Article Text

Abstract
Background: Non-alcoholic fatty liver disease is a common liver injury, but the pathophysiological mechanisms leading to the development of non-alcoholic steatohepatitis (NASH) remain unclear. The pathological roles of the nuclear receptor constitutive androstane receptor (CAR), a key regulator of drug-metabolising enzymes, in the development of NASH were investigated.
Methods and results: CAR+/+ and CAR−/− mice were given a methionine and choline-deficient (MCD) diet to establish a dietary model of NASH. Increases in serum alanine aminotransferase (ALT) and in infiltration of inflammatory cells were dominant in CAR+/+ mice at 8 weeks. There was no significant difference in the lipid concentration of the liver—namely, the first hit between CAR+/+ and CAR−/− mice. The index of lipid peroxidation increased in liver of the CAR+/+ mice, as demonstrated by 8-iso-prostaglandin F2α (F2-isoprostanes). Western blotting analysis showed that nuclear translocation of CAR occurred in CAR+/+ mice fed the MCD diet. As a result, the CAR activation caused the lipid peroxidation—namely, the second hit. The expressions of cytochrome P450 (CYP)2B10, 2C29, 3A11 all increased considerably in the CAR+/+ mice. Furthermore, α smooth muscle actin immunohistochemistry and Sirius red staining showed an increase in the degree of fibrosis in CAR+/+ mice fed the MCD diet at 16 weeks. The mRNA expressions of collagen α1(1) and the tissue inhibitor of metalloproteinase-1 were found to be elevated in CAR+/+ mice.
Conclusion: CAR caused the worsening of the hepatic injury and fibrosis in the dietary model of NASH. Our results suggest that the CAR nuclear receptor may thus play a critical role in the pathogenesis of NASH.
- ALT, alanine aminotransferase
- CAR, constitutive androstane receptor
- CYP, cytochrome P450
- F2-isoprostane, 8-iso-prostaglandin F2α
- iNOS, inducible nitric oxide synthase
- MCD, methionine and choline-deficient
- NASH, non-alcoholic steatohepatitis
- NF-κB, nuclear factor-κB
- 8-OHdG, 8-hydroxy-2′-deoxyguanosine
- RXR, retinoid X receptor
- PXR, pregnane X receptor
- αSMA, α smooth muscle actin
- TCPOBOP, 1,4 bis[2-(3,5-dichloropyridyloxy)]benzene
- TNFα, tumour necrosis factor α
Statistics from Altmetric.com
- ALT, alanine aminotransferase
- CAR, constitutive androstane receptor
- CYP, cytochrome P450
- F2-isoprostane, 8-iso-prostaglandin F2α
- iNOS, inducible nitric oxide synthase
- MCD, methionine and choline-deficient
- NASH, non-alcoholic steatohepatitis
- NF-κB, nuclear factor-κB
- 8-OHdG, 8-hydroxy-2′-deoxyguanosine
- RXR, retinoid X receptor
- PXR, pregnane X receptor
- αSMA, α smooth muscle actin
- TCPOBOP, 1,4 bis[2-(3,5-dichloropyridyloxy)]benzene
- TNFα, tumour necrosis factor α
Non-alcoholic steatohepatitis (NASH) is a common liver injury, in which the histopathological abnormalities mimic those of alcoholic steatohepatitis.1 The histopathological features include steatosis, evidence of liver-cell injury, a mixed inflammatory lobular infiltrate and variable degrees of fibrosis.1–,3 The pathophysiological mechanisms leading to the development of NASH remain unclear.1–,3 A two-hit theory has been proposed, with hepatic steatosis as the first hit and the triggering host or environmental factors as the second hit, precipitating a cascade of events leading to cell necrosis, inflammation and fibrosis.4–,6
The nuclear receptor constitutive androstane receptor (CAR) is a key regulator of such xenobiotic-metabolising enzymes as cytochrome P450 (CYP),7–,9 UDP-glucuronosyltransferase10,11 and multidrug resistance-associated protein.12,13 CAR, which acts as a heterodimer with the retinoid X receptor (RXR), binds to a nuclear receptor-binding site NR1 within the 51 bp phenobarbital-responsive enhancer module, thereby activating the xenobiotic-metabolising enzyme genes.7–,9 CAR is known to upregulate CYP2B and 3A, which are considered to play an important role in drug metabolism. As a second hit, the environmental exposure to hepatotoxins has recently been implicated in NASH.14 Furthermore, some chemicals have been known to induce NASH clinically, hence drug-induced NASH. Because of its nature as a xenobiotic sensor for environmental pollution or drugs, CAR may play a role in the CYPs associated with NASH. Some other nuclear receptors, such as PPARα and PPARγ, are known to be involved in the pathogenesis of NASH.15,16
As a result, the nuclear receptor may play a critical role in the pathogenesis of NASH because of its ability to regulate genes. Nuclear receptors that regulate drug or xenobiotic-metabolising enzymes, in particular, may also play a pathological role in the development of NASH. However, the precise relationship between CAR and NASH is still not fully understood. In this study, we investigated the pathogenesis of CAR in the development of NASH using CAR null mice who received a methionine and choline-deficient (MCD) diet.
MATERIALS AND METHODS
Materials
1, 4 bis[2-(3, 5-dichloropyridyloxy)]benzene (TCPOBOP) was purchased from Sigma Chemicals (St Louis, Missouri, USA). The MCD diet and control diets were both purchased from Funabashi Farm (Chiba, Japan). All other chemicals were obtained from commercial sources at the highest grade of purity available.
Animals and treatment
The CAR+/+ and CAR−/− mice used in this study were generated as described previously.17 All mouse procedures were performed in accordance with the guidelines for animal care and use established by Gunma University Graduate School of Medicine, Maebashi, Gunma, Japan. Germline transmission of the disrupted allele was detected by polymerase chain reaction.18 Mice from each genotype were randomly divided into experimental groups and fed either a plain MCD diet or an MCD diet supplemented with choline bitartrate (2 g/kg) and dl-methionine (3 g/kg) designated as a control diet. Figure 1⇓ shows a schematic presentation of the treatment protocol. During the experimental period, the individual body weights and food intake were recorded 3 times per week. Male mice at 5–6 weeks of age were fed an experimental diet for 8 or 16 weeks (n = 6 per treatment group) and were then killed, and their sera and livers were collected. Each experiment was performed independently at least three times. To activate the CAR, TCPOBOP was given twice a week in olive oil at a dose of 3 mg/kg body weight from 6 to 8 weeks of feeding. In the control mice, 100 μl of olive oil per 25 g of body weight was injected. The serum alanine aminotransferase (ALT), total bilirubin, blood urea nitrogen, triglyceride and total cholesterol levels were all measured with an autoanalyser. For the histological examinations, liver tissue specimens were fixed in 10% formalin, embedded in paraffin wax and then stained with H&E. Blinded investigators (NS and KS) evaluated the slides and assigned a score for steatosis and inflammation as follows: steatosis—grade 0, none present; grade 1, steatosis of <25% of parenchyma; grade 2, steatosis of 26–50% of parenchyma; grade 3, steatosis 51–75% of parenchyma; and grade 4, steatosis >76% of parenchyma. For inflammation—grade 0, no inflammatory foci; grade 1, <5 inflammatory foci/hpf; and grade 2, >5 inflammatory foci/hpf. An immunohistochemical analysis for 8-hydroxy-2′-deoxyguanosine (8-OHdG), nitrotyrosine, nuclear factor-κB (NF-κB) and α smooth muscle actin (αSMA) was performed by the avidin–biotin–peroxidase complex method (Vectastain ABC kit, Vector Laboratories, Burlingame, California, USA) using anti- 8-OHdG (CHEMICON International, Temecula, California, USA), nitrotyrosine (CHEMICON International), NF-κB (Santa Cruz Biotechnology, Santa Cruz, California), αSMA (DAKO, Glostrup, Denmark) antibodies. The sites of peroxidase binding were determined using the diaminobenzidine method. At a magnification of ×200, 10 areas of αSMA positive cells were measured in a blinded fashion for each group. Sirius red staining was performed according to the usual method and the area of the Sirius red positive area was measured using the NIH image software program (National Institute of Health, Maryland, USA) in nine microscopic fields at a ×200 magnification and the mean (standard deviation (SD)) was calculated.
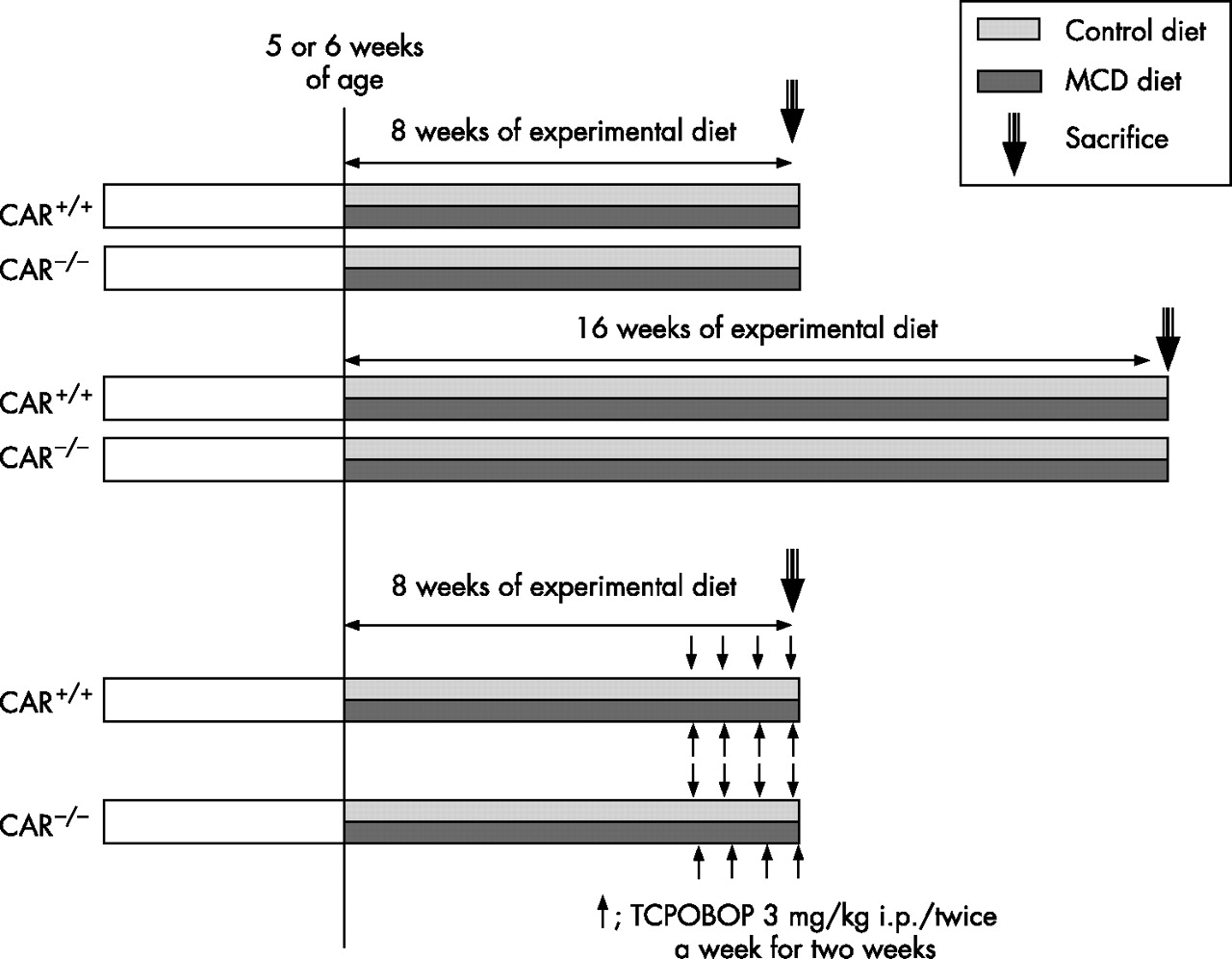
Treatment protocol.
Reverse transcription-polymerase chain reaction
Total RNA extraction from liver and the subsequent synthesis of first strand cDNA were performed using TRIzol reagent (Invitrogen, Carlsbad, California, USA) and the SuperScript preamplification system (Invitrogen), respectively. cDNAs were amplified using the following sets of primers: CYP1A2 mRNA, 5′-CCA AGG AGC GCT GTA TC-3′ and 5′-AAG CCG AAG AGC ATC AC-3′; CYP2A5 mRNA, 5′-CTC TTC TTT GCT GGC ACA-3′ and 5′-TCC GTA TAG GGC ATC TTC ATT-3′; CYP2B10 mRNA, 5′-CAC CAC GCT CCA CTA TGG CT-3′ and 5′-CTG TGT GGC ACT CCA ATA GGT ATA A-3′; CYP2D9 mRNA, 5′-ATT CCC GAT ACT CTT GCG-3′ and 5′-CAG GAA GGC ATC AGT CAA A-3′; CYP2E1, 5′-CCC AGG ACC TTT CCC AAT TC-3′ and 5′-TGA CCC AGG TGC AGT GTG AA-3′; CYP2C29 mRNA, 5′-AAA CAG GTA AAC CAC ATT GAA C-3′ and 5′-GTC AAT CTC TTC CTG GAC TTT AG-3′; CYP3A11 mRNA, 5′-GGG TGC TCC TAG CAA TCA GCT-3′ and 5′-GTG CCT AAA AAT GGC AGA GGT T-3′, CYP4A10, 5′-GTG CTG AGG TGG ACA CAT TCA T-3′ and 5′-TGT GGC CAG AGC ATA GAA GAT C-3′; inducible nitric oxide synthase (iNOS), 5′-GAG ATT GGA GGC CTT GTG-3′ and 5′-TCA AGC ACC TCC AGG AAC GT-3′; tumour necrosis factor α (TNFα), 5′-CTG TGA AGG GAA TGG GTG TT-3′ and 5′-GGG GGC TCT GAG GAG TAG AC-3′; tissue inhibitor of metalloproteinase-1, 5′-CAT GGA AAG CCT CTG TGG ATA TG-3′ and 5′-GAT GTG CAA ATT TCC GTT CCT T-3′; collagen α1(1), 5′-CCT CAG GGT ATT GCT GGA CAA C-3′ and 5′-ACC ACT TGA TCC AGA AGG ACC TT-3′; matrix metalloproteinase-13, 5′-ACT TAA CTT ACA GGA TTG TGA ACT ATA CTC CT-3′ and 5′-TGT CAG CAG TGC CAT CAT AGA TT-3′. One-twentieth of each cDNA synthesised from 5 μg of RNA was subjected to real time polymerase chain reaction using SYBR green dye (SYBR®PE Applied Biosystems, Foster City, California).
Hepatic lipid concentration and lipid peroxidation
Total liver lipids were extracted from 50 mg of liver homogenate using methanol and chloroform, and then were followed by a reaction with a vanillin-phosphoric acid reagent.19 The total triglycerides were determined using the triglyceride GPO-Trinder kit (Sigma-Aldrich).
To determine an index of lipid peroxidation, 8-iso-prostaglandin F2α (F2-isoprostane) levels were measured in 100 mg liver homogenate using the Direct 8-iso-Prostaglandin F2α Enzyme Immunoassay Kit (Assay Designs, Ann Arbor, Michigan, USA) following the manufacturer’s instructions.
Western blotting analysis
Nuclear extracts were resolved on a sodium dodecyl sulfate 10% polyacrylamide gel, transferred to a polyvinylidene difluoride membrane and incubated with anti-CAR antibody (Santa Cruz Biotechnology). Microsome extracts were also resolved on a 10% sodium dodecyl sulfate polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and incubated with anti-CYP2B1, CYP2C6, CYP3A2 (Daiichi Pure Chemicals, Tokyo, Japan) and CYP2E1 (BIOMOL International, LP, Plymouth meeting, Pennsylvania, USA) antibodies. After incubation with the secondary antibodies, the immunoreactive bands were visualised using an enhanced chemiluminescence system (Amersham, Buckinghamshire, UK).
Data analysis
All experimental data are shown as the mean (SD). The differences were determined by a one-way factorial analysis of variance for each group. The level of significance for all statistical analyses was set at p<0.05.
RESULTS
Body and liver weights of the mice
The mice fed the MCD diet lost weight in comparison to those fed the control diet in both CAR+/+ and CAR−/− mice (table 1⇓); this is consistent with the findings of a previous study,6 despite a higher food intake relative to their body weight. Despite the extent of weight loss, the general condition of the animals remained good and their behaviour appeared normal throughout the experimental period. There was no difference in the body weight between the CAR+/+ and CAR−/− mice fed the MCD diet for 8 weeks, although relative liver weight (liver weight/body weight) was raised considerably more in CAR+/+ mice (table 1⇓). As a result, livers of the CAR+/+ mice fed the MCD diet were considerably larger than livers of the CAR−/− mice.
The body weight, relative liver weight, and serum and hepatic lipids in constitutive androstane receptor (CAR)+/+ and CAR−/− mice fed the methionine and choline-deficient or control diet for 8 weeks
Serum alanine aminotransferase level and histological change with the MCD diet
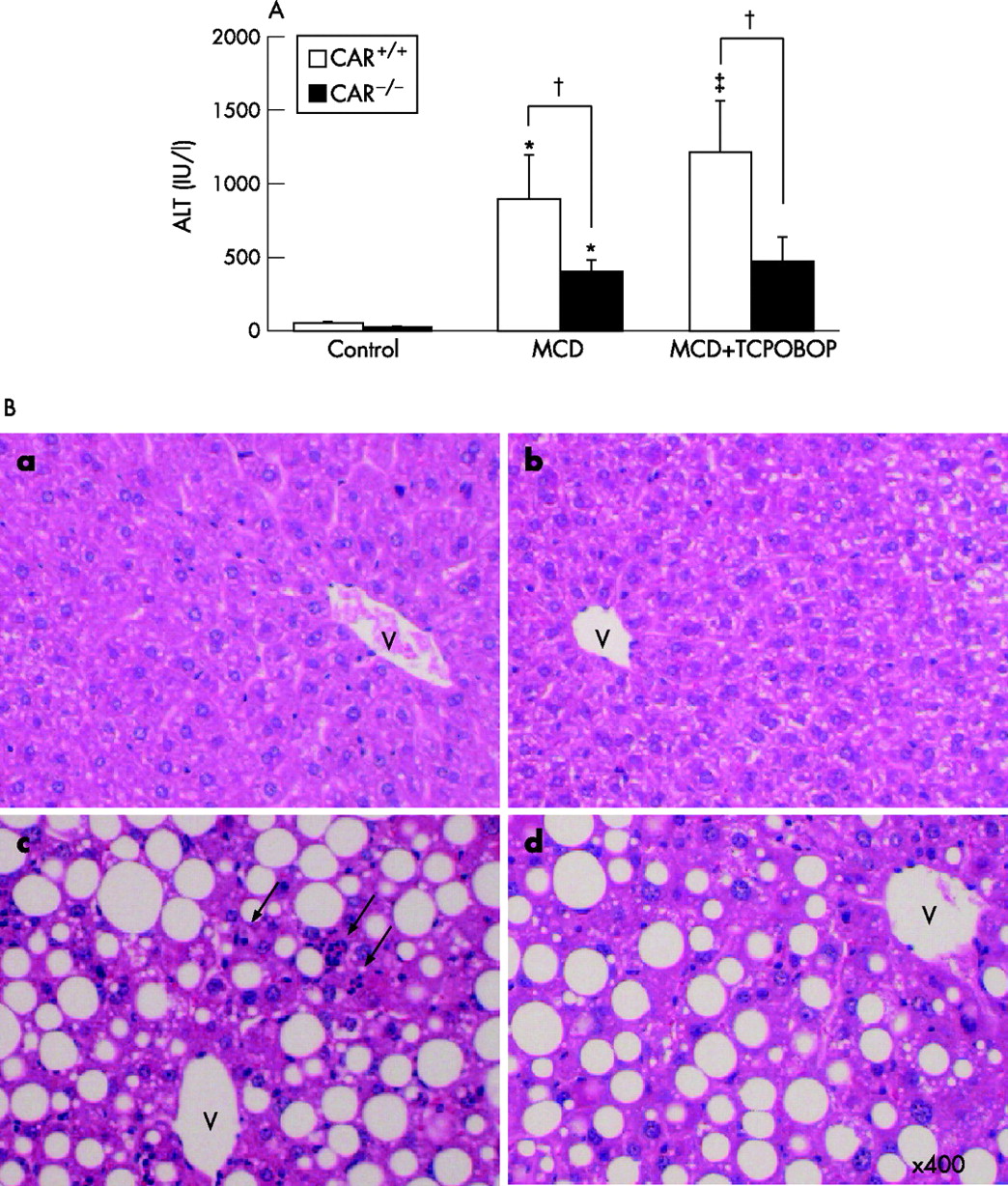
Consumption of the MCD diet resulted in a major increase in the serum ALT levels in comparison to the controls after receiving this dietary regimen for 8 weeks (fig 2A⇓). This elevation in the ALT level was significantly dominant in the CAR+/+ mice in comparison to CAR−/− mice. The administration of TCPOBOP, a CAR agonist, significantly raised the ALT level in the CAR+/+ but not in the CAR−/− mice (fig 2A⇓). H&E histological slides were evaluated by blinded investigators and analysed using a semiquantitative score for steatosis and inflammation. Consistent with serum ALT levels, inflammatory cell infiltration and hepatocyte necrosis dissecting liver parenchyma in the CAR+/+ mice were more severe than in the CAR−/− mice (fig 2B⇓). Inflammation score for the CAR−/− mice was lower than the CAR+/+ mice (0.8 (0.3) vs 1.7 (0.3); p<0.01). Both the CAR+/+ and CAR−/− mice fed the MCD diet had marked steatosis compared with the mice fed the control diet. Livers showed lipid droplets as clear macrovacuoles affecting all but zone 1 hepatocytes (periportal hepatocytes) (fig 2B⇓). Staining with oil red O confirmed the lipid content of these vacuoles (data not shown). There were no significant changes in steatosis score between the CAR+/+ and CAR−/− mice fed the MCD diet (2.3 (0.2) vs 2.2 (0.3)).

Alanine aminotransferase (ALT) elevation and inflammatory cell infiltration with the methionine and choline-deficient (MCD) diet for 8 weeks. (A) The ALT levels. 1, 4 bis[2-(3, 5-dichloropyridyloxy)]benzene (TCPOBOP) was administered as described in the Materials and methods. Data are the mean. *p<0.01 in comparison to control diet; †p<0.01 in comparison between constitutive androstane receptor (CAR)+/+ and CAR−/− mice; ‡p<0.05 in comparison to MCD diet without TCPOBOP. (B) The histological findings in liver (H&E staining); a, CAR+/+ mice, control diet; b, CAR−/− mice, control diet; c, CAR+/+ mice, MCD diet; d, CAR−/− mice, MCD diet. The administration of the MCD diet to both groups produced severe macrovesicular and panlobular steatosis. The infiltration of the inflammatory cells and hepatic necrosis was more prominent in liver of CAR+/+ than of CAR−/− mice. The arrows indicate the inflammatory cells infiltrations. V, central vein. Magnification, ×400.
Lipid content and lipid peroxidation in liver
Table 1⇑ shows the lipid contents of liver. Consistent with the morphological changes of lipid depositions, the total hepatic lipid content increased in the MCD diet in comparison with the control diet (table 1⇑). The concentrations of triglyceride increased threefold in liver obtained from the MCD diet-fed mice compared with those fed the control diet. However, there was no significant change in the hepatic triglyceride levels between CAR+/+ and CAR−/− mice fed the MCD diet. As a result, there was no difference in the first hit between the CAR+/+ mice and CAR−/− mice.
The index of lipid peroxidation—namely, the F2-isoprostane, increased threefold in liver prepared from the MCD diet-fed mice in comparison to the controls in the CAR+/+ mice after 8 weeks of treatment. The F2-isoprostane significantly increased in the CAR+/+ mice fed the MCD diet in comparison to the CAR−/− mice fed the MCD diet (fig 3A⇓). Immunohistochemistry for 8-OHdG, an index of oxidative DNA damage, showed increased accumulation in the nuclei of the hepatocytes in the CAR+/+ mice in comparison to the CAR−/− mice (fig 3B⇓).

(A) An index of lipid peroxidation measured with 8-iso-prostaglandin F2α (F2-isoprostane) in liver samples treated with each diet for 8 weeks. The data are presented as mean (SD). *p<0.01 in comparison to control diet; †p<0.01 in comparison between constitutive androstane receptor (CAR)+/+ and CAR−/− mice. (B) 8-hydroxy-2′-deoxyguanosine (8-OHdG) staining. a, CAR+/+ mice, control diet; b, CAR−/− mice, control diet; c, CAR+/+ mice, methionine and choline-deficient (MCD) diet; and d, CAR−/− mice, MCD diet. The arrows indicate the positive staining of 8-OHdG to the nuclei. Magnification, ×400.
Nuclear translocation of CAR with MCD diet
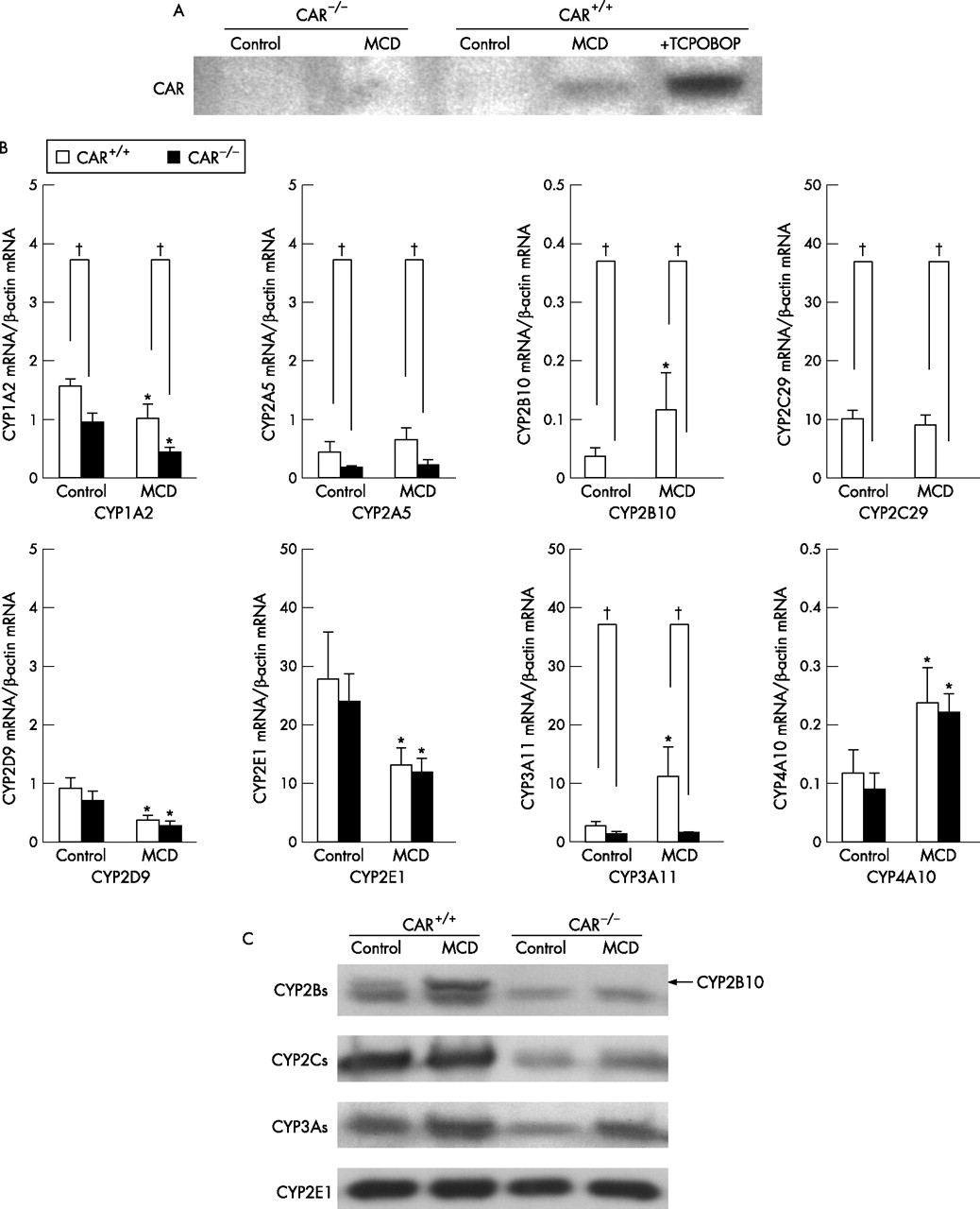
It is well known that the first step in inducing of CYPs is translocation of CAR into nuclei. To evaluate the nuclear translocation of CAR, the nuclear extracts were subjected to western blotting analysis. Nuclear content of CAR increased in CAR+/+ mice fed the MCD diet for 8 weeks (fig 4A⇓). Thus, CAR activation step occurred in CAR+/+ mice fed the MCD diet.

Nuclear translocation of constitutive androstane receptor (CAR) and hepatic mRNA and protein levels fed the methionine and choline-deficient (MCD) diet or the control diet for 8 weeks. (A) Western blotting analysis of nuclear CAR. Nuclear extracts were prepared and subjected to an analysis with anti-CAR antibody as indicated in the materials and methods. (B) mRNA expression of cytochromes P450 (CYPs). Total liver RNA was prepared from CAR+/+ or CAR−/− mice and subjected to a real-time polymerase chain reaction analysis with the indicated primers in the materials and methods. *p<0.01 in comparison to control diet, †p<0.01 in comparison between CAR+/+ and CAR−/− mice. (C) The CYP2B, CYP2C, CYP3A and CYP2E protein expression. Microsome extracts were prepared from liver and then were subjected to a western blotting analysis as described in the Materials and methods. The arrow indicates the corresponding band for CYP2B10.
Hepatic gene and protein expressions in CAR+/+ and CAR−/− mice fed the MCD diet
To characterise the hepatic gene expression related to NASH in CAR+/+ and CAR−/− mice fed the MCD or control diet, we determined the levels of liver CYPs, iNOS and TNFα mRNA levels. As a result, no differences were observed in the CYP2E1, CYP2D9 and CYP4A10 expressions between the CAR+/+ and CAR−/− mice fed the MCD diet (fig 4B⇑). The mRNA expression of CYP2B10, CYP2C29 and CYP3A11 increased in the CAR+/+ mice fed the MCD diet in comparison to the CAR−/− mice. The mRNA expression of CYP1A2 and CYP2A5 decreased in the CAR−/− mice in comparison to the CAR+/+ mice, although the differences were smaller than those of CYP2B10, CYP2C29 and CYP3A11.The protein levels of CYP2B10, CYP2C29, CYP3A11 and CYP2E1 were also determined by western blotting (fig 4C⇑). The protein levels of CYP2B10, CYP2Cs and CYP3As increased in the CAR+/+ mice fed the MCD diet in comparison to the CAR−/− mice. In addition, no significant change was seen in the protein levels of the CYP2E1 expression between CAR+/+ and CAR−/− mice.
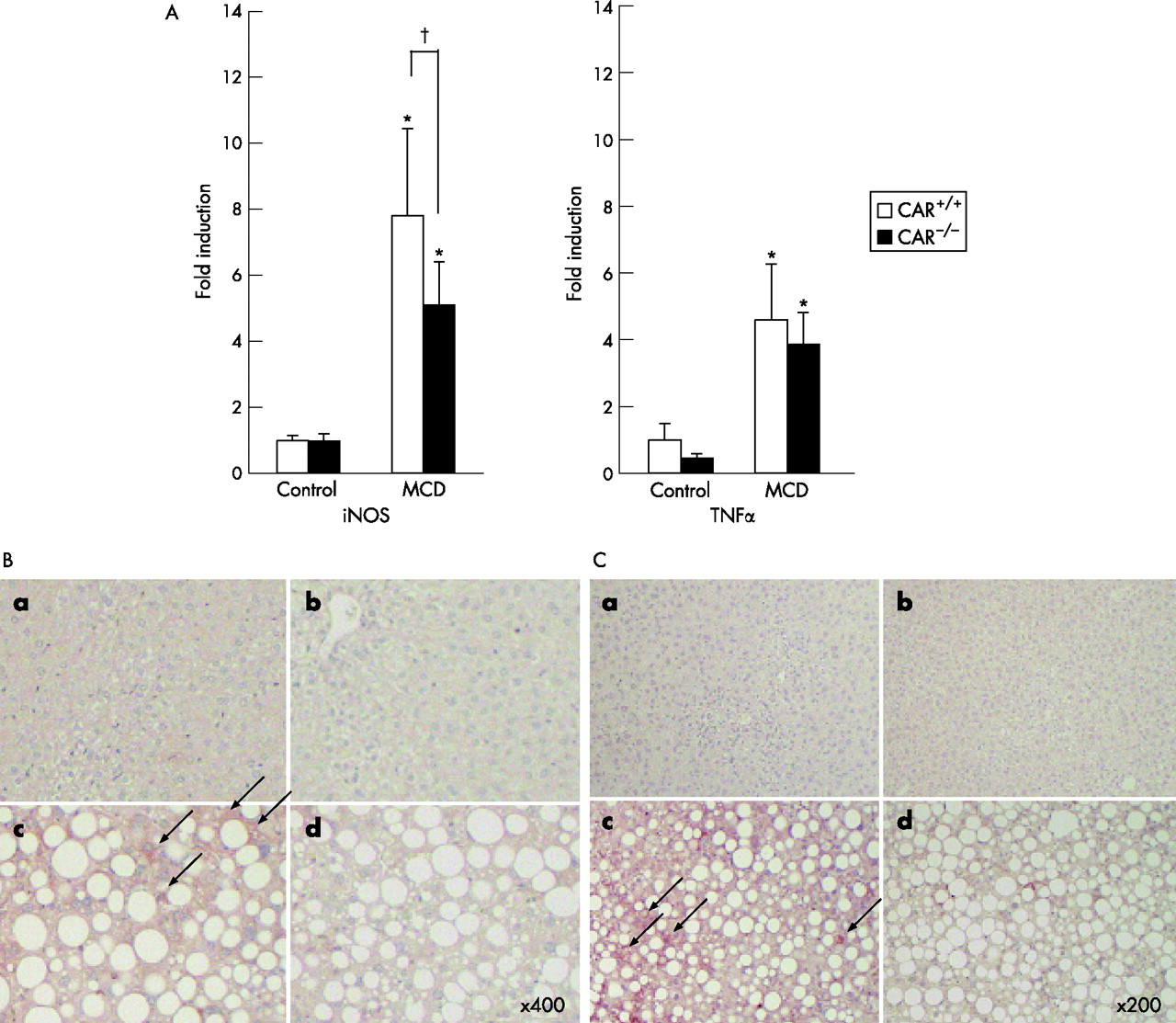
The promoter region of iNOS, one of the second hit candidates of NASH, have nuclear receptor binding sites including CAR/RXR and pregnane X receptor (PXR)/RXR.20 The mRNA expression of iNOS was repressed in the CAR−/− mice in comparison with the CAR+/+ fed the MCD diet (fig 5A⇓). Furthermore, immunohistochemistry for nitrosylation showed an increased amount of protein nitrosylation in CAR+/+ than in CAR−/− mice (fig 5B⇓). The mRNA expression of TNFα was slightly repressed in CAR−/− than in CAR+/+ mice fed the MCD diet (fig 5A⇓), although it did not reach statistical significance. As NF-κB is activated in the presence of proinflammatory stimuli, and its activation results in an increased expression of various proinflammatory and immune response genes including TNFα, immunohistochemistry for NF-κB was performed. It showed increased nuclear and cytosolic expressions of activated NF-κB in CAR+/+ mice fed the MCD diet (fig 5C⇓).

(A) mRNA expression of inducible nitric oxide synthase (iNOS) and tumour necrosis factor α (TNFα). *p<0.01 in comparison to control diet, †p<0.01 in comparison between constitutive androstane receptor (CAR)+/+ and CAR−/− mice. (B) Nitrotyrosine staining. a, CAR+/+ mice, control diet; b, CAR−/− mice, control diet; c, CAR+/+ mice, methionine and choline-deficient (MCD) diet; d, CAR−/− mice, MCD diet. Magnification, ×400. (C) Nuclear factor-κB staining. a, CAR+/+ mice, control diet; b, CAR−/− mice, control diet; c, CAR+/+ mice, MCD diet; d, CAR−/− mice, MCD diet. Magnification, ×200.
Long-term treatment with the MCD diet caused hepatic fibrosis and an elevation in the mRNA expression of the gene related to liver fibrosis
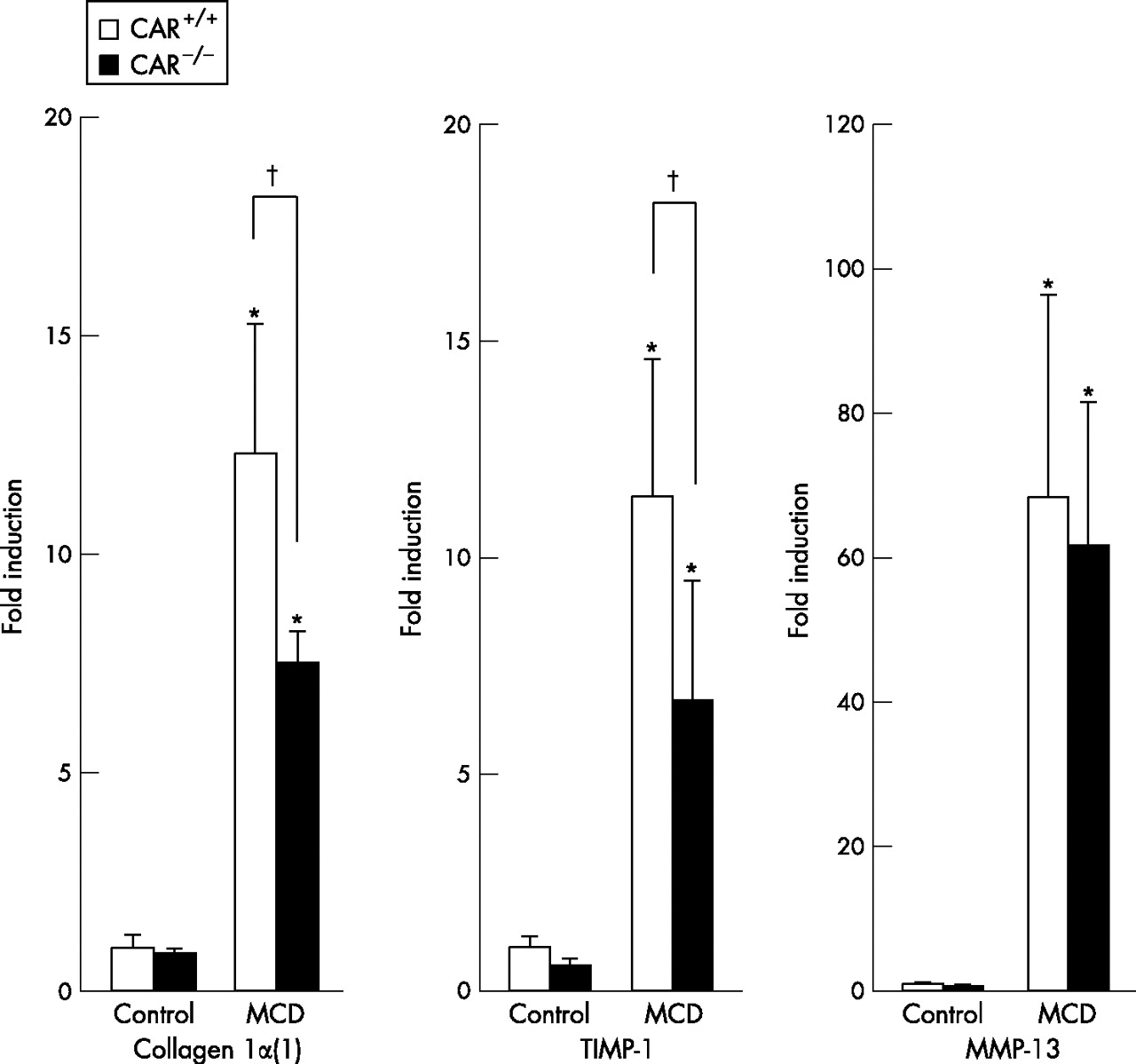
To evaluate the effect of CAR on hepatic fibrosis in a NASH model with MCD diet, long term effect of MCD diet was evaluated in CAR+/+ and CAR−/− mice. At 16 weeks after consuming the MCD diet, both perivenular and pericellular fibrosis was more clearly observed in CAR+/+ than in CAR−/− mice. Sirius red (fig 6A⇓,B) and immunohistochemistry for αSMA (fig 6C⇓,D) showed an increase in the fibrotic changes in CAR+/+ than in CAR−/− mice. Furthermore, the mRNA expressions of fibrogenic markers, collagen α1(1) and tissue inhibitor of metalloproteinase-1 levels were considerably raised in CAR+/+ than CAR−/− (fig 7⇓). There was no difference in the mRNA expression of the anti-fibrogenic marker matrix metalloproteinase-13 between CAR+/+ and CAR−/− mice (fig 7⇓).

Long-term treatment with methionine and choline-deficient (MCD) diet for 16 weeks. (A) Sirius red staining; a, constitutive androstane receptor (CAR)+/+ mice, control diet; b, CAR−/− mice, control diet; c, CAR+/+ mice, MCD diet; d, CAR−/− mice, MCD diet. (B) The quantification of sirius red positive areas using the NIH image software program. Nine microscopic fields at ×200 magnification were measured and the mean (SD) was shown. *p<0.01 in comparison to control diet, †p<0.01 in comparison between CAR+/+ and CAR−/− mice. (C) α smooth muscle actin (αSMA) staining; a, CAR+/+ mice, control diet; b, CAR−/− mice, control diet; c, CAR+/+ mice, MCD diet; d, CAR−/− mice, MCD diet. (D) The number of αSMA positive cells. Ten areas of αSMA positive cells were counted at ×200 magnification and the mean (SD) was calculated. Magnification, ×200. *p<0.01 in comparison to control diet, †p<0.01 in comparison between CAR+/+ and CAR−/− mice.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hepatic mRNA expressions relating to fibrosis. Total liver RNA was prepared from constitutive androstane receptor (CAR)+/+ or CAR−/− mice treated with each diet for 16 weeks, and then were subjected to a real-time polymerase chain reaction analysis with the indicated primers in the materials and methods. The fold induction of the mRNA level from the CAR+/+ mice fed the control diet was indicated. *p<0.01 in comparison to control diet, †p<0.01 in comparison between CAR+/+ and CAR−/− mice.
DISCUSSION
In this study, we showed that CAR worsened the dietary model of NASH with the MCD diet, associated with an elevation in the lipid peroxidation and the induction of CYPs and iNOS. There was also no change in the lipid concentration of liver between CAR+/+ and CAR−/− mice. As a result, there was no change in the first hit. Lipid peroxidation caused the inflammation to develop into NASH. Furthermore, the long-term treatment caused an elevation of the fibrogenic gene expression and fibrosis. Although a study by Leclercq et al6 showed an elevation of CYP2E1 mRNA and protein, and indicated CYP2E1 played a major role in the MCD diet NASH model, there were no significant differences in the CYP2E1 and CYP4A expression between CAR+/+ and CAR−/− mice in our study. In comparison to the findings of their study, the development of NASH with MCD diet occurred more slowly in this strain of mice (C3H). CYPs are well known to have species or sex differences. Such species or sex differences may thus have caused the differences between our study (C3H male mice) and their study (C57BL6/J female mice). CYP2E1 has been proposed to be a potential inducer of lipid peroxidation in NASH, as its expression and activity have been shown to be upregulated in rat and mouse models of NASH6,21,22 and in humans.23,24 However, Leclercq et al6 showed that the development of experimental NASH could not be prevented in CYP2E1 knockout mice. They suggested that hepatic CYP4A might be a source of lipid peroxidation and a substitute for CYP2E1 if the latter is absent.6 As a result, CYPs have low substrate specificities and other CYPs may be substituted for repressed CYP. Not only CYP2E1 but also CYP1A, CYP2B and CYP3A have been reported to be induced by alcoholic hepatitis.25 Kono et al25 showed that CYP2B and CYP3A both produce reactive oxygen species, which causes alcoholic hepatitis, while CYP2E1 plays only a small role in the mechanism of early alcoholic liver injury. As a result, many CYPs other than CYP2E1 are also related to NASH or alcoholic hepatitis. Although one of the CYPs related to NASH was knocked out in mice, the CYPs network can be substituted for repressed CYP. CYPs network is closely associated with each other and thus overlaps with the substrates. Therefore, nuclear receptors regulating CYPs seem to play an important role in the pathogenesis of NASH.
Nuclear receptors PPARα and PPARγ have been reported to be involved in the pathogenesis of NASH.15,16 CAR also belongs to the nuclear receptor superfamilies forming a heterodimer with RXR in the same manner as PPARα and PPARγ. PPARα is highly expressed in liver and its activation by agonists leads to augmented fatty acid oxidation while also protecting against steatosis. PPARγ, which is transcriptionally upregulated in steatosis, activates lipogenic enzymes and exacerbates steatosis. However, the roles of these nuclear receptors in the pathogenesis of NASH are more complicated and are not simple to understand. Ip et al26 recently showed that the massive induction of CYP4A after the administration of a PPARα agonist in MCD mice did not lead to an increased lipid peroxidation. Interestingly, in the study by Ip et al, the pretreatment of MCD mice with a PPARα agonist not only induced CYP4A, but also prevented the occurrence of steatosis.26,27 As a result, CYPs or lipid metabolising enzymes are closely associated with each other while also interacting with each other. To understand these networks of CYPs and the lipid metabolic enzymes regulated by nuclear receptors is thus considered to be one way to show the pathogenesis of NASH.
Human beings are exposed to various types of environmental pollution or drugs throughout their lifetime. Environmental pollution or drugs are one of the causes of NASH,14 and they are potent inducers of nuclear receptor CAR and CYPs. In our study, the MCD diet caused an elevation in the ALT levels in CAR+/+ than in CAR−/− mice without a CAR inducer. Environmental stimulation may also cause the up regulation of CAR, due to the constitutively active nature of CAR. If environmental pollution or drugs cause NASH, then an inverse agonist of CAR, such as androstanol, may be a potentially effective treatment strategy for NASH.
In this study, the short-term administration of CAR agonist, TCPOBOP caused the severe liver injury in CAR+/+ mice fed the MCD diet. TCPOBOP itself did not have any toxicity in mice fed the control diet. Treatment with the MCD diet resulted in the nuclear translocation of CAR while also increasing the expression of CAR target genes in CAR+/+ mice. The administration of TCPOBOP enhanced this phenomenon more clearly. Preliminarily, we also tried to evaluate the parameters of liver fibrosis with long-term administration of TCPOBOP. However, the CAR activation with TCPOBOP caused the marked liver enlargement in the CAR+/+ mice. As shown in table 1⇑, MCD diet itself caused liver enlargement in the both mice, more prominent in the CAR+/+ mice. We tried to inject the same dosage of TCPOBOP intraperitoneally with this experiment for several weeks. However, the marked liver enlargement in CAR+/+ mice fed MCD diet prevented us from continuing the injections long term to evaluate the fibrosis. Because TCPOBOP enhanced liver injury after short-term administration, we thus think that the long-term administration of a small amount of TCPOBOP may worsen the degree of liver fibrosis as a result of liver injury.
CAR is known not only as a CYP regulating receptor, but also as a regulator of many other enzymes.18 Since the iNOS-induced production of nitric oxide is known to influence inflammation and apoptosis, iNOS is therefore considered to play a role in the pathogenesis of NASH. In addition, CAR/RXR and PXR/RXR binding sites have also reported in the promoter region of the human iNOS gene.20 In the natural iNOS promoter context, the DR4-type response element specifically mediates the down regulation of the promoter activity by androstanol through CAR/RXR heterodimers and the upregulation by the xenobiotic drug clotrimazole through PXR/RXR heterodimers.20 In our study, iNOS was induced with the MCD diet to a greater content than in the controls, and it was also elevated in CAR+/+ mice in comparison to the level in CAR−/− mice fed the MCD diet. CAR and PXR have a rather broad, overlapping set of ligands and they also recognise similar DNA binding sites in the promoter regions of regulating genes. Because PXR is also a key regulator of the CYPs and drug metabolising enzymes, PXR may also contribute to the pathogenesis of NASH. Although PXR null or CAR/PXR double null mice were not available for this study, further studies using PXR null mice should provide us with valuable insight into the precise understanding of the pathogenesis of NASH.
In conclusion, the nuclear receptor CAR worsened the dietary model of NASH inducing lipid peroxidation. Our results suggest that the nuclear receptor CAR may thus play a critical role in the pathogenesis of NASH. Further study evaluating the role of CAR and PXR in NASH is thus called for in the future.
REFERENCES
Footnotes
Published Online First 1 September 2006
Funding: This work was supported in part by a Grant-in Aid for Scientific Research (No 18590716) from the Ministry of Education, Science, Sports and Culture of the Japanese Government.
Competing interests: None.