


Abstract
CYP3A4, the most abundant form of cytochrome P450 in the human adult liver, shows wide interindividual variation in its activity. This variability is thought to be caused largely by transcriptional and genetic factors, yet the underlying mechanisms are poorly understood. The purpose of this study was to clarify the mechanisms controlling the CYP3A4 gene transcription and to search for genetic polymorphisms in the 5′-flanking region of the CYP3A4 gene. Transient transfection of human hepatoma HepG2 cells and of normal human hepatocytes with a series of CYP3A4 promoter-luciferase reporter plasmids revealed that a region from –11.4 to –10.5 kilobases, designated the constitutive liver enhancer module of CYP3A4 (CLEM4), was important for the constitutive activation of the CYP3A4 gene. Gel shift assay using nuclear extracts prepared from HepG2 cells showed that HNF-1α, HNF-4α, USF1, and AP-1 interacted with CLEM4. Furthermore, the introduction of mutations into their binding sites demonstrated that essentially all sites were required for the maximal enhancer activity. Screening for genetic polymorphisms within CLEM4 in genomic DNA from French persons, we identified the novel variant, TGT insertion between –11,129 and –11,128 (–11,129_–11,128insTGT), whose allele frequency was 3.1%. The –11,129_–11,128insTGT resulted in the disruption of USF1 binding and a 36% reduction of the enhancer activity. These results suggest that CLEM4 is a constitutive enhancer of the CYP3A4 gene in the liver and that –11,129_–11,128insTGT may at least partly contribute to the interindividual variability of CYP3A4 expression.
Cytochromes P450 play important roles in the metabolism of various xenobiotics and endogenous compounds (Wrighton and Stevens, 1992). The most abundant form of P450 in the human liver is CYP3A, which accounts for ∼30% of the total P450s (Shimada et al., 1994). The human CYP3A subfamily consists of four members, CYP3A4, CYP3A5, CYP3A7, and CYP3A43, which are aligned in tandem on 7q22.1 (Gellner et al., 2001). Although the nucleotide sequences of CYP3A cDNAs resemble one another, their expression patterns are quite different. CYP3A4 is predominantly expressed in human adult liver and intestine (Beaune et al., 1986; Watkins et al., 1987), whereas CYP3A7 is expressed in human fetal liver (Komori et al., 1990). CYP3A5 is polymorphically expressed in various tissues, especially the adult liver, kidney, and lung (Aoyama et al., 1989; Schuetz et al., 1992; Anttila et al., 1997). CYP3A43 is highly expressed in the adult prostate and is detectable at a low level in several other tissues (Gellner et al., 2001). These facts imply that the transcription of the human CYP3A genes is regulated through different mechanisms.
CYP3A4 is responsible for the metabolism of more than 30% of clinically used drugs, such as the immunosuppressant cyclosporin, the calcium blocker nifedipine, and the antibiotic erythromycin (Li et al., 1995). Moreover, CYP3A4 catalyzes the 6β-hydroxylation of testosterone and cortisol (Brian et al., 1990) and is involved in the bioactivation of environmental carcinogens, including aflatoxin B1 (Shimada and Guengerich, 1989). Thus, CYP3A4 has been considered a key enzyme in the metabolism of endo- and xenobiotics. Notably, expression and activity of CYP3A4 in the liver show wide interindividual variation (Westlind et al., 1999). Because this variation must severely affect the efficacy and toxicity of drugs, assessment and prediction of the expression level of CYP3A4 are important for patients subjected to drug therapy. Although genetic factors may serve as important determinants of the CYP3A4 interindividual variation, the underlying mechanisms have not yet been clarified.
Thus far, 24 polymorphisms have appeared in the CYP3A4 allele nomenclature (http://www.imm.kise/CYPalleles/cyp3a4.htm). Among them, five polymorphisms from CYP3A4*1B to CYP3A4*1F are regulatory variants; others from CYP3A4*2 to CYP3A4*19 are coding variants (Lamba et al., 2002a). The CYP3A4*1B A392G transition in the nifedipine-specific element has been studied most extensively. Although the association of CYP3A4*1B with a prostate cancer (Rebbeck et al., 1998) and treatment-related leukemia risk (Felix et al., 1998) were initially reported, subsequent in vitro and in vivo studies argued against the functional significance of this variant (Westlind et al., 1999; Lamba et al., 2002b). Other coding variants, whose allele frequencies are less than 5%, were also insufficient to explain the large interindividual variation of CYP3A4 activity (Lamba et al., 2002b). These findings have prompted many investigators to seek other causal polymorphisms for CYP3A4 expression variance in the trans-acting factors, such as the Pregnane X receptor (PXR) gene. However, the variants causing amino acid substitution of the PXR protein were also insufficient to account for the variation of CYP3A4 expression (Zhang et al., 2001). Thus, the large interindividual variability of CYP3A4 expression, most of which is estimated to be under transcriptional and genetic controls, remains unclear.
CYP3A4 is inducible by a number of clinically important drugs including rifampicin, clotrimazole, and dexamethasone, sometimes causing severe drug-drug interactions (Thummel and Wilkinson, 1998). Recently, PXR has been identified as a receptor for these inducers in the induction of the CYP3A4 gene (Lehmann et al., 1998). Liganded PXR forms a heterodimer with the retinoid X receptor and targets the PXR-responsive elements located in the proximal promoter and a region located at approximately –8 kb upstream of the CYP3A4 gene, referred to as the proximal PXR response element and the xenobiotic-responsive enhancer module (XREM), respectively (Lehmann et al., 1998; Goodwin et al., 1999). In addition, these PXR-responsive elements are a target of another orphan nuclear receptor, the constitutive active receptor (CAR) (Goodwin et al., 2002). The nuclear translocation of CAR is induced by some xenobiotics, such as phenobarbital (Kawamoto et al., 1999). Although PXR and CAR are critical determinants of CYP3A4 expression levels with respect to xenobiotic induction, other trans-acting factors and the corresponding cis-acting elements may also serve as modifiers of the CYP3A4 gene transcription. First, the gene disruption of neither PXR nor CAR affects the constitutive expression of murine Cyp3a genes (Xie et al., 2000; Ueda et al., 2002), although the up-regulation of Cyp3a11 in the livers from PXR-null mice has also been reported (Staudinger et al., 2001). Second, the XREM and proximal PXR response element are both highly conserved in the CYP3A7 gene, the expression pattern of which in the liver is quite different from that of the CYP3A4 gene (Bertilsson et al., 2001).
The sequences of the 5′-flanking region between the CYP3A4 and CYP3A7 genes are highly similar. Indeed, the region up to –8.8 kb is more than 90% identical to each other, whereas the identities fall to ∼25% in the region farther upstream (Bertilsson et al., 2001). Recently, we analyzed the 5′-flanking region up to –5.5 kb of the CYP3A4 and CYP3A7 genes in human hepatoma HepG2 cells (Saito et al., 2001). Whereas the CYP3A7 promoter conferred high transcriptional activity, the CYP3A4 promoter could not activate the transcription. Thus, it seemed reasonable to assume that an unknown enhancer(s) responsible for the constitutive expression of the CYP3A4 gene was expected to exist in additional 5′-upstream regions, accounting for interindividual variation of CYP3A4 expression.
In this study, we analyzed the region farther 5′-upstream up to –13.0 kb of the CYP3A4 gene in HepG2 cells. We identified a novel enhancer from –11.4 to –10.5 kb, designated the constitutive liver enhancer module of CYP3A4 (CLEM4), responsible for the constitutive activation of the CYP3A4 gene. In addition, we found the genetic polymorphism, three-nucleotide TGT insertion between –11,129 and –11,128, in CLEM4. This variant affected the enhancer activity of the CYP3A4 gene in vitro.
Materials and Methods
Cell Culture. Human hepatoma HepG2 cells were purchased from Riken (Tsukuba, Japan). HepG2 cells were maintained in Dulbecco's modified Eagle's medium (Nissui, Tokyo, Japan) supplemented with 10% fetal bovine serum (Cambrex Bioscience Walkersville, Inc., Walkersville, MD), nonessential amino acids (ICN Pharmaceuticals Biochemicals Division, Aurora, OH), and 1 mM sodium pyruvate (Invitrogen, Carlsbad, CA) at 37°C in 5% CO2. Normal human hepatocytes were purchased from KAC (Osaka, Japan) and cultivated in Williams' E medium with GlutaMAX 1 added with 100 IU/ml penicillin, 100 μg/ml streptomycin, 4 μg/ml bovine insulin, 50 μM hydrocortisone hemisuccinate, and 0.1% (w/v) bovine serum albumin (BPI, Rennes, France).
Construction of Reporter Plasmids. The oligonucleotide primers (Sigma-Genosys, The Woodlands, TX) used for the synthesis of DNA fragments or for a site-directed mutagenesis are shown in Table 1. A reporter plasmid p3A4/–10.5, which contained a 5′-flanking region from nucleotides –10,467 to +103 (abbreviated to –10,467/+103) relative to the transcriptional start site of the CYP3A4 gene, was constructed by ligation of an XhoI/HindIII DNA fragment (–10,467/–3211) from pBS32B (Saito et al., 2001), a HindIII/HindIII fragment (–3210/+103) from p3A4/–5.5 (Saito et al., 2001), and a HindIII/XhoI fragment from a control luciferase reporter plasmid, Basic Vector 2 (Toyoinki, Tokyo, Japan). To construct a reporter plasmid p3A4/–13.0, a region (–12,931/–10,468) was amplified by PCR using 3A4–13s as a sense primer, 3A4–10.5as as an antisense primer, and the bacterial artificial chromosome clone (clone ID, H_NH0249F04) as a template. After the synthesized fragment was inserted into the EcoRV site of the pBluescript, this plasmid was digested with restriction enzymes SmaI and XhoI. The resultant fragment was inserted into the SmaI/XhoI site of the p3A4/–10.5. Reporter plasmids p3A4/–8.0 and p3A4/–7.5, which contained the 5′-flanking regions (–8020/+103 and –7492/+103) of the CYP3A4 gene, were generated by the cleavage of the p3A4/–10.5 with restriction enzymes ApaI and SpeI, respectively. To generate an internal deletion construct p3A4–11.4 to 10.5Δ, a region (–12,931/–11,381) was amplified by PCR using 3A4–13s as a sense primer and 3A4–11.4asXhoI as an antisense primer. The synthesized fragment was digested with the restriction enzyme XhoI. The resultant fragment was inserted into the SmaI/XhoI site of p3A4/–10.5. For the construction of reporter plasmids p3A4–13.0/–10.5, p3A4–11.2/–10.5, and p3A4–10.9/–10.5, PCR was conducted using 3A4–13s, 3A4–11.2s, and 3A4–10.9s as a sense primer and 3A4–10.5as as an antisense primer. After the synthesized fragments were inserted into the EcoRV site of the pBluescript, these plasmids were digested with restriction enzymes SmaI and XhoI. The resultant fragments were inserted into the SmaI/XhoI site of p3A4/–360 (Saito et al., 2001). To generate a reporter plasmid p3A4–11.4/–10.5, PCR was performed using 3A4–11.4sKpnI as a sense primer and 3A4–10.5asXhoI as an antisense primer. The synthesized fragment was digested with restriction enzymes KpnI and XhoI and then inserted into the KpnI/XhoI site of p3A4/–360. Reporter plasmids p3A4mtA:HNF-4, p3A4mtB:HNF-1, p3A4mtC:HNF-4, p3A4mtD:E-box, p3A4mtE:E-box, p3A4mtF:CRE, and p3A4mtG:E-box were produced by PCR-based site-directed mutagenesis using 3A4–11,357sHNF-4mt/3A4–11,357asHNF-4mt, 3A4–11,336sHNF-1mt/3A4–11,336asHNF-1mt, 3A4–11,172sHNF-4mt/3A4–11,172asHNF-4mt, 3A4–11,140sE-boxmt/3A4–11,140asE-boxmt, 3A4–11,085sE-boxmt/3A4–11,085asE-boxmt, 3A4–11,042sCREmt/3A4–11,042asCREmt, 3A4–10,991sE-boxmt/3A4–10,991asE-boxmt as mutated primers. A reporter plasmid p3A4insTGT was constructed by PCR-based site-directed mutagenesis using 3A4insTGTmt-s/3A4insTGTmt-as as mutated primers.
Primers
Transient Transfection and Dual-Luciferase Assay. HepG2 cells (2 × 105 cells/well) and normal human hepatocytes (7.5 × 105 cells/well) were plated onto a 12-well plate and then transfected with a reporter plasmid (0.45 μg) and pRL-SV40 (0.05 μg) (Promega, Madison, WI) as an internal control by FuGENE 6 (Roche Diagnostics, Indianapolis, IN). After 36 h, the cells were washed with phosphate-buffered saline and subjected to a dual-luciferase assay according to the manufacturer's instructions (Promega).
Gel Shift Assay. Nuclear extracts were prepared from HepG2 cells according to the method of Dignam et al. (1983). A gel shift assay was performed using double-stranded synthetic oligonucleotides labeled with [γ-32P]ATP (Amersham Biosciences Inc., Piscataway, NJ) and T4 polynucleotide kinase (Takara, Tokyo, Japan). The binding reaction was carried out with a reaction mixture (10 μl) containing 25 mM Hepes, pH 7.9, 4% Ficoll, 40 mM KCl, 0.5 mM dithiothreitol, 0.1 mM EGTA, 1 mM MgCl2, 5% glycerol, 0.5 μg of poly(dI-dC), 10 μg of nuclear extracts, and 5 fmol of a 32P-labeled probe DNA. The mixture was incubated at 24°C for 30 min. The samples were resolved on a 4% nondenaturing polyacrylamide gel in 0.5× Tris-borate/EDTA at 100 V at room temperature and visualized by BAS-2500 Imaging Analyzer (Fuji Film, Tokyo, Japan). Antibodies to HNF-1α (sc-6547X), HNF-1β (sc-7411X), HNF-4α (sc-6556X), HNF-4γ (sc-6558X), USF1 (sc-229X), CREB1 (sc-186X), CREB2 (sc200X), ATF1 (sc-270X), Fos (sc-253X), and Jun (sc-44X) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The supershift assay was performed using these antibodies. After incubation of probe DNAs with nuclear extracts as described above, the antibodies were added to the reaction mixture and incubated at 24°C for 1 h. The products were then analyzed by a gel shift assay. The sequences of oligonucleotides used as probes are shown in Table 2.
Probes
Subjects. Genomic DNA was isolated from Japanese volunteers (Ariyoshi et al., 2002) and French persons in a hospital-based lung cancer case-control study (Loriot et al., 2001). Informed consent was obtained from all participants. The study was approved by the Ethical Committee of the Faculty of Pharmaceutical Sciences, Hokkaido University.
PCR and Sequencing of Genomic DNA Samples. The 5′-flanking region from –12.6 to –10.5 kb of the CYP3A4 gene was successfully amplified by PCR with 3A4–12.6sKpnI, 5′-ACCTGGTACCCCAGTGAGACAG-3′ as a sense primer and 3A4–10.5asXhoI as an antisense primer. The reaction mixture contained 50 ng of genomic DNA, 1× LA-PCR buffer II, 2.5 mM MgCl2, 0.2 mM dNTPs, 0.4 μM concentrations of each primer, and 1.0 U of LA-Taq DNA polymerase in a total volume of 25 μl. The PCR conditions consisted of an initial denaturation at 94°C for 3 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 57°C for 30 s, and extension at 72°C for 2.5 min. The PCR product with the expected size of 2133 bp was directly sequenced in the forward and reverse directions using the amplification primers 3A4–11.8sKpnI, 5′-ACCAGGTACCGCATGTTGTCACTCATAAG-3′ and 3A4–10.5asXhoI (BigDye Terminator v3.0; Applied Biosystems, Foster City, CA) and run on an ABI PRISM 3100 sequencer (Applied Biosystems) following the manufacturer's protocol.
Genotyping for Regulatory CYP3A4 Variant Identified in This Study. An allele-specific PCR-based method was developed to genotype samples with the three-nucleotide TGT insertion, –11,129_–11,128insTGT. The primers used for the first PCR were 3A4–11.9s, 5′-CAATCCAAAAGCCCATCAAT-3′ and 3A4–10.9asXhoI, 5′-TGAGCTCGAGAGTGACCTACGG-3′. The first PCR was conducted under the same conditions as described above except for the extension at 72°C for 1 min. The first PCR products with the expected size of 1069 bp were subsequently used as a template for the second PCR. The primers used for the second PCR were 3A4–11.9s as a sense primer and 3A4insTGTwt-as, 5′-TCTAGCCATTAGAACCACAT-3′ or 3A4insTGTmt-as, 5′-TCTAGCCATTAGAACCACAA-3′ as antisense primers (underlined letters indicate mutated base). In the case of the second PCR, the reaction mixture contained the first PCR product (1 μl), 1× LA-PCR buffer II, 1.5 mM MgCl2, 0.2 mM dNTPs, 0.4 μM concentrations of each primer, and 1.0 U of LA-Taq DNA polymerase in a final volume of 25 μl. PCR was conducted with an initial denaturation at 94°C for 1 min, followed by 17 cycles of reactions composed of denaturation at 94°C for 15 s, annealing at 60°C for 15 s, and extension at 72°C for 25 s. The expected size of the PCR product was 855 bp.
Results
Identification of a Far Upstream Enhancer of theCYP3A4Gene. We demonstrated in a previous study that the 5′-flanking region up to –5.5 kb of the CYP3A4 gene had no promoter activity (Saito et al., 2001). Thus, it was supposed that an enhancer region(s) necessary for the CYP3A4 gene transcription is located in the region farther upstream of –5.5 kb. To test this possibility, we constructed the luciferase reporter plasmid containing the CYP3A4 promoter up to –13 kb and then transfected the plasmid into HepG2 cells (Fig. 1A). The p3A4/–13.0 showed a ∼16-fold increase of the luciferase activity compared with the p3A4/–5.5 and Basic Vector 2. Successive deletions of the promoter to –10.5, –8.0, and –7.5 kb resulted in luciferase activity reductions of 41, 56, and 12%, respectively (Fig. 1A). These results indicate that an enhancer(s) responsible for the constitutive activation of the CYP3A4 gene is located within the regions from –13.0 to –10.5 kb and from –8.0 to –7.5 kb.

Identification of a far upstream enhancer of the CYP3A4 gene. A, the construction of deletion mutants is described under Materials and Methods. The numbers given to deletion mutants indicate the 5′-end of the 5′-flanking sequence of the CYP3A4 gene counted negatively from the transcriptional start site. An open box with the p3A4–11.4 to 10.5Δ indicates an internal deletion from –11.4 to –10.5 kb. B, the construction of deletion mutants is described under Materials and Methods. The numbers given to deletion mutants indicate the 5′-end of the 5′-flanking sequence of the CYP3A4 gene counted negatively from the transcriptional start site. Open boxes represent the CYP3A4 proximal promoter region from nucleotide –360 to +103. In both A and B, HepG2 cells (2 × 105 cells) were transiently transfected with FuGENE 6 with a reporter plasmid (0.45 μg). Cells were harvested 36 h after DNA transfection, and then luciferase activity was assayed. The luciferase activity represents the average ± S.D. from at least three independent experiments. The mean value obtained with the p3A4/–13.0 (A) and the p3A4–11.4/–10.5 (B) was defined as 100%. C, the construction of deletion mutants is described under Materials and Methods. The numbers given to deletion mutants indicate the 5′-end of the 5′-flanking sequence of the CYP3A4 gene counted negatively from the transcriptional start site. An open box with the p3A4–11.4 to 10.5Δ indicates an internal deletion from –11.4 to –10.5 kb. Normal human hepatocytes (7.5 × 105 cells) were transiently transfected with FuGENE 6 with a reporter plasmid (0.45 μg) and pRL-TK (0.1 μg) as an internal control. The luciferase activity represents the average ± S.D. from at least three independent experiments. The mean value obtained with the p3A4/–13.0 was defined as 100%.
To further characterize the region from –13.0 to –10.5 kb, we inserted this region upstream of the CYP3A4 proximal promoter from –360 to +103 (Fig. 1B). The region from –13.0 to –10.5 kb enhanced the CYP3A4 proximal promoter by 21-fold. Thus, the region from –13.0 to –10.5 kb acts as an enhancer. Although deletion to –11.4 kb increased enhancer activity by 25-fold, further deletion to –11.2 and –10.9 kb led to 45 and 13% reductions of the enhancer activity relative to that with the p3A4–11.4/–10.5 (Fig. 1B). Furthermore, the internal deletion of the p3A4/–13.0 construct from –11.4 to –10.5 kb (designated p3A4/–11.4–10.5Δ) decreased the luciferase activity to 28% compared with that of the p3A4/–13.0 (Fig. 1A). These results indicate that the region from –11.4 to –10.5 kb is responsible for the enhancer activity.
The enhancer activity of the region from –11.4 to –10.5 kb was further assessed using normal human hepatocytes (Fig. 1C). The luciferase activity of the p3A4/-13.0 was ∼5 fold higher than that of Basic Vector 2. The internal deletion of the region from –11.4 to –10.5 kb (p3A4/–11.4–10.5Δ) decreased the activity to nearly the same level as that of Basic Vector 2 (∼20%). This result indicates that the region from –11.4 to –10.5 kb acts indeed as an enhancer of the CYP3A4 gene transcription in the liver.
Binding of HNF-1α, HNF-4α, USF1, and AP-1 to the Far Upstream Enhancer of theCYP3A4Gene. Searching the consensus sequences of transcription factors, we found several putative binding sites for liver-enriched transcription factors and ubiquitous transcription factors within the region from –11.4 to –10.9 kb. We designated these sites as A:HNF-4, B:HNF-1, C:HNF-4, D:E-box, E:E-box, F:CRE, and G:E-box (Fig. 2A). To examine whether or not nuclear proteins interact with these putative sites, a gel shift assay was performed using nuclear extracts prepared from HepG2 cells. As shown in Fig. 2B, DNA-protein complex I was formed with A:HNF-4. The formation of complex I was eliminated by the addition of a 100 M excess of the unlabeled A:HNF-4 and HNF-4 consensus sequence (HNF-4) but not mtA, which had two nucleotide mutations within the HNF-4 binding site of A:HNF-4. Moreover, antibodies to HNF-4α but not HNF-4γ supershifted complex I, suggesting that the A:HNF-4 is recognized by HNF-4α. We confirmed that C:HNF-4, another putative HNF-4 site, was also recognized by HNF-4α (Fig. 2B). Complex II but not III formed with C:HNF-4 was disrupted by the addition of the HNF-4 consensus site and supershifted by antibodies to HNF-4α.
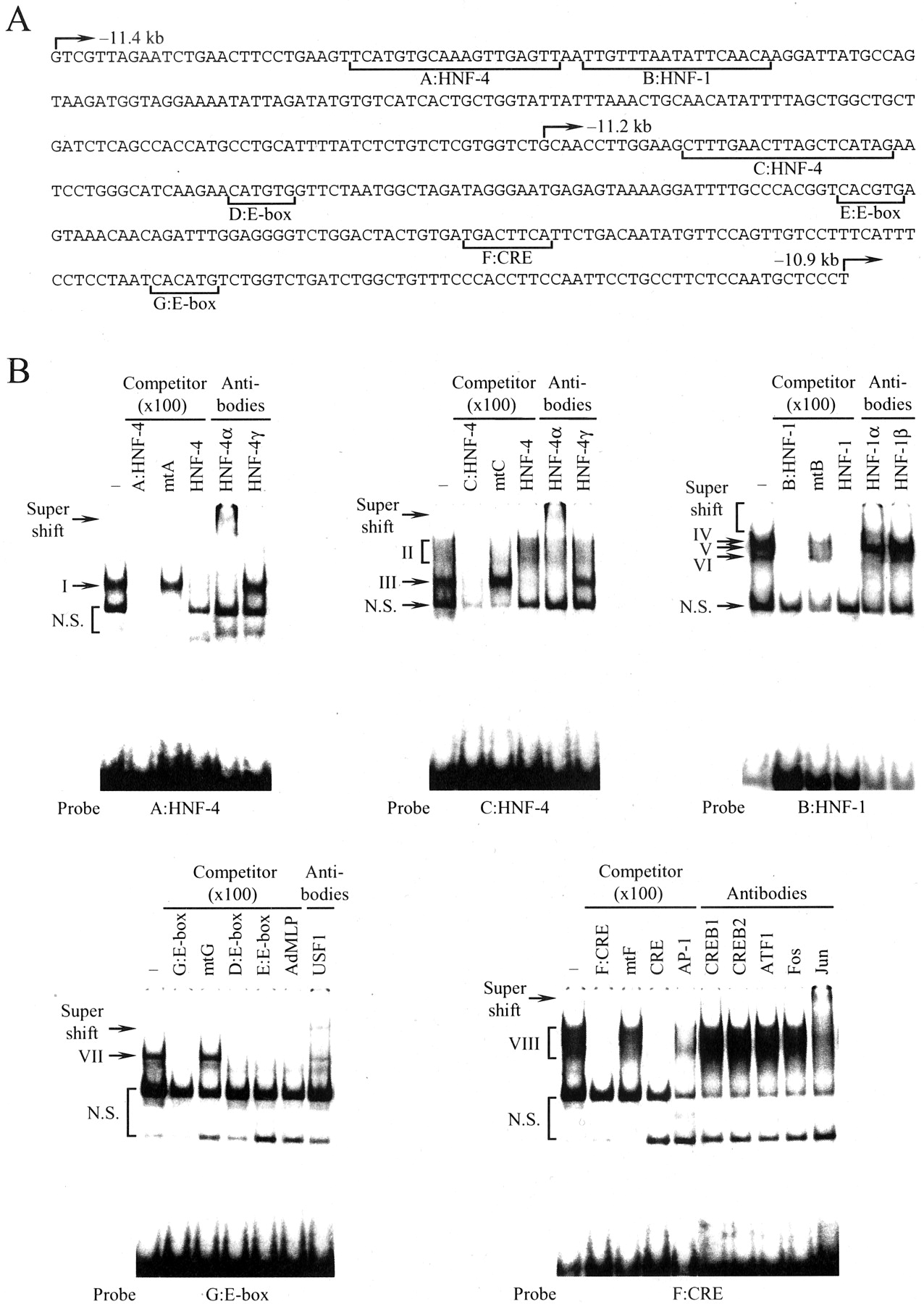
Nuclear proteins interacting with the far upstream enhancer of the CYP3A4 gene. A, nucleotide sequences from –11.4 to –10.9 kb of the CYP3A4 gene. Arrows indicate the 5′-end of the 5′-flanking sequence of the CYP3A4 gene in deletion constructs p3A4–11.4/–10.5, p3A4–11.2/–10.5, and p3A4–10.9/–10.5 as shown in Fig. 1B. Putative binding sites for HNF-1 and HNF-4, E-box-like element, and CRE-like element are indicated by brackets and designated A:HNF-4, B:HNF-1, C:HNF-4, D:E-box, E:E-box, F:CRE, and G:E-box. B, gel shift assay with A:HNF-4, B:HNF-1, C:HNF-4, F:CRE, and G:E-box. Nuclear extracts (10 μg) from HepG2 cells were incubated with the 32P-labeled probe in the presence or absence of various competitors and antibodies. Arrows and brackets indicate the DNA-protein complexes or super shift bands. N.S., nonspecific band.
With the B:HNF-1 as a probe, three DNA-protein complexes, IV, V, and VI, were detected (Fig. 2B). Unlabeled B:HNF-1 and the HNF-1 consensus site (HNF-1) destroyed the formation of these three complexes, whereas the mutant B:HNF-1 (mtB) was ineffective. Addition of antibodies to HNF-1α but not HNF-1β supershifted complexes IV and VI. These results indicate that HNF-1α binds to the B:HNF-1 and that complex V is an HNF-1-related factor(s).
Complex VII was formed with the G:E-box (Fig. 2B). We observed similar DNA-protein complexes with the D:E-box and E:E-box as a probe (data not shown). E-box is targeted by several transcription factors belonging to a basic helix-loop-helix family. In the liver, the predominant member of this family is USF (Shih et al., 1995). To determine whether USF is a component of these DNA-protein complexes, competition assay was performed using G:E-box as a probe and unlabeled G:E-box, D:E-box, E:E-box, or USF-binding sequence of the adenovirus major late promoter as a competitor. Except for mtG, which had mutations within the E-box-like sequence in the G:E-box, complex VII disappeared because of these competitors. Furthermore, the addition of antibodies to USF1 resulted in the appearance of a supershifted complex. These results suggest that USF1 interacts with the E-box-like elements found in the CYP3A4 enhancer.
To characterize a nuclear factor(s) binding to F:CRE, gel shift assay was performed with F:CRE as a probe and several competitors or antibodies (Fig. 2B). Complex VIII, seen as a broad band, was formed with F:CRE (Fig. 2B). Complex VIII appeared specifically with the sequence because it was eliminated by the addition of unlabeled F:CRE (but not mtF) that had two nucleotide mutations in its sequence. Although the CRE consensus sequence destroyed the formation of complex VIII, antibodies to CREB1, CREB2, and ATF1, which are members of the CREB/ATF family, were not effective. It is reported that CRE is also recognized by several transcription factors, including AP-1, which is a homo- or heterodimer of the Jun and Fos family (Hai and Curran, 1991). The addition of AP-1 consensus sequence partially disrupted the formation of complex VIII. Antibodies to Jun family super shifted complex IX, whereas antibodies to Fos family did not affect it. These results indicate that AP-1 consisting of Jun family members, but not CREB/activation transcription factor family members, is a component of complex VIII.
Functionalcis-Acting Elements in the Far Upstream Enhancer of theCYP3A4Gene. To examine the functional significance of the identified transcription factor binding sites, the same mutations used in a gel shift assay were introduced into these sites of the p3A4–11.4/–10.5 reporter plasmid by site-directed mutagenesis (Fig. 3). HepG2 cells were transiently transfected with these reporter plasmids. The introduction of mutations into A:HNF-4, B:HNF-1, and C:HNF-4 reduced the enhancer activity by 19, 30, and 44%, respectively, relative to the level seen with the p3A4–11.4/–10.5. With mutations into the D:E-box, E:E-box, and G:E-box that interacted with USF1, the enhancer activities were 33, 43, and 47% lower, respectively. In addition, the introduction of mutations into the F:CRE that was recognized by Jun family members decreased the activity by 80%. These results suggest that all of these sites for HNF-1, HNF-4, USF1, and AP-1 are required for the maximal enhancer activity of the CYP3A4 gene and that F:CRE interacting with AP-1 plays the most important role in the activation.

Mutational analysis of binding sites for HNF-1α, HNF-4α, USF1, and AP-1 in the enhancer activity. The construction of the mutant plasmids is described under Materials and Methods. HepG2 cells (2 × 105 cells) were transiently transfected with FuGENE 6 with a reporter plasmid (0.45 μg). The values represent the averages ± S.D. from at least three independent experiments. The mean value obtained with the p3A4–11.4/–10.5 was defined as 100%.
Novel Polymorphism in the Far Upstream Enhancer of theCYP3A4Gene. To investigate whether or not genetic polymorphisms contributing to interindividual variation of the CYP3A4 expression level exist in the identified enhancer, the region from –11.8 to –10.5 kb of the CYP3A4 gene was sequenced in genomic DNA from Japanese and French subjects. We found a novel polymorphism in samples from French subjects. This polymorphism, identified in the present study, was the three-nucleotide TGT insertion between –11,129 and –11,128 (–11,129_–11,128insTGT), was present within the D:E-box site in the enhancer from –11.4 to –10.5 kb. To rapidly screen the variant and confirm our direct-sequencing results, a genotyping method, allele-specific PCR for –11,129_–11,128insTGT, was developed (Fig. 4). As shown in Table 3, we applied the method to genomic DNA samples from 511 French and 131 Japanese persons (total, 1284 chromosomes). In French subjects, the allele frequency of –11,129_–11,128insTGT was 3.1%. The observed genotype frequency in French subjects did not deviate from that expected by Hardy-Weinberg equilibrium. We found no linkage disequilibrium of the variant with known CYP3A4 variant alleles, including CYP3A4*1B (data not shown). In contrast with the French subjects, the –11,129_–11,128insTGT variant allele was not found in the Japanese subjects examined. These results suggest that the –11,129_–11,128insTGT variant allele is relatively rare in French persons and extremely rare or null in Japanese persons.

Allele-specific PCR-based diagnosis of –11,129_–11,128insTGT variant allele. W, wild homozygotes; H, heterozygotes; wt, PCR product obtained by wild-type allele-specific primer; mt, PCR product obtained by mutant allele-specific primer.
Frequencies of the genetic polymorphism –11129_–11128insTGT in the 5′-flanking region of the CYP3A4 gene in French and Japanese subjects
The position relative to the transcriptional start site is indicated.
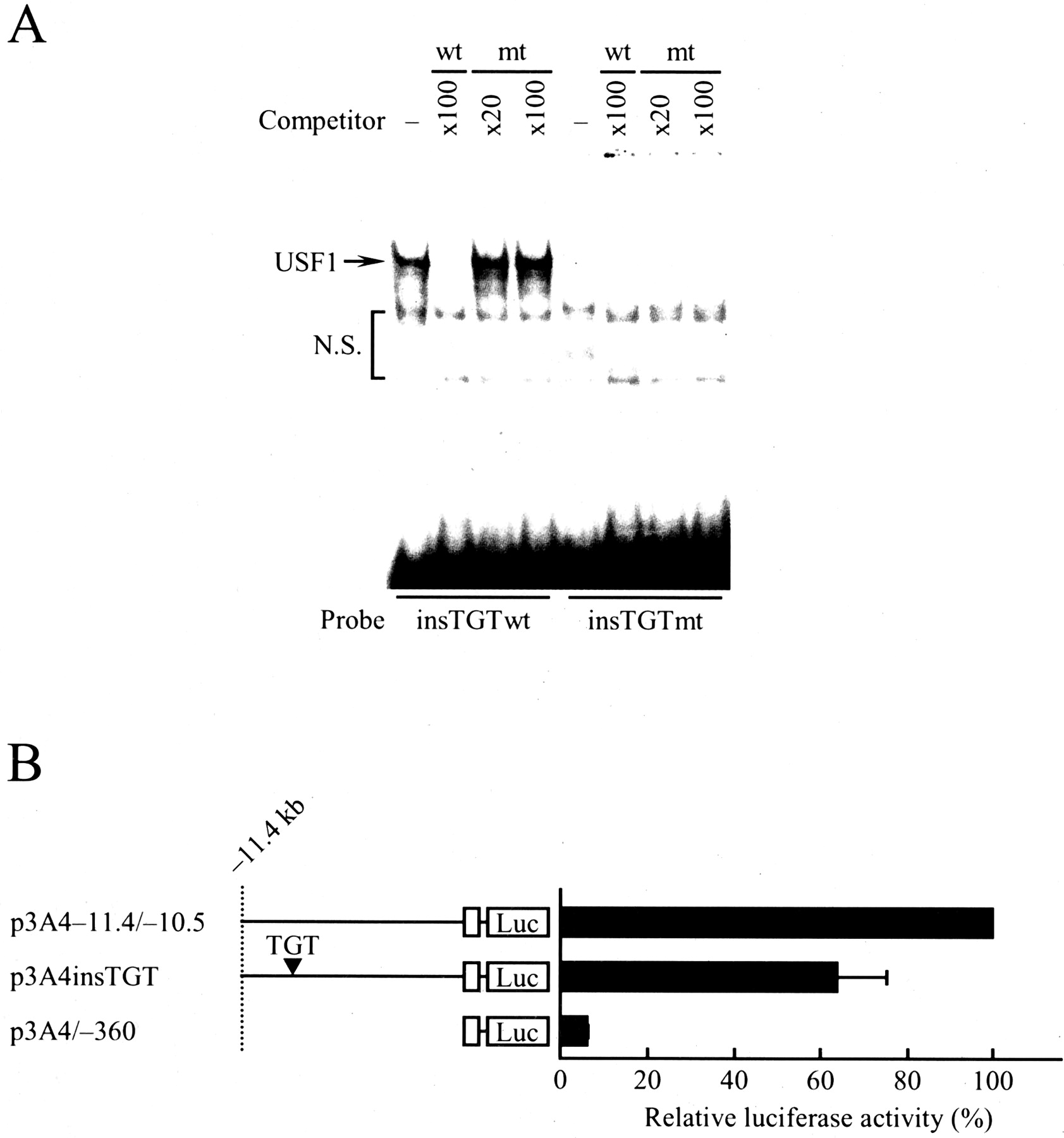
Finally, we examined functional alterations of the enhancer caused by the variant using gel shift assay (Fig. 5A) and transient transfection assay (Fig. 5B). As shown in Fig. 5A, USF1 complex that formed with insTGTwt (same as D:E-box) as a probe was not competed out by an excess of the unlabeled mutant D:E-box containing TGT insertion (insTGTmt). Furthermore, no USF1 complex appeared with the insTGTmt as a probe. These results suggest that the variant –11,129_–11,128insTGT causes the disruption of USF1 binding and thereby may alter the enhancer activity of the CYP3A4 gene. To test this possibility, a reporter plasmid carrying the variant p3A4insTGT was constructed and then transiently transfected into HepG2 cells (Fig. 5B). The insertion of three nucleotides TGT between –11,129 and –11,128 reduced the enhancer activity to 64% compared with that of the p3A4–11.4/–10.5. Taken together, these results suggest that the variant allele affects the enhancer activity of the CYP3A4 gene.

Functional characterization of –11,129_–11,128insTGT variant allele. A, gel shift assay with wild-type and mutant –11,129_–11,128insTGT (insTGTwt and insTGTmt). Nuclear extracts (10 μg) from HepG2 cells were incubated with the 32P-labeled insTGTwt or insTGTmt. Competition analysis was carried out by the addition of a 20 or 100 M excess of the unlabeled oligonucleotides as indicated. DNA-protein complex is indicated by an arrow. N.S., nonspecific band. B, effects of –11,129_–11,128insTGT on the enhancer activity. The construction of reporter plasmids is described under Materials and Methods. The p3A4insTGT had TGT insertion from nucleotides –11,129 to –11,128 with the p3A4–11.4/–10.5. HepG2 cells (2 × 105 cells) were transiently transfected by FuGENE 6 with a reporter plasmid (0.45 μg). The position relative to the transcription initiation site is indicated. Open boxes show the CYP3A4 proximal promoter region from nucleotide –360 to +103. The values represent the averages ± S.D. from at least three independent experiments. The mean value obtained with the p3A4–11.4/–10.5 was defined as 100%.
Discussion
Hepatic CYP3A4 expression shows large interindividual differences, but the underlying mechanisms for these differences are poorly understood. In this study, we analyzed the 5′-flanking region up to –13.0 kb of the CYP3A4 gene in HepG2 cells and identified the novel polymorphic enhancer, designated CLEM4, involved in the constitutive expression of the CYP3A4 gene. Interestingly, CLEM4 was located in the region far upstream from –11.4 to –10.5 kb relative to the transcription start site of the CYP3A4 gene. Corresponding regions of other human CYP3A genes, CYP3A5, CYP3A7, and CYP3A43, show low homologies less than 50%. Thus, CLEM4 was peculiar to the CYP3A4 gene. Previously, Goodwin et al. (1999) investigated the same promoter region of the CYP3A4 gene in HepG2 cells and found no promoter activity without inducers. This inconsistency may be attributable to two possibilities. First, the nucleotide sequences of CLEM4 and/or other regions in the CYP3A4 gene in the luciferase reporter constructs examined may differ between the previous and present studies, although both CYP3A4 clones were obtained from genomic DNA in white persons. Our data show high prevalence of genetic polymorphisms in the CYP3A4 gene in white persons and support this possibility. Second, the difference of cell culture conditions between the two may affect the CYP3A4 gene transcription in HepG2 cells.
CLEM4 consists of an array of cis-acting elements encompassing ∼900 bp. Several transcription factors, including HNF-1, HNF-4, USF1, and AP-1, interacted with CLEM4 and are likely to activate the enhancer cooperatively. Among them, HNF-4 and its binding sites may be important for the hepatic expression of the CYP3A4 gene. The importance of HNF-4 for the liver-enriched expression of the rat and murine CYP3A genes has been well documented. Although the functional HNF-4-binding site is located within the proximal promoter region in the rodent CYP3A genes, the element is not conserved in the corresponding region of the human CYP3A4 gene. However, a recent study using the adenoviral expression of HNF-4 antisense RNA in primary cultured human hepatocytes revealed that HNF-4 was also critical for the hepatic expression of the human CYP3A4 gene (Jover et al., 2001). Because CLEM4 is the sole enhancer containing functional HNF-4 sites identified so far in the CYP3A4 promoter, we believe that CLEM4 is a bona fide enhancer of the CYP3A4 gene in the liver. During the preparation of this manuscript, Tirona et al. (2003) identified the functional HNF-4 site within XREM. Although their data suggest that this HNF-4 site is involved in the induction of CYP3A4 through the cooperative interaction with the adjacent PXR sites, we cannot exclude the possibility that the HNF-4 site identified by Tirona et al. (2003) is also involved in the constitutive expression of the CYP3A4 gene.
Interindividual variability of hepatic CYP3A4 activity is estimated to be largely controlled by genetic factors using a repeated drug administration (Ozdemir et al., 2000). In addition, high correlation between CYP3A4 protein level and its enzyme activity in the liver samples obtained so far strongly indicates that regulatory polymorphisms rather than structural polymorphisms in the CYP3A4 gene contribute to CYP3A4 variability (Westlind et al., 1999; Sy et al., 2002). Sequencing the region encompassing CLEM4 from –11.8 to –10.5 kb in DNA from 511 French persons, we identified the novel polymorphism that altered the CYP3A4 gene transcription in vitro. The allele frequency of –11,129_–11,128insTGT was 3.1% in French subjects, which was relatively high among the known CYP3A4 variants. Nevertheless, because homozygosity with respect to –11,129_–11,128insTGT occurs statistically in only (3.13%)2 = 0.098% of the population, a large population will be required to address whether these persons show low CYP3A4 content in the liver. In addition, we cannot exclude the possibility that the allele frequency of –11,129_–11,128insTGT as 3.1% deviates from that of healthy volunteers, because our French subjects came from a hospital-based lung cancer case-control study. Investigation of the genotype-phenotype relationship using liver banks from Japanese and white volunteers is now in progress.
This study also suggests that the variant –11,129_–11,128insTGT shows ethnic difference in its frequency. The variant allele was not found in 131 Japanese populations examined. Large ethnic differences have also been reported in other CYP3A4 alleles (Lamba et al., 2002a): CYP3A4*1B in white persons (2–9.6%), African Americans (35–67%), and Japanese (0%); CYP3A4*2 in white persons (2.7%), African Americans (0%), and Chinese (0%); CYP3A4*18 in white persons (0%), African Americans (0%), and Chinese (10%). However, marked interindividual variability in the hepatic expression of CYP3A4 has been reported in wide ethnic groups. Thus, a genetic basis of CYP3A4 variability may differ among ethnic populations. A part of hepatic CYP3A4 variation, which cannot be fully explainable by genetic polymorphisms in the CYP3A4 gene itself, may also be attributable to the variation of HNF-1 and/or HNF-4 levels. Recently, we demonstrated that the interindividual variation of hepatic dihydrodiol dehydrogenase 4 and UDP-glucuronosyltransferase 2B7 mRNA levels correlated highly with the variation of HNF-1 plus HNF-4 and HNF-1 mRNA levels, respectively (Toide et al., 2002; Ozeki et al., 2002). Because CLEM4 contained both functional HNF-1 and HNF-4 recognition sites, amounts of these transcription factors may also modulate the CYP3A4 expression level. In addition, a recent study suggests that hepatic CYP3A4 mRNA level significantly correlates with the mRNA levels of PXR in white persons (Westlind-Johnsson et al., 2003). Collectively, various factors, including genetic polymorphisms in the CYP3A4 promoter, genetic polymorphisms in transcription factor genes, and their combination, should be considered to fully understand a genetic basis for the variability of CYP3A4 expression.
In summary, we identified a novel enhancer of the CYP3A4 gene, referred to as CLEM4, that conferred the constitutive activation of the CYP3A4 gene in the liver. CLEM4 was polymorphic, suggesting that the regulatory variant identified in this study may at least partly contribute to interindividual variability of hepatic CYP3A4 level.
Acknowledgments
We thank Drs. Marie-Anne Loriot and Philippe Beaune (U490 INSERM, Paris, France) and Dr. Isabelle Stücker (U170 INSERM, Villejuif, France) for generously providing French genomic DNA and for valuable discussions.
Footnotes
-
This work was supported in part by Grants-in-Aid (99–2) from the Organization for Pharmaceutical Safety and Research and the Ministry of Education, Science, Sports and Culture of Japan. This work was also supported in part by Grants-in-Aid from the Core Research for Evolutional Science and Technology and an SRF Grant for Biomedical Research in Japan.
-
ABBREVIATIONS: P450, cytochrome P450; PXR, pregnane X receptor; kb, kilobase(s); XREM, xenobiotic responsive enhancer module; CAR, constitutive active receptor; USF, upstream stimulatory factor; CLEM4, constitutive liver enhancer module of CYP3A4; CRE, cAMP response element; CREB, cAMP response element-binding protein; AP-1, activating protein; mt, polymerase chain reaction product obtained by mutant allele-specific primer; PCR, polymerase chain reaction; wt, polymerase chain reaction product obtained by wild type allele-specific primer; LA-PCR, long and accurate polymerase chain reaction.
- Received June 25, 2003.
- Accepted October 17, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}