


Abstract
Arylamine N-acetyltransferase-1 (NAT1) is a polymorphically expressed enzyme that is widely distributed throughout the body. In the present study, we provide evidence for substrate-dependent regulation of this enzyme. Human peripheral blood mononuclear cells cultured in medium supplemented withp-aminobenzoic acid (PABA; 6 μM) for 24 h showed a significant decrease (50–80%) in NAT1 activity. The loss of activity was concentration-dependent (EC50 ∼ 2 μM) and selective because PABA had no effect on the activity of constitutively expressed lactate dehydrogenase or aspartate aminotransferase. PABA also induced down-regulation of NAT1 activity in several human cell lines grown at confluence. Substrate-dependent down-regulation was not restricted to PABA. Addition of other NAT1 substrates, such as p-aminosalicylic acid, ethyl-p-aminobenzoate, or p-aminophenol to peripheral blood mononuclear cells in culture also resulted in significant (P < .05) decreases in NAT1 activity. However, addition of the NAT2-selective substrates sulfamethazine, dapsone, or procainamide did not alter NAT1 activity. Western blot analysis using a NAT1-specific antibody showed that the loss of NAT1 activity was associated with a parallel reduction in the amount of NAT1 protein (r 2 = 0.95). Arylamines that did not decrease NAT1 activity did not alter NAT1 protein levels. Semiquantitative reverse transcriptase polymerase chain reaction of mRNA isolated from treated and untreated cells revealed no effect of PABA on NAT1 mRNA levels. We conclude that NAT1 can be down-regulated by arylamines that are themselves NAT1 substrates. Because NAT1 is involved in the detoxification/activation of various drugs and carcinogens, substrate-dependent regulation may have important consequences with regard to drug toxicity and cancer risk.
Acetylation is a major route of biotransformation for several therapeutic arylamine and hydrazine drugs. It also plays an important role in the bioactivation of numerous potential human carcinogens in the diet, cigarette smoke, and the environment (Kato, 1986; Minchin et al., 1992; Hein et al., 1993; Hein et al., 1994; Zenser et al., 1996). In humans, these acetylation reactions are catalyzed by two closely related cytosolic enzymes, N-acetyltransferase-1 (NAT1; EC 2.3.1.5) and N-acetyltransferase-2 (NAT2; EC 2.3.1.5). Despite sharing an 87% sequence homology, NAT1 and NAT2 differ considerably with regard to substrate specificity (Minchin et al., 1992; Hein et al., 1993), tissue distribution (Pacifici et al., 1986), and developmental pattern (Pacifici et al., 1986; Pariente-Khayat et al., 1991). NAT1 appears to be located in almost every tissue examined (Blondheim, 1955; Pacifici et al., 1986; Grant et al., 1989; Coroneos and Sim, 1991), whereas NAT2 is primarily located in the liver (Deguchi, 1992) and colon epithelium (Ilett et al., 1994). A notable difference between the two isozymes is the presence of NAT1 in fetal and neonatal tissue (Vest and Salzberg, 1965; Pacifici et al., 1986). By contrast, NAT2 is not detectable until about 12 months after birth (Pariente-Khayat et al., 1991). Recent studies have shown that NAT1 is competitively inhibited by folic acid and folate analogs (Sim and Ward, 1995; Ward et al., 1995), with the folate catabolitep-aminobenzoylglutamate identified as a selective endogenous substrate for the NAT1 isozyme (Minchin, 1995). These observations suggest that NAT1 may have important cellular functions in addition to its role in xenobiotic metabolism.
NAT1, like NAT2, exhibits a genetic polymorphism in humans (Vatsis and Weber, 1993; Vatsis et al., 1995; Butcher et al., 1998; Hughes et al., 1998; Lin et al., 1998) that affects its functional activity (Butcher et al., 1998). Nevertheless, interindividual variation in activity can range over 1 to 2 orders of magnitude (Weber and Vatsis, 1993; Vatsis and Weber, 1994). This variability has been observed within a single phenotype (Butcher et al., 1998), suggesting that nongenetic factors may contribute to overall activity in vivo.
Many enzymes that exhibit genetic polymorphism also are regulated by environmental factors. Regulation can include both substrate-dependent activation/induction and down-modulation (Guzelian, 1988; Song et al., 1989; Johnson, 1991; Ramsay, 1998). In the present study, we have investigated the effects of various arylamine compounds on the activity of human NAT1 in peripheral blood mononuclear cells (PBMC) and several human cell lines. We found that substrates for the enzyme induced a time-dependent loss in NAT1 activity, which correlated with a loss in enzyme protein. The results provide evidence for substrate-dependent regulation of NAT1, which may have important implications for the cellular function(s) of this enzyme.
Experimental Procedures
Materials.
Ficoll-Paque and enhanced chemiluminescence detection reagent were purchased from Amersham Australia (Sydney, Australia). Cell culture media were purchased from Life Technologies (Melbourne, Australia), and fetal calf serum was obtained from WA Serum Laboratories (Perth, Australia).p-Aminobenzoic acid (PABA), p-aminosalicylic acid (PAS), p-aminophenol (PAP),N-acetyl-p-aminophenol (NAPAP), sulfamethoxazole (SMX), sulfamethazine (SMZ), ethyl-p-aminobenzoate (EPAB), dithiothreitol (DTT), cycloheximide (CHX), and phorbol 12-myrisate 13-acetate (PMA) were obtained from Sigma (Castle Hill, Australia). Acetyl-coenzyme A (AcCoA) was purchased from Boehringer-Mannheim (New South Wales, Australia). Polymerase chain reaction (PCR) primers were obtained from Bresatec (Castle Hill, Australia), and Taq DNA polymerase was purchased from Fisher Biotech (Adelaide, Australia).
Isolation and Culture of PBMC.
Blood was collected from healthy volunteers and anticoagulated with EDTA. PBMC were isolated by mixing whole blood with an equal volume of PBS, which was then layered on an equal volume of Ficoll-Paque and centrifuged (16–18°C) at 400g for 20 min. The PBMC layer was collected, washed once with PBS, and resuspended in basal medium Eagle (BME), RPMI 1640, or Dulbecco's modified Eagle's medium (DMEM) culture medium. Media were supplemented with 10% fetal calf serum, gentamicin (50 μg/ml), and benzylpenicillin (80 μg/ml). PBMC were cultured at 37°C in an atmosphere of 5% CO2/95% air at a density of 1 × 106 cells/ml. The sodium salts of PABA and PAS and the hydrochloride of PAP were dissolved in PBS. SMX, SMZ,N-acetyl-p-aminobenzoic acid (NAPABA), and NAPAP were dissolved in ethanol. EPAB was dissolved in dimethyl sulfoxide. The final concentration of ethanol or dimethyl sulfoxide in the culture medium was 0.1% and did not affect NAT1 activity. Cell viability was assessed by trypan blue exclusion and was >95% for all treatments.
Assay of NAT1 Activity.
PBMC were washed with PBS to remove any residual PABA and resuspended in 0.8 ml of 20 mM Tris/1 mM EDTA buffer, pH 7.4, containing 1 mM DTT and disrupted at 4°C using a cell sonicator (Branson Sonifier B250; duty cycle 100%, output = 5; 3 × 5 s bursts). The cell lysate was then centrifuged for 3 min at 16,000g (4°C), and the supernatant was retained for assay of NAT1 activity. Protein concentration was determined by the method of Bradford (1976). Reaction vials contained 5 to 10 μg of cell lysate protein, 440 μM PABA, and 1.1 mM AcCoA in a 150-μl total volume. In some experiments, the concentrations of AcCoA and PABA were varied from 0 to 640 μM so that K mand V max values could be estimated. Reactions were started by the addition of AcCoA, then incubated at 37°C for 30 min and terminated by the sequential addition of trichloroacetic acid (25%; 25 μl) and 25 μl 2 M NaOH. After centrifugation at 16,000g for 3 min, NAPABA in the supernatant was quantified by HPLC (Chan et al., 1988). NAT1 activity was normalized to lysate protein concentration, and, under these conditions, the rate of PABA acetylation was linear with respect to time and protein concentration. There was no formation of NAPABA in the absence of added PABA, indicating that PABA present in the culture medium did not interfere with the determination of NAT1 activity.
Assay of Lactate Dehydrogenase (LDH) and Aspartate Aminotransferase (AST).
LDH and AST assays were performed on PBMC lysates (prepared as above) using diagnostic kits obtained from Randox Laboratories (Antrim, UK). Assays were run on a Cobas Mira Analyzer (Roche Diagnostic Systems, Sydney, Australia) according to the protocols supplied by the manufacturer.
Western Blots for NAT1.
Cell lysates (15 μg) were electrophoresed on 12% (w/v) SDS-polyacrylamide gels, transferred to nitrocellulose membranes (100 mA, overnight, 4°C), and immunoblotted using a polyclonal NAT1-specific antibody (Stanley et al., 1996). Briefly, membranes were blocked for 1 h at room temperature with 5% (w/v) skim milk powder in PBS and then washed for 1 h with 0.05% (v/v) Tween 20 in PBS. After washing, membranes were incubated at room temperature with NAT1 antibody (diluted 1:4000 with 0.05% (v/v) Tween 20 in PBS) for 2 h, washed for 30 min, incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG [diluted 1:10,000 with 0.05% (v/v) Tween 20 in PBS] for 1 h, and then washed for another 30 min. NAT1 was visualized using enhanced chemiluminescence detection. Protein content was estimated from Western blots by densitometry using ImageQuant Ver. 1.1 (Molecular Dynamics, Sunnyvale, CA).
Extraction of Total RNA and Reverse Transcription (RT).
Total RNA was extracted from PBMC cultured in the absence or presence of PABA using a High Pure RNA Isolation Kit (Boehringer-Mannheim, Castle Hill, Australia) according to the manufacturer's instructions. Total RNA was serially diluted with RNase-free water over the range 16.6 to 500 ng/4 μl and reverse transcribed using avian myeloblastosis virus-RT (Promega, Sydney, Australia). RT reactions contained 4 μl of diluted RNA, 5 mM MgCl2, 1 mM dNTPs, 1 U/μl rRNasin ribonuclease inhibitor, 0.5 μg oligo(dT)15 primer, and 15 U avian myeloblastosis virus-RT in 1× RT buffer (10 mM Tris-HCl, pH 8.8, 50 mM KCl, and 0.1% Triton X-100). Reactions were incubated at 42°C for 30 min, and then the RT was inactivated by heating at 98°C for 5 min.
Semiquantitative PCR of NAT1 cDNA.
cDNA from cells grown in the absence or presence of PABA was amplified by PCR using specific primers for NAT1 (forward primer, 5′-GGG AGG GTA TGT TTA CAG CA-3′; reverse primer, 5′-TTT AGA TAG TGG TAC AAA CCC-3′) or β-actin (forward primer, 5′-TCA CCC ACA CTG TGC CCA TCT ACG A-3′; reverse primer, 5′-CAG CGG AAC CGC TCA TTG CCA ATG G-3′). PCR were carried out in 1× PCR buffer containing 5 μl of cDNA mix, 2 mM MgCl2, 0.5 U Taq DNA polymerase, and 12.5 pmol of each primer in a final volume of 20 μl. Samples were amplified using a Perkin Elmer Thermal Cycler (model 9600) and the following conditions; denaturation at 95°C for 5 min, followed by 25 (β-actin) or 34 (NAT1) cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 45 s. PCR products were then separated on 0.8% agarose gels, and quantities were estimated by densitometry using ImageQuant Ver. 1.1 (Molecular Dynamics).
Data Analysis and Statistics.
Data are expressed as mean ± S.E. Statistical comparisons between different treatments were assessed by Student's t tests. Values ofP < .05 were considered significant (SigmaStat Ver. 1.01; Jandel Scientific, San Rafael, CA).
Results
Effect of PABA on NAT1 Activity in Cultured Cells.
Cultured human PBMC were used to investigate the effect of PABA on NAT1 activity. Cells were cultured in BME for up to 3 days in the absence or presence of PABA (6 μM), and NAT1 activity (measured as theN-acetylation of PABA in vitro) was assessed. The NAT1 activity of cells cultured in the presence of PABA was significantly (P < .05) reduced with time in culture compared with cells grown in the absence of PABA (Fig.1A). Near maximal loss occurred within 24 h and was typically between 50 and 80% of the initial value, but showed marked variation between cell preparations. PBMC also were grown in DMEM or RPMI 1640 culture media. Cells cultured in DMEM showed no loss of NAT1 activity with time in culture. However, cells grown in RPMI 1640 showed a similar decrease in NAT1 activity to that observed with culture in BME supplemented with PABA. Comparison of the constituents of the different media showed that DMEM, like BME, was PABA-free whereas RPMI 1640 contained around 6 μM PABA. When cells were cultured in DMEM supplemented with 6 μM PABA, NAT1 activity decreased with a similar profile to that seen with culture in RPMI 1640 or BME containing 6 μM PABA (Fig. 1B). This PABA-induced down-regulation of NAT1 activity was dose-dependent, with a half-maximal effect at around 2 μM (Fig. 1C). Moreover, PABA (25 μM for 24 h) was able to down-regulate NAT1 activity in a number of different human cell lines (Table 1).

Effect of PABA on the activity of NAT1 in PBMC. A, time-dependent change in NAT1 activity of PBMC in the absence (●) and presence (○) of 6 μM PABA. Cells were cultured in BME for up to 3 days. At each time point, the cells were washed to remove residual PABA and supernatants were prepared. NAT1 activity was determined using 440 μM PABA and 1.1 mM AcCoA. Data are presented as mean ± S.E. (n = 3), and an asterisk indicates significant difference (P < .05) compared with cells cultured in the absence of PABA. B, activity of NAT1 in cells cultured in various media in the absence (■) and presence (▪) of 6 μM PABA. PBMC were cultured for 24 h in each medium, and NAT1 activity was determined. Data are presented as mean ± S.E. (n = 3), and an asterisk indicates significant difference (P < .05) compared with freshly isolated cells (control). C, concentration-dependent down-regulation of NAT1 activity by PABA. Cells were cultured in BME as described above with PABA (0–10 μM) for 24 h, after which NAT1 activity was determined. D, effect of mitogen stimulation on PABA-dependent down-regulation of NAT1. PBMC were cultured in BME in the presence (●) of 6 μM PABA. At t = 0, some cells were treated with 100 ng/ml PMA and PABA (○). In these cells, down-regulation of NAT1 was evident for the initial 24 h, after which activity returned to control values. This induction was paralleled by an increase in cell proliferation as measured by [3H]thymidine incorporation into DNA (data not shown). Cells treated with only 100 ng/ml PMA are also shown (▪).
Effect of PABA on NAT1 activity in human cell lines
Treatment of PABA down-regulated PBMC with the mitogen PMA (100 ng/ml) restored NAT1 activity to initial values. However, PMA treatment of PBMC that were not down-regulated with PABA had no significant effect on NAT1 activity (Fig. 1D). To determine whether the loss of NAT1 activity caused by PABA was a specific effect, activities of the constitutively expressed housekeeping enzymes LDH and AST were measured in cells cultured in the absence or presence of 6 μM PABA. After culture for 24 h in media containing PABA, NAT1 activity was reduced to 29 ± 9% of that measured in cells grown in PABA-free medium, but LDH and AST activities were not changed (99 ± 4 and 101 ± 7%, respectively).
Kinetic Studies.
N-Acetylation by NAT1 proceeds via sequential interaction with cofactor (AcCoA) and substrate (PABA). To determine whether the loss of PBMC NAT1 activity after culture in the presence of PABA was due to a decreased ability of the enzyme to interact with either substrate or cofactor, the kinetic parametersK m and V maxwere measured. Compared with cells grown in PABA-free media, cells grown in the presence of 6 μM PABA showed a significant (P < .05) reduction inV max for both PABA (23.2 ± 0.9 and 15.3 ± 0.4 nmol/min/mg protein, respectively) and AcCoA (18.9 ± 0.5 and 6.8 ± 0.2 nmol/min/mg protein, respectively). However, the K m values for both PABA and AcCoA were not significantly different for cells cultured in the absence or presence of PABA (142 ± 14 and 145 ± 10 μM for PABA and 189 ± 5 and 181 ± 13 μM for AcCoA, respectively).
Effect of Other Arylamines on NAT1 Activity.
A number of arylamines and related compounds were examined for their ability to down-regulate NAT1 activity in cultured PBMC (Fig.2A). These included the NAT1 substrates PAS, PAP, EPAB, and SMX. Incubation of PBMC for 24 h in the presence of the PABA-like arylamines (PAS, PAP, and EPAB; all 25 μM) significantly (P < .05) decreased NAT1 activity. SMX (25 μM), which is structurally dissimilar to PABA, had no significant effect on NAT1 activity. The NAT1 acetylation products NAPAP (25 μM) and NAPABA (25 μM), and the NAT2-selective substrates SMZ, dapsone (DAPS), or procainamide (PA) (all 25 μM), also had no effect on enzyme activity. These data indicated that only arylamines that are themselves NAT1 substrates had the ability to down-regulate NAT1 activity.

Effects of various arylamines on NAT1 activity in PBMC. A, PBMC were incubated for 24 h with 25 μM arylamine, the cells were lysed, and NAT1 activity was determined. The compounds studied were selective NAT1 substrates (■) or selective NAT2 substrates (▪). Other compounds are shown as (▨). Data are expressed as mean ± S.E. (n = 3). Above the graph is a representative Western blot of cytosols from the treated cells using a NAT1-specific antibody (Stanley et al., 1996). The relative amount of NAT1 present was quantified by densitometry. B, correlation between the activity of NAT1 after treatment with arylamines and the relative amount of NAT1 protein present in the cytosol. The correlation coefficent (r) was 0.97 (P < .05).
Effect of PABA on NAT1 Activity In Vitro.
The apparent down-regulation of NAT1 activity in cultured cells by PABA and some related arylamines may be the result of direct inactivation during catalysis. To investigate this possibility, cell lysates were incubated at 37°C for 4 h in the presence of 1.1 mM AcCoA and 25 μM PABA, PAS, PAP, or EPAB. Preincubated cell lysates were then desalted using P-6 DG spin columns (Bio-Rad, Hercules, CA). NAT1 activity in control lysates (16.9 ± 0.3 nmol/min/mg protein) was not significantly different to that in lysates after pretreatment with PABA (17.0 ± 0.2 nmol/min/mg protein), PAS (16.9 ± 0.3 nmol/min/mg protein), PAP (16.6 ± 0.2 nmol/min/mg protein), or EPAB (16.8 ± 0.3 nmol/min/mg protein). These data show that arylamines that down-regulate NAT1 activity in cultured cells are not able to directly inactivate NAT1 in vitro.
Effect of Arylamines on NAT1 Protein Levels.
To determine whether the loss of NAT1 activity observed after incubation of PBMC with the arylamines was due to a change in NAT1 protein levels, cell lysates were subjected to Western blot analysis using a NAT1-specific antibody. There was a reduction in the amount of NAT1 protein in cell lysates from PBMC treated with arylamines that decreased NAT1 activity (Fig. 2A). Furthermore, the magnitude of the decrease in NAT1 protein was strongly correlated with NAT1 activity (r 2 = 0.95, Fig. 2B). NAT1 protein concentration was unaltered in cells treated with arylamines that did not down-regulate NAT1 activity. These data indicate that the arylamine-induced down-regulation of NAT1 activity was due to a parallel reduction in the amount of NAT1 protein. To investigate the possible mechanism of protein loss, PBMC were incubated for 24 h in the presence of the protein synthesis inhibitor CHX (10 μg/ml) with and without PABA (25 μM). Incubation with CHX alone resulted in a significant (P < .05) reduction in NAT1 activity and NAT1 protein, similar to that seen when cells were cultured in the presence of PABA (25 μM). However, there was no significant additional loss of NAT1 activity or NAT1 protein when cells were treated with CHX and PABA simultaneously (Fig.3). These results indicate that translational blockade leads to a loss in NAT1 protein within the cell.

Effect of cycloheximide (CHX) on NAT1 activity in PBMC. Cells were cultured for 24 h in the presence of PABA (25 μM) and/or CHX (10 μg/ml). Treatment with CHX alone decreased NAT1 activity to a similar extent as that seen with PABA. Cotreatment with CHX and PABA resulted in no additional loss in NAT1 activity. Western blot analysis using a NAT1-specific antibody showed that the decrease in NAT1 activity correlated with a loss in enzyme protein. Data are presented as mean ± S.E. (n = 3), and an asterisk indicates significant difference (P < .05) compared with control.
Effect of PABA on NAT1 mRNA Levels.
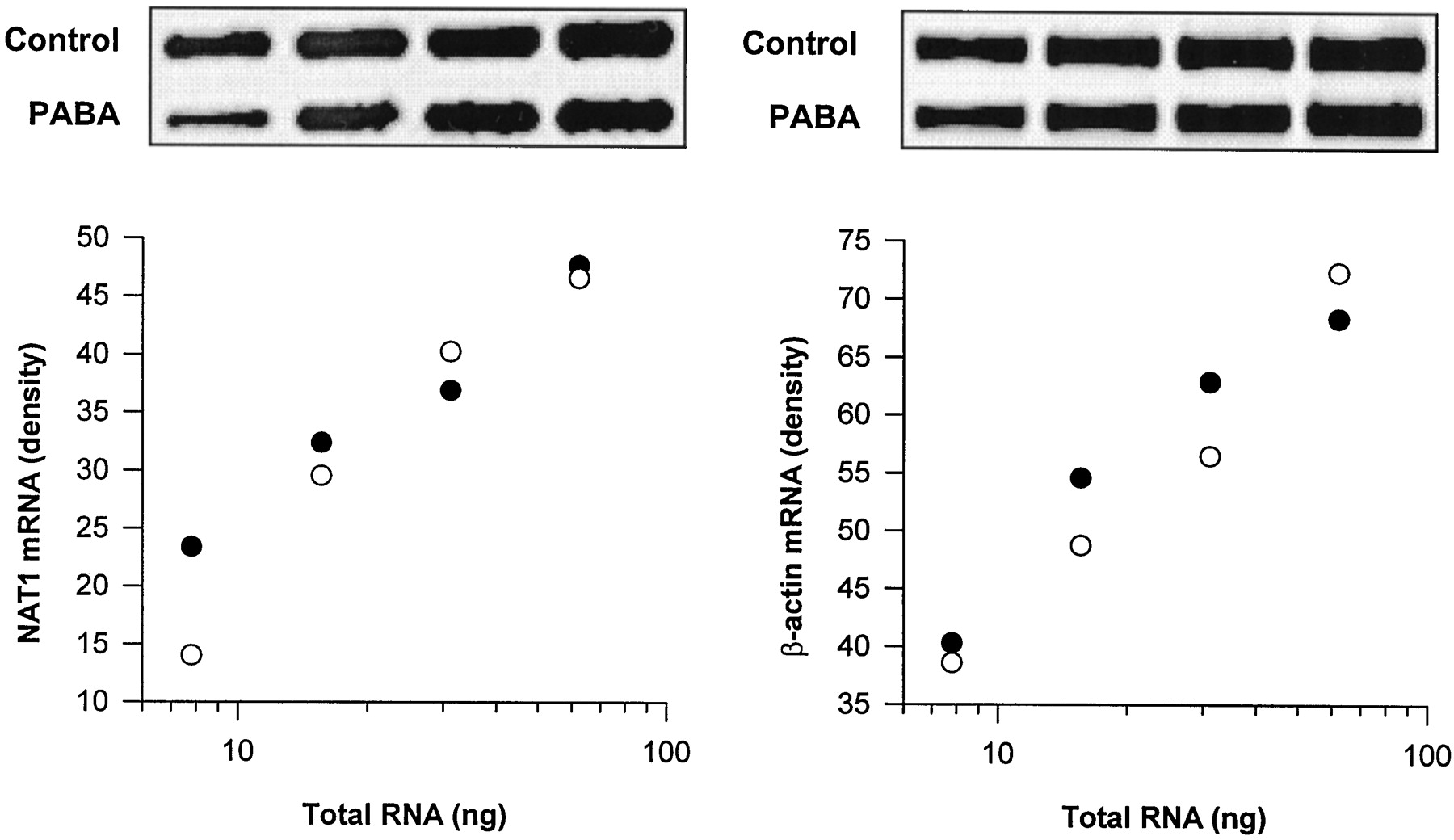
NAT1 mRNA levels from cells grown in the absence or presence of PABA were estimated by semiquantitative PCR to determine whether the arylamine-induced loss of NAT1 protein was due to a change in the amount of NAT1 mRNA present. Dilutions of total RNA were reverse transcribed and then subjected to PCR using NAT1- or β-actin (internal control)-specific primers. PCR product was quantified by densitometry, and standard curves for NAT1 and β-actin were constructed (Fig. 4). We observed no difference in the levels of NAT1 mRNA from cells grown in PABA-free or PABA-supplemented medium. The relative NAT1/β-actin mRNA ratios were 0.27 ± 0.01 (n = 4) for cells grown in PABA-free medium and 0.26 ± 0.03 (n = 4) in PABA-supplemented medium.

Effect of PABA on NAT1 mRNA levels in PBMC. Total RNA was extracted from cells cultured in PABA-free (●) or PABA-supplemented (○) media and reverse transcribed. The cDNA were amplified by PCR using NAT1-specific (left, 34 cycles) or β-actin-specific (internal control, right, 25 cycles) primers, and the relationship between RNA and PCR product was plotted after quantitation by densitometry. The linear relationship indicates that the PCR procedure was carried out under normal exponential amplification conditions. The densities of the PCR products were used to calculate a ratio of NAT1 to β-actin for each of the treatment conditions. There was no significant difference between the NAT1/β-actin ratios for PBMC grown in PABA-free or PABA-supplemented media (0.27 ± 0.1 and 0.26 ± 0.3, respectively). The panels above each of the plots show corresponding PCR product densities after electrophoresis on agarose.
Discussion
The ability of NATs to bioactivate a range of carcinogens has prompted many researchers to investigate their potential role in various types of cancers. Whereas earlier studies were centered on the polymorphic NAT2 isozyme, more recent studies have investigated the role that NAT1 may play, because it has a widespread tissue distribution and is now known to be polymorphically expressed (Vatsis and Weber, 1993; Vatsis et al., 1995; Butcher et al., 1998; Hughes et al., 1998; Lin et al., 1998). To date, the regulation of human NAT1 expression at the genomic level has not been reported. In the present study we show that several arylamines, which are themselves NAT1 substrates, have the ability to down-regulate human NAT1.
Because many polymorphic metabolizing enzymes are subject to substrate-dependent regulation, we investigated the effect of PABA, a selective NAT1 substrate, on NAT1 activity in human PBMC. When cells were cultured in media supplemented with PABA, they rapidly lost their ability to N-acetylate PABA in vitro. Also, the loss of NAT1 activity caused by PABA appeared to be a specific effect because the levels of the constitutively expressed housekeeping enzymes LDH and AST were unaltered. Kinetic studies indicated that the decrease in NAT1 activity was due to a loss in the amount of active enzyme present and not to a reduced ability of the enzyme to interact with either cofactor or substrate.
Interestingly, NAT1 activity could be restored to initial levels by treatment with PMA. However, PMA was unable to induce NAT1 activity in cells where no prior down-regulation had occurred. PABA also was able to down-regulate NAT1 activity in THP-1 monocytic cells, MCF-7 breast carcinoma cells, Jurkat lymphocytic cells, and CEM lymphoblastic leukemia cells. However, a significant reduction in NAT1 activity only was observed when the cell lines were near confluence and growth arrest was evident. Little or no loss of activity occurred when the cells were in the exponential growth phase. Thus, the ability of PABA to down-regulate NAT1 activity appears to be growth dependent and may indicate a role for NAT1 in cell proliferation.
Substrate-dependent down-regulation of NAT1 was not specific to PABA. Decreased NAT1 activity also was observed for other arylamines that are NAT1 substrates, with one exception. SMX had no effect on NAT1 activity, which may be due to the fact that it is structurally very different to PABA. Further studies are required to determine why SMX failed to alter NAT1 activity. The NAT2-selective substrates SMZ, PA, and DAPS had no effect on NAT1 activity, as was the case for the NAT1 acetylated products NAPABA and NAPAP.
The possibility of direct inactivation of NAT1 during catalysis was investigated by pretreating cell lysates with arylamine substrates (PABA, PAS, PAP, and EPAB) and cofactor. Although these arylamines down-regulated NAT1 activity in cultured cells, they did not directly inactivate NAT1 in vitro. However, this does not preclude the possibility that a metabolite formed by intact PBMC can inactivate NAT1 and enhance its degradation.
The mechanism of substrate-induced NAT1 down-regulation was investigated by Western blot analysis using a NAT1-specific antibody. Substrate-dependent down-regulation of NAT1 activity was due to a parallel loss of NAT1 protein. Treatment of PBMC with the protein synthesis inhibitor CHX also resulted in a decrease in both NAT1 activity and protein. Interestingly, there was no additional loss of activity or protein when cells were cotreated with CHX and PABA. These data suggest that the effects of CHX and PABA on NAT1 activity are similar and that PABA-induced loss of NAT1 protein is the result of an inhibition of NAT1 translation. In support of this, PABA had no measurable effect on NAT1 mRNA levels. However, at this stage we do not know what effects PABA and other NAT1 substrates may have on protein stability.
The mechanism of substrate-dependent down-regulation of NAT1 and the intracellular consequences of this process remain to be elucidated. It is possible that NAT1 substrates affect the intracellular equilibrium of AcCoA and cells respond by down-regulating pathways that use this cofactor. If this is the case, then NAT1 may act as a sensor of intracellular AcCoA concentration. Further studies are necessary to establish the exact mechanism of NAT1 modulation.
We conclude that NAT1 is subject to substrate-dependent down-regulation that is due to a loss of NAT1 protein. Because NAT1 activity could be restored by treatment with the mitogen PMA, it also appears likely that environmental factors can modulate NAT1 expression. Further investigations of the phenomenon are necessary as they may give insight into the role of NAT1 in both xenobiotic activation and cellular metabolism.
Acknowledgments
We thank Prof. Edith Sim for the NAT1 antibody used in these studies.
Footnotes
- Received July 26, 1999.
- Accepted November 17, 1999.
-
Send reprint requests to: Dr. R. F. Minchin, Department of Pharmacology, University of Western Australia, Nedlands, Western Australia, 6907, Australia. E-mail:rminchin{at}receptor.pharm.uwa.edu.au
-
This work was supported by a grant from the Medical Research Foundation, Royal Perth Hospital. N.J.B. is supported by the Elizabeth Stalker McEwan Trust.
Abbreviations
- NAT1
- N-acetyltransferase-1
- AcCoA
- acetyl-coenzyme A
- AST
- aspartate aminotransferase
- BME
- Basal Medium Eagle
- CHX
- cycloheximide
- DAPS
- dapsone
- DMEM
- Dulbecco's modified Eagle's medium
- DTT
- dithiothreitol
- EPAB
- ethyl-p-aminobenzoate
- LDH
- lactate dehydrogenase
- NAPABA
- N-acetyl-p-aminobenzoic acid
- NAPAP
- N-acetyl-p-aminophenol
- NAT2
- N-acetyltransferase-2
- PA
- procainamide
- PABA
- p-aminobenzoic acid
- PAP
- p-aminophenol
- PAS
- p-aminosalicylic acid
- PBMC
- peripheral blood mononuclear cells
- PMA
- phorbol 12-myristate 13-acetate
- SMX
- sulfamethoxazole
- SMZ
- sulfamethazine
- PCR
- polymerase chain reaction
- RT
- reverse transcription
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}