Article Text

Abstract
The human gastrointestinal (GI) tract microbiota plays a pivotal role in our health. For more than a decade a major input for describing the diversity of the GI tract microbiota has been derived from the application of small subunit ribosomal RNA (SSU rRNA)-based technologies. These not only provided a phylogenetic framework of the GI tract microbiota, the majority of which has not yet been cultured, but also advanced insights into the impact of host and environmental factors on the microbiota community structure and dynamics. In addition, it emerged that GI tract microbial communities are host and GI tract location-specific. This complicates establishing relevant links between the host’s health and the presence or abundance of specific microbial populations and argues for the implementation of novel high-throughput technologies in studying the diversity and functionality of the GI tract microbiota. Here, we focus on the recent developments and applications of phylogenetic microarrays based on SSU rRNA sequences and metagenomics approaches exploiting rapid sequencing technologies in unravelling the secrets of our GI tract microbiota.
Statistics from Altmetric.com
The human gastrointestinal (GI) tract consists of different and connected organs that are involved in supplying the human body with nutrients and energy sources by the conversion and absorption of food. The human GI tract has a well-known anatomical architecture and is approximately 7 m long with a surface area of approximately 300 m2 in adults.1 Since it is continuously exposed to the outside environment, the GI tract has several protection systems. These include the low pH in the stomach, the coverage of the complete GI tract with a mucus layer, an enormous army of immune cells that lie beneath this mucus layer, and the presence of commensal microbes that abundantly colonise the GI tract. These microbial communities are collectively called commensal microbiota or GI tract microbiota. This GI tract microbiota is distributed along the entire GI tract, with the density and diversity increasing from the stomach to the colon. The acknowledged functions of the GI tract microbiota include the conversion of indigestible food components2 ,3 and the production of essential vitamins and co-factors. However, our understanding of the GI tract microbiota is fragmented, which is due to the limited accessibility of the different parts of the GI tract and the immensely complex and diverse community structure of the GI tract microbiota which differs between individuals, intestinal location and age groups.4
THE GASTROINTESTINAL TRACT MICROBIOTA
The myriad of microbial cells in the GI tract, which outnumber our body cells by a factor of at least 10, has a large species diversity and consists of cultivated species as well as those that have not yet been cultured. A recent estimation of the cultivable fraction of the GI tract microbiota includes 442 bacterial, three archaeal, and 17 eukaryotic species.5 However, it is evident that any figures that report the known diversity of the GI tract microbiota are continuously outdated due to the steady reporting of novel intestinal inhabitants. The majority of the currently available cultivated representatives were discovered following the introduction of anaerobic cultivation technologies that are still being used as standard tools.6 Novel bacteria that are still being isolated from the human GI tract include a wide variety of butyrate-producing bacteria belonging to the phylum Firmicutes7 ,8 and bacteria belonging to the phylum Bacteroidetes,9–12 which are abundant GI tract phyla. In addition, the use of alternative carbon sources brought the discovery of the first GI tract representatives of the phyla Verrucomicrobia and Lentisphearae, Akkermansia muciniphila and Victivallis vadensis, respectively.13 ,14 A muciniphila, especially, is a relevant isolate since it is a specialist in mucin degradation and a very common and abundant inhabitant of the GI tract which has previously not been noted because of its small size and specific carbon source requirements.13 ,15 ,16
Recently, novel culturing methods have been developed which exploit microbeads or multiplexed solid surfaces and allow unconventional and high-throughput culturing approaches.17 ,18 Such methods enable the simultaneous single-cell cultivation of thousands of microbes, which is essential for an appropriate study of the complex and dense GI tract microbiota. Application of high-throughput culturing is expected to expand our knowledge of this neglected yet important element of the human body. However, in the course of evolution many GI tract microbes have developed intimate relations with the host and with each other, which makes microbes dependent on the metabolic activity of another member of the ecosystem and, therefore, almost impossible to grow into pure culture.19 As an alternative, the use of small subunit ribosomal RNA (SSU rRNA) and its corresponding gene has become established for the classification and phylogenetic analysis of microbes. The SSU rRNA gene includes approximately 1500 bp, which is a sufficient size for comparative sequence analysis. SSU rRNA is present in literally all organisms, and due to its conserved function its sequence has remained relatively conserved throughout evolution. Nevertheless, it contains nine variable regions that can be used for the identification and differentiation of specific microbial species. In fact, all life forms can be classified using the SSU rRNA sequences and this has resulted in the discovery of the Archaea as the third domain of life (Bacteria and Eucarya are the other two domains) and the construction of the revolutionary “tree of life”.20 ,21 SSU rRNA and its corresponding gene can be obtained directly from any environmental sample without cultivation procedures and this allows the detection of basically all members of an ecosystem, including those that cannot be cultured yet. Currently, over 400 000 SSU rRNA sequences are available in DNA databases, which is far more than for any other gene (http://www.arb-silva.de (accessed 11 July 2008)). The first breakthrough discovery of the SSU rRNA based analyses was that cultivation represents only a small fraction (estimated to be between 10 and 50%, depending on the study) of the true microbial diversity within the GI tract (table 1).
Exploring the diversity of an ecosystem by comparative sequence analysis is based on identification of species-level phylogenetic types, ie, phylotypes. Since variation of the SSU rRNA gene sequence is present within the members of the same species,40 ,41 and sometimes even between different copies of the SSU rRNA gene within the same microbial genome,42 phylotypes are defined as groups of SSU rRNA gene sequences with a certain level of similarity. However, the cut-off value of SSU rRNA sequence similarity that is used for phylotype definition is not consistent between different studies and varies between 97 and 99%. The higher the cut-off value, the higher is the number of distinct phylotypes that will be found in the same clone library.43 For diversity studies the SSU rRNA sequences can be targeted using different, often complementary, approaches. Commonly applied approaches include cloning and subsequent sequencing of SSU rRNA genes, fingerprinting approaches such as denaturing gradient gel electrophoresis (DGGE) and fluorescence in situ hybridisation (FISH), which allows visualisation of specific microbes. These approaches are relatively low throughput and the choice of approach will mainly depend on the question to be answered. Cloning of SSU rRNA genes delivers the most detailed phylogenetic information, fingerprinting is most often used to compare microbial communities and monitor their dynamics, while FISH is the preferred approach to quantify specific microbial populations. The combination of these approaches has greatly advanced the phylogenetic framework of the enormous microbial diversity and novel insights into its ecology. It has to be realised that neither of the methods for studying GI tract microbiota gives absolutely accurate information about the diversity of this ecosystem, including the “gold standard” cloning and sequencing-based studies. For example, the Actinobacteria, which belong to the high G+C Gram-positive bacteria, are frequently undetected or under-represented in the clone libraries, although they represent a dominant fraction of GI tract microbiota, as has been demonstrated by FISH using SSU rRNA-targeted oligonucleotide probes.44 ,45
The composition of GI tract microbiota is most often studied by analysing faecal samples, but the microbiota of other intestinal samples, such as specimens from the human colon and ileum, have also been characterised.27 ,32 ,33 ,46 ,47 Fingerprinting studies based on DGGE analysis of SSU rRNA gene amplicons indicated that the dominant community in faecal samples in about half of the subjects studied does not necessarily represent that found in other parts of the GI tract, including the colonic mucosa.48 ,49 This most likely reflects the fact that the number of microbes that are associated with the mucosa is small compared to the number of microbes present in the colon lumen. Although most likely primed by mucosa-associated microbes, the composition of the latter can be affected by the growth on specific substrates that reach the colon. If these substrates become limited at the end of the colon, this could also explain why certain abundant microbial species in faeces are not viable any more and this argues for studying samples derived from several GI tract regions.50
Studies that employ SSU rRNA gene sequence analysis are rapidly expanding our knowledge about the diversity of the GI tract microbiota, and only a decade after their introduction, the number of molecularly detected GI tract phylotypes has by far outnumbered the cultivated GI tract species (fig 1). An intensive survey of the publications focusing on the diversity of the human GI tract microbiota demonstrated that from more than 1200 microbes described, only 12% were recovered by application of both molecular and cultivation-based approaches, while the vast majority (∼75%) was detected solely as an SSU rRNA sequence.51 These values have to be taken with caution as it has also to be realised that most of the SSU rRNA sequences are retrieved from intestinal samples, mostly faeces, from only a dozen individuals. Considering the location and individual-specific microbial composition of the GI tract, this indicates that the 1200 species described only reflect a fraction of the true microbial diversity, which is estimated to consist of up to 1000 microbes per individual and more than 5000 microbes in total.23 ,33 ,51 ,52 This implies that we are still at the beginning of describing the GI tract microbial diversity. Hence, there is a need to develop and apply high-throughput approaches in further culturing studies, SSU rRNA sequencing and microbial diversity analysis, notably at different locations in the GI tract. These are essential to enable linking microbial populations to host-related factors, such as host genotype, age, health status, geographical location, ethnic origin, and diet.

PHYLOGENETIC MICROARRAY ANALYSIS OF GASTROINTESTINAL TRACT MICROBES
As indicated above, the human GI tract contains a microbiota, the diversity of which is beyond our imagination given the total number of microbes, and their location- and individual-specific composition. This complicates establishing links between members of the microbiota and GI tract disorders. At the moment, causal effects have been determined only for the well-studied and cultured pathogens such as Helicobacter pylori, Listeria monocytogenes, Clostridium difficile and members of the Enterobacteriaceae.53 In addition, correlations have been suggested between the health status of individuals and the composition and activity of their microbiota. Irritable bowel syndrome (IBS) and intestinal bowel diseases (IBDs), such as Crohn’s disease and ulcerative colitis, have frequently been associated with a rather unstable and disturbed microbiota composition in contrast to healthy individuals.54–57 These differences have to be taken with caution since they can be influenced by several factors, including the use of medicine that may affect the microbiota. In addition, the unstable microbiota of patients with IBD can be caused by the disease status of the patients as differences in community structures were observed in patients who relapsed versus those who were in remission.54–57 Last but not least, the stability of the microbiota in healthy individuals decreases when the time span between the sampling increases, and differs between microbial populations.5 It was demonstrated that a significant fraction of microbial phylotypes is continuously present in the GI tract of a person over a 10 year time span, which indicates that the microbiota consists of a stable individual core of colonising microbes surrounded by temporal visitors.5 Despite these individual differences, there are indications that some microbial phylotypes are shared by different people and this led to the hypothesis that besides the individual core of microbes representing the stable colonisers in healthy individuals, humans also share a common core of microbes in their GI tract.58 This hypothesis expands a previously proposed hypothesis that the human gastrointestinal microbiota is diverse but it is dominated by a limited number of bacterial species in everybody.59–61
As it is not yet possible to define the microbiota of a healthy intestinal tract, it is equally difficult to define the microbiota associated with an intestinal disorder. Another factor that complicates such an association is the fact that these GI tract disorders have a largely undefined, complex and possibly heterogeneous aetiology in which, in addition to the microbiota, host genetics and environmental factors also play a role. Moreover, our inability to cultivate all members of the microbiota makes it impossible to formulate hypotheses about the role of uncultured microbes in health and disease as a (partial) SSU rRNA sequence provides no information about the function of an organism. Last but not least, the individual-specific composition of the GI tract microbiota is also an important factor that complicates the establishment of links between microbes and the health status of the host.22 ,62 Since different people are recruited by the different research groups, for which each of them has a favourite target microbe or methodology for microbiota analysis, comparisons between different studies are basically impossible. Moreover, as the number of persons tested is still low, it is evident that high-throughput analyses of the microbiota using standardised methods are needed to make statistically relevant links between the presence or quantity of uncultured bacterial populations and GI tract disorders.
The most commonly used high-throughput analytical method is DNA microarrays. DNA microarrays are basically glass surfaces, each the size of a microscopic slide, that are spotted with thousands of covalently linked DNA probes. These DNA microarrays can be hybridised with DNA or RNA and the most current applications include monitoring gene expression (transcriptional profiling) and detecting DNA sequence polymorphisms or mutations in genomic DNA. However, it is also possible to use DNA microarrays for diversity analysis (fig 2). Guschin and colleagues63 described the first phylogenetic microarray (or diversity microarray) in which oligonucleotides complementary to SSU rRNA gene sequences of nitrifying bacteria were used to detect and identify these bacteria in environmental samples. Thereafter, DNA microarray technology has been implemented in a variety of ecological studies in which not only SSU rRNA genes, but also antibiotics resistance genes were used as targets.64–69 Recently, the great potential of using phylogenetic microarrays to detect thousands of microbes simultaneously was demonstrated by DeSantis and colleagues.70 This study illustrated that the use of phylogenetic microarrays is more powerful for the analysis of microbial community structure than a canonical clone library approach.70 Besides this general phylogenetic microarray, there are also specific microarrays that focus on the microbial communities in specific ecosystems5 ,71 ,72 (table 2). These include a phylogenetic microarray that targets the microbiota of the oral cavity and two microarrays that focus on the microbiota of the human GI tract.
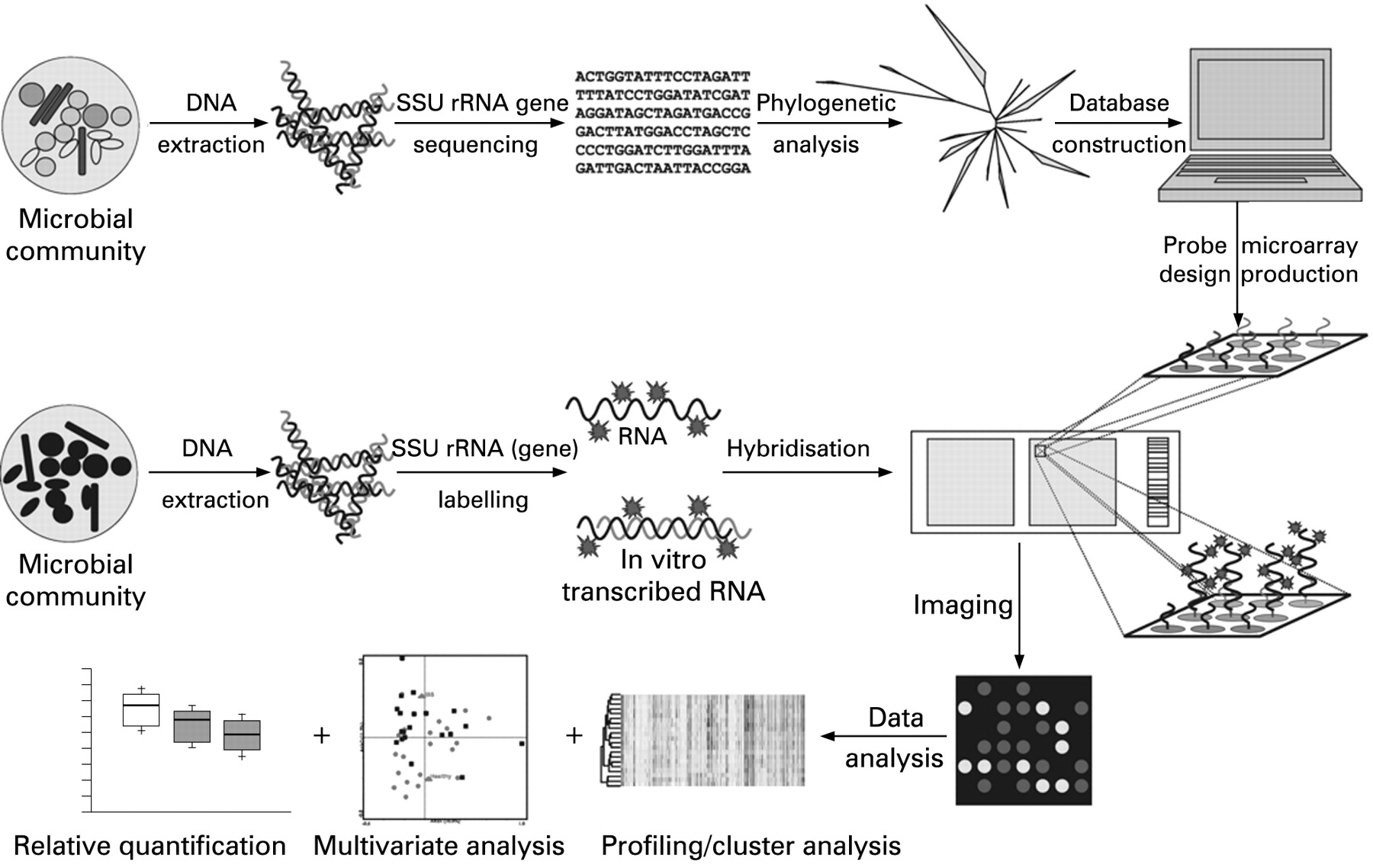
The first studies in which phylogenetic microarrays were used to characterise the GI tract microbiota have illustrated the power of such an approach to gain insights into the structure and population dynamics in the GI tract (box 1). In one study 14 newborn babies were monitored over a year and the results showed that the microbiota of these infants is relatively simple and individual-specific, but chaotic in the early months of life followed by a similar pattern of development towards a more complex adult-like microbiota.77 These results confirm and expand earlier studies performed using DGGE analysis.25 The direct phylogenetic identification enabled by the microarray brought to light another remarkable observation: bifidobacteria were found to be only a minor fraction of the total microbiota of the infants analysed. This contrasts with observations in several previous studies which showed that bifidobacteria are the most dominant group in infants.78 ,79 Although such a surprising finding could be partially explained by technical biases (eg, inadequate primer sequence or inefficient cell lysis), a biological explanation is also possible as infants from different studies originated from different continents and received different paediatric practices and diets.77 The disparity of findings of different studies illustrates our present inability to define the normal GI tract microbiota even of its simplified form, which at present is in early infancy.
Besides focusing on the GI tract microbiota in healthy individuals, the first phylogenetic microarray studies on people with a GI tract disorder have also been performed.5 As indicated before, the common GI tract disorders, such as IBD and IBS, are complex since they are influenced by the microbiota and host genetic and environmental factors80–83 and, therefore, comprehensive and high-throughput approaches are needed to gain insight into this complexity. The phylogenetic microarray analysis demonstrated that persons having a GI tract disorder had distinct microbiotas compared to healthy individuals and that the severity of the disease is correlated with the significance of the difference with the healthy group.5 A striking observation was that patients with IBS had increased heterogeneity among the microbiota composition compared to healthy individuals.5 This could be explained by the fact that IBS is known as a heterogeneous disorder based on varying clinical symptoms.84 Despite this increased heterogeneity of the GI tract microbiota, a remarkable observation is that, in IBS, members of the Firmicutes were affected the most dramatically. This is in line with the previous phylogenetic microarray studies which indicated that the Firmicutes are more strongly affected by environmental changes than are Bacteroidetes and Actinobacteria.5 This suggests that the alterations in the Firmicutes could be candidate biomarkers for IBS. The Firmicutes is the most diverse and abundant phylum within the GI tract microbiota consisting of a wide variety of uncultured organisms. As a result, the functionality of the majority of Firmicutes is not known and it is difficult to hypothesise what it might be. Therefore, explaining the observed trends is only possible for a few microbial groups, such as Roseburia spp., which are among the butyrate-producing isolates from the gut.85 For instance, Roseburia spp. were found to be increased in patients suffering from diarrhoea-predominant IBS,5 which adds to the controversy about the effect of butyrate on human health.86 ,87 With respect to these GI tract disorders it is evident that obtaining more insight into the function of Firmicutes is crucial. To overcome the problem of the fact that the majority of Firmicutes will remain uncultured in the near future, culture-independent approaches to study functionality are needed.
The first applications of phylogenetic microarrays have already provided novel insights into the microbiota composition and dynamics in relation to health and disease and these approaches are promising for future research. However, the application of phylogenetic microarrays also has its limitations, as does any other microbiological approach. Phylogenetic microarray analyses are dependent on the isolation of nucleic acids and subsequent polymerase chain reaction (PCR) amplification of SSU rRNA genes, which are vulnerable to technical biases. These are, unfortunately, general drawbacks of culture-independent technologies and should be minimised as much as possible. Furthermore, phylogenetic microarrays have a dynamic range that only covers the dominant microbes present in the GI tract. On the other hand, at present, there are numerous group- and species-specific quantitative PCR (qPCR) assays that are useful for quantification of different phylogenetic groups belonging to the human GI tract microbiota.88–90 These specific PCR protocols can be combined with phylogenetic microarray analysis, which will allow determination of the diversity and relative abundance within low abundant groups. This could be of special interest for studying the population dynamics of pathogens or probiotics as they are usually present in low numbers.
In conclusion, the first phylogenetic microarray-based studies provided novel insights into the GI tract ecology by demonstrating correlations between certain microbial populations and host factors. To determine the significance of a certain correlation, the next questions to be addressed are whether this correlation reflects a causal relation and, if so, what mechanisms underlie the observed effects. Answering this type of question cannot be done by SSU rRNA gene-based approaches, but need the integration of functional-based approaches, including the application of the so-called meta-“omics” approaches in GI tract research, as will be discussed in the next section.
METAGENOMICS AND OTHER COMMUNITY-BASED APPROACHES
SSU rRNA-based approaches are fruitful for describing the microbial diversity in the human GI tract and finding potential links between microbes and a certain health status. However, the results obtained with such approaches cannot be interpreted beyond the description of the microbial diversity, since potential functions of these microbes cannot be extracted from SSU rRNA data. This means that a correlation between a disease status and the presence of a microbe cannot explain whether its presence is the result or the cause of the disorder. This means that other approaches are needed to gain insight into the potential roles of microbes in the human GI tract and how they are related to health and disease. In this respect, much insight has been gained from isolates that are known as pathogens and have been well-characterised in the laboratory. Animal and in vitro models have shed some light on the strategies these microbes have to interact with the host and, moreover, the availability of their genome sequences allows the discovery of genes involved in these interactions. Besides pathogens, commensal bacteria and their role in the GI tract have also been studied in a similar way. The pioneering work of Gordon and co-workers91–94 and that of others showed that microbial colonisation improves nutritional and defensive functions of host. Moreover, the impact of the host on the microbe was also studied.94 ,95 These so-called reductionist approaches provide insight into new genes and functions of individual organisms, which serve as models for community based studies.
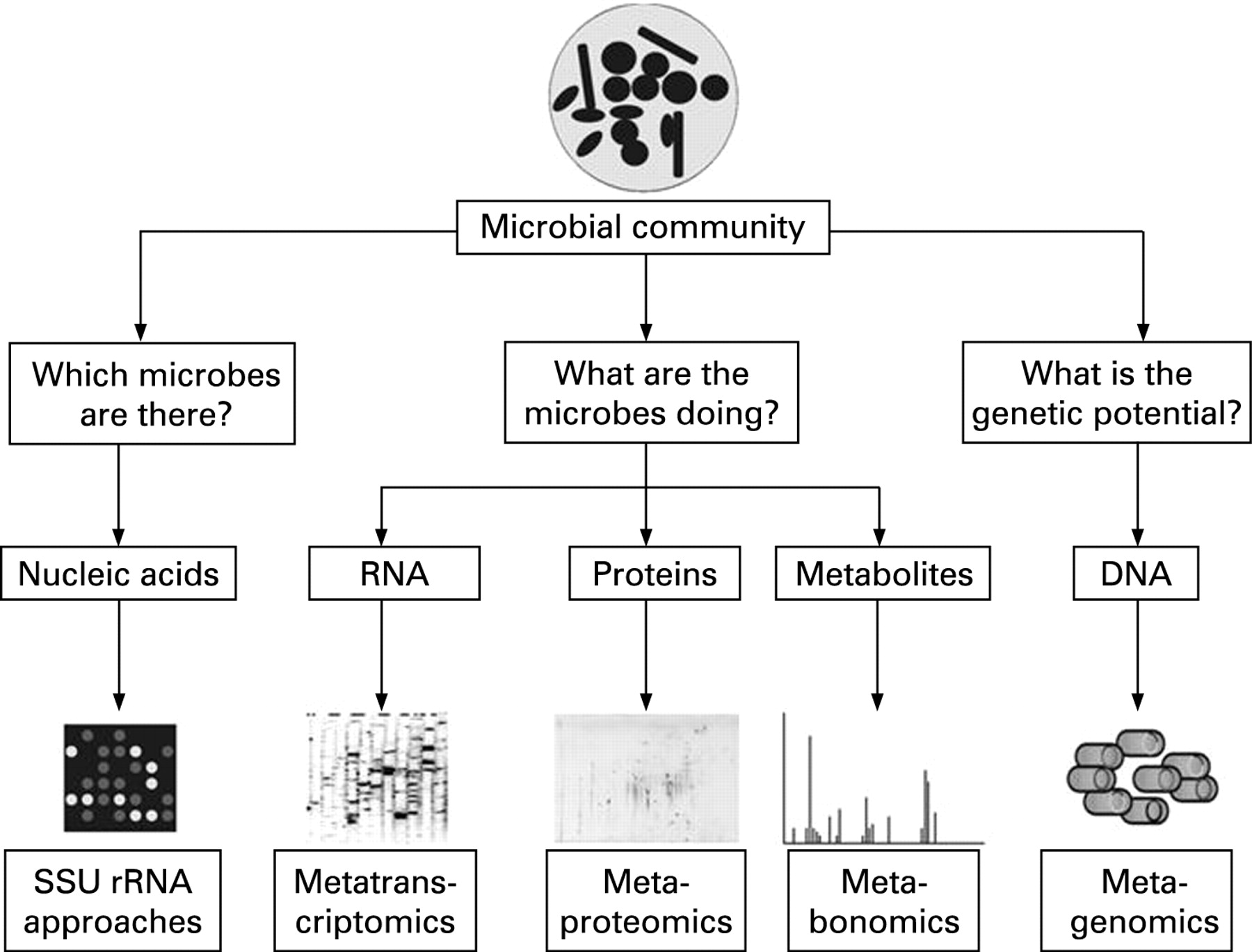
Model systems have provided detailed insights into mechanisms that underlie the communication between host and microbe. However, there is still a huge gap between understanding these interactions in a model system and that in a complex ecosystem, such as the human GI tract. One way to gain insight into potential functions and activities of microbes without the need of cultivation is by performing metagenomics and other community approaches (box 1). Metagenomics is a DNA-based approach to gain insights into the genetic potential of microbial communities, while the other meta-“omics” approaches focus on activity biomarkers, such as messenger RNA, proteins or metabolites (fig 3). Metagenomics is defined as the study of collected genomes from an ecosystem that can be used to study the phylogenetic, physical and functional properties of microbial communities.96 Metagenomics is performed by extracting DNA from the microbial community followed by cloning of the DNA fragments in a suitable host (usually E coli) using a vector, such as fosmid or bacterial artificial chromosome vectors. This results in a metagenomic library that can be used for sequence-driven or function-driven analysis. Sequence-driven analyses are basically performed to obtain a snapshot of the genetic diversity of an ecosystem, while function-driven analyses are done to screen the library for novel enzymes or particular functions of interest.
{kind=link}
{kind=link}
{kind=link}
Box 1 Novel insights that have been gained from high-throughput analysis of the human gastrointestinal (GI) tract microbiota
The human GI tract microbiota is composed of numerous uncultured microbes and predominated by Firmicutes, Bacteroidetes and Actinobacteria
In healthy adults the human GI tract microbiota fluctuates around a stable individual core of phylotypes that are affected by host genetics, environmental and stochastic factors
In infants the GI tract microbiota is succeeding from an unstable chaotic community towards a stable adult community
A reduction in the abundance and diversity of Firmicutes is frequently associated with intestinal bowel diseases and irritable bowel syndrome
The human GI tract microbiome is enriched in functions that are essential to the human host
The human GI tract microbiome from healthy human adults consists of a functionally uniform core and is a hotspot for gene transfer
Function-driven metagenomics offers great possibilities to discover new classes of genes with specific functions. The screening for these features requires functional transcription and translation of the genes, which are located in the metagenome clone, in the host that was used for the metagenome library construction. E coli is the most commonly used host for construction of the metagnomic library and it is predicted that it can express up to 40% of the functional potential from randomly cloned environmental DNA.97 Function-driven metagenomic studies based on enzyme activity assays have already led to the discovery of novel activities, such as β-glucanases in the colon of mice,98 bacterial hydrolases in rumen,99 and antibiotic resistance in the oral cavity.100 Recently, a phenotyping approach was used to screen for metagenomic clones that contain capacities to modulate the growth of epithelial cells in vitro.101 This approach showed that the identification of potentially novel mechanisms of host–microbe interactions in the GI tract is possible. These types of screening methods can be very useful to screen for beneficial or harmful functions that are present in the GI tract microbiota. Subsequently, the expression of the genes that are responsible for the function under study can be monitored in situ and serve as potential biomarkers for intervention studies.
It has to be realised that functional screening is quite laborious since relatively large metagenomic libraries have to be screened to obtain a handful of positives per enzyme screen.99 To overcome this drawback, a high-throughput screening approach, termed substrate-induced gene-expression screening was developed, which allowed fluorescence-activated cell sorting, in which catabolic genes were activated by various substrates.102 Although this approach looks promising for high-throughput screening of human GI tract microbiota to discover novel enzyme-encoding genes, the positive identification relies on several host requirements, which include the recognition and transport of the inducing substrate and the ability of the host to express the corresponding genes located on the cloned insert.
Sequence-driven metagenomics approaches are used to create a catalogue of the genetic potential that is present in an ecosystem, which can be fruitful to gain insight into the functionality of the particular ecosystem. Sequence-driven metagenomics has already been applied to study a variety of ecosystems and the first metagenomic approach that was performed on the human GI tract described the diversity of the viral community in human faeces.103 This study revealed that the viral community in faeces is very diverse consisting of approximately 1200 viral genotypes from which the majority of viral sequences had the highest sequence similarity to phages that are known to infect Gram-positive bacteria. As mentioned previously, Gram-positive bacteria, which include the phyla Firmicutes and Actinobacteria, were identified as dominant members of the GI tract microbiota in SSU rRNA gene-based studies. The first prokaryotic sequence-driven metagenomic approach focused on the SSU rRNA sequences that are represented in the metagenomic libraries from pooled faecal samples from healthy individuals and from patients with Crohn’s disease.104 This study demonstrated that Firmicutes are significantly reduced in complexity in patients with Crohn’s disease compared to healthy subjects. This is in line with SSU rRNA gene-based observations, including those in which phylogenetic microarrays were used.5 In another study the “mobile metagenome” of the human GI tract microbiota was investigated. In this study transposon-aided capture was developed and applied to the study of the plasmid-encoded genes that are present in the human GI tract.105 In addition to genes involved in replication and mobilisation of the plasmids, genes encoding for phosphoesterase or phosphohydrolase enzymes could be detected. Besides these targeted sequence-driven metagenomic approaches, large-scale sequence-driven metagenomic approaches have also been applied to investigate the genetic potential in the GI tract ecosystem.106 ,107 The first study demonstrated that many genes in the colonic microbiota represent functions that are essential to the host, such as vitamin production, which have often been ascribed to the microbiota.106 Remarkably, sequences affiliated to Bacteroidetes, one of the dominant microbial groups in the colon, were not detected in this library, which indicated that cell lysis, DNA extraction and cloning procedures can have a major impact on the genetic diversity that is represented in a library. Recently, a comparative metagenomics approach was described to compare and contrast the genetic diversity in the human colon of 13 subjects, including adults and infants.107 This study indicated that the human microbiota is a hot spot for horizontal gene transfer. Moreover, this study demonstrated that the genomic features of infants were less complex and individual-specific, while those of adults were more complex with high functional uniformity between individuals. The latter finding confirms the proposal that all human GI tract microbiotas share a common core of microbes, despite their individual-specific composition.58
Sequence-driven approaches result in a wealth of scattered pieces of sequences and this means that special tools for post-metagenomic analysis are indispensable. Although software packages have been developed to re-assemble the genomic fragments that are sequenced, a major limitation of metagenomics is that the genetic diversity in an ecosystem is enormous. Despite the continuous development of novel, relatively cheap, sequencing technologies, such as 454 pyrosequencing,108 which will result in increased numbers of metagenomic sequences, at the moment it is almost impossible to obtain reasonable coverage for complex ecosystems such as the human GI tract microbiota. Nevertheless, this raw sequence information can be analysed by comparative metagenomics and these types of analyses can already reveal the identification of genes that are specific or enriched in a certain ecosystem or niche.109 ,110
It has to be realised that the detection of genes in a metagenomic library does not necessarily mean that these are functionally important. Therefore, metagenomics should be considered more as catalogues for activity-based approaches and that other meta-“omics” approaches, which use RNA, proteins and metabolites as targets, are better approaches to gain insight into the activity and functionally of the microbes in an ecosystem (fig 3). These meta-“omics” approaches are still in their infancy,111 but we expect that they will be extensively used in the near future.
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
For more than a decade it has been recognised that a major part of the human GI tract microbiota has not been characterised by cultivation. This has led to the implementation of sequence analysis of SSU rRNA and its corresponding gene in studying the human GI tract microbiota diversity and this has provided tremendous expansion of our knowledge about the ecology of the GI tract. However, these approaches have indicated that the GI tract microbiota is individual- and location-specific, and that its diversity is enormous with thousands of novel microbial species to be discovered.51 This argues for the introduction of novel high-throughput and comprehensive technologies for studying the microbiota in the human GI tract, as described in this paper.
The implementation of high-throughput phylogenetic microarrays allows the simultaneous analysis of thousands of microbes in a single experiment and, therefore, is very attractive for studying the population dynamics of the GI tract microbiota in health and disease. This resulted in the discovery that the development of the microbiota in infants is initially chaotic but stabilises towards an adult-like community after 1 year.77 In addition, phylogenetic microarray analysis indicated that the human microbiota fluctuates around an individual core of stable colonisers.5 Last, but not least, significant links have been established between the presence or abundance of specific groups of microbes and GI tract disorders such as IBS and IBD.5 Therefore, it is already evident that the implementation of phylogenetic microarrays in GI tract research will increase our knowledge in the near future. Nevertheless, the up-to-date status of phylogenetic microarrays will always depend on the discovery of novel GI tract inhabitants and, therefore, ongoing sequencing of SSU rRNA gene libraries and cultivation of the novel GI tract inhabitants are indispensable.
It is evident that phylogenetic microarrays will enable us to make correlations between microbial groups and characteristics of the host. However, this will not lead to extrapolation of microbial functions as the majority of GI tract microbes are only known as a partial SSU rRNA gene. Therefore, metagenomics and other meta-“omics” approaches are needed to gain insight into the genetic potential and activity of GI tract microbiota. The field of these meta-“omics” is hardly 5 years old and their application in the study of the human GI tract is even younger. Therefore, the analysis and interpretation of data derived from meta-“omics” approaches are still in infancy. We expect many novel technological procedures to be developed and improved in the coming years, not only with respect to wetlab technologies, such as pyrosequencing and functional screening tools, but also in the field of bioinformatics to analyse the enormous mass of data that is obtained with such approaches. Nevertheless, the first applications of meta-“omics” approaches have already demonstrated their power as discussed in this paper and we expect this field to grow explosively in the near future.
Only an integration of all reductionist and meta-“omics” approaches in the near future will provide adequate understanding of GI tract microbiota, as these approaches complement each other by delivering different pieces of the GI tract puzzle. For example, the analysis of different cell cultures will result in the discovery of novel genes and functions of individual organisms and therefore, serve as milestones for meta-“omics” approaches. This integration is essential to explain data derived from meta-“omics” analysis, since the limitations in our predictive capacity was demonstrated in the first metaproteomics study of the human GI tract microbiota.112
Overall, it will be a challenging future for GI tract researchers. With the introduction of the novel high-throughput technologies that are described here, it will be possible, for the first time, to obtain statistically relevant links between microbial phylotypes and activities, and human health. Ultimately, this will lead to the discovery of biomarkers that will help us to understand and predict the microbial life in our intestine, which is the dream of a GI tract microbial ecologist.
REFERENCES
Footnotes
Competing interests: None.