocation of cPKCβ-II by TUDCA to hepatocyte membranes in IPRL, IRHC and Ntcp-transfected HepG2 hepatoma cells (data not shown; see also Beuers et al14). Thus, cPKCα is most likely the PKC isoform involved in the anticholestatic action of TUDCA in experimental cholestasis as suggested previously.14 20 36
A role of cPKCα as a mediator of the choleretic effect of TUDCA in IPRL was recently questioned and the cPKC agonist thymeleatoxin was shown to induce cholestasis in IPRL.37 In contrast to thymeleatoxin, TUDCA interacts with various hepatocellular (and cholangiocellular) signalling cascades.9 Their impact on control of diverse liver cell functions by TUDCA in health and under cholestatic conditions remains to be further unravelled. Our previous data were in line with the assumption that bile flow and organic anion secretion under normal “non-cholestatic” conditions are modulated by TUDCA mostly independent of Ca2+ and Ca2+-dependent cPKCα (fig 1C; see also Beuers et al12) whereas Ca2+-dependent cPKCα appears to play a key role in the anticholestatic action of TUDCA in TLCA-induced cholestasis.20
A relevant effect of TUDCA on hepatocellular PKA activity has not been observed15 although PKA is a well-known mediator of apical secretion in hepatocytes.38 In line with previous findings, we were unable to observe an effect of TUDCA on cAMP levels and PKA activity as reflected by unchanged CREB phosphorylation in liver tissue. In addition, the selective PKA inhibitor H89 alone did not affect TUDCA-induced bile secretion in IPRL in the present study. It should be kept in mind, however, that global determination of CREB phosphorylation and cAMP in liver tissue may trivialise the complexity of PKA action and cAMP signals in microdomains of the apical plasma membrane.39 This assumption is strengthened by the recent finding that type II inositol-1,4,5-trisphosphate receptor (InsP3) isoforms are concentrated in the pericanalicular region in a lipid raft-dependent way.40 41 Type II InsP3 are essential for hepatocellular Ca2+ wave formation and, possibly, Ca2+/cPKCα/PKA interaction. Thus, more sophisticated methodological approaches will be needed to further elucidate the possibly permissive role of PKA in the anticholestatic action of TUDCA in the pericanalicular zone of hepatocytes.9
Under non-cholestatic conditions, an integrin-dependent dual signalling pathway involving the MAPK Erk1/2 and p38MAPK plays a role in mediating the choleretic effect of TUDCA in IPRL.16–19 We were not able to show a role of MAPK for the anticholestatic action of TUDCA in TLCA-induced cholestasis when either Erk1/2 or p38MAPK or Erk1/2 and p38MAPK concomitantly were inhibited.30 Thus, different signalling pathways mediate the choleretic and the anticholestatic action of TUDCA in hepatocytes.9
A cooperative stimulation of secretory activity by PKC/PKA as assumed in the present model for the anticholestatic action of TUDCA has previously been described in different cell types.28 29 Apical carriers and their anchoring proteins42 43 are potential targets of a concerted action of PKA and cPKCα at the canalicular hepatocyte membrane for TUDCA-induced carrier insertion as well as of nPKCϵ in TLCA-induced carrier retrieval.9 The deduced amino acid sequences of the conjugate and bile salt export pumps, Mrp2/Abcc2 and Bsep/Abcb11, respectively, show numerous potential serine/threonine phosphorylation sites for PKC and PKA. We found that Mrp2/Abcc2 can be phosphorylated in vitro in rat hepatocytes by PKC agonists or after immunoprecipitation from HepG2 hepatoma cells by recombinant activated cPKCα, nPKCϵ, and PKA. Interestingly, Mrp2/Abcc2 transport activity has recently been shown to be stimulated by cPKCα in a baculovirus coexpression system in vitro.44 These findings are in line with those previously observed for Bsep/Abcb1131 which also can be phosphorylated by cPKCα. Identification of key serine/threonine phosphorylation sites required for apical insertion and retrieval of Mrp2/Abcc2 or BSEP/ABCB11 – as demonstrated for the sodium taurocholate cotransporting polypeptide, Ntcp/Slc10a1, in the basolateral membrane45 – will be a prerequisite to further unravel the cholestatic and anticholestatic post-translational effects of bile acids in hepatocytes.
In conclusion, the well-known anticholestatic effect of TUDCA is largely blocked by selective pharmacological intervention in the present study, indicating that TUDCA exerts its post-translational anticholestatic effect mainly by a cooperative cPKCα-/PKA-dependent mechanism in the experimental model of TLCA-induced cholestasis.
REFERENCES
Footnotes
RW and SH contributed equally to this study.
Data in this study were reported, in part, at the Annual Meeting of the European Association for the Study of the Liver, Barcelona, 13 April 2007, and at the Biannual International Bile Acid Meeting, Freiburg, 7 October 2006, and were published in part in abstract form in J Hepatol 2007;46(Suppl l):S119.
Funding: This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG Be 1242/5-5).
Competing interests: None.