


Abstract
UDP-glucuronosyltransferases (UGTs) play important roles in the metabolism, detoxification,and clearance of many different xenobiotics, including drugs and endogenous compounds. Structural information about these membrane-bound enzymes of the endoplasmic reticulum is limited. We do not know the identity or the location of the key residues for catalysis and binding of the aglycone substrate and the cosubstrate UDP-glucuronic acid (UDPGA). One suggestion was that His371 (UGT1A6 numbering) is the “catalytic base” that deprotonates the phenol group. We have now re-examined this hypothesis by analyzing the activities of the corresponding mutants, 6H371A (in UGT1A6) and the 9H369A (in UGT1A9). The Km values of mutant 6H371A for scopoletin and UDPGA were higher by 4- and 11-fold, respectively, than in UGT1A6. The Kd for the enzyme-UDPGA complex, derived from bisubstrate kinetics, was about 9-fold higher in 6H371A than in UGT1A6, indicating severely impaired cosubstrate binding by the mutant. The effect of mutation on Vmax was large in UGT1A6 but variable in UGT1A9, suggesting that His371 does not play the catalytic role previously hypothesized. In both UGTs, the E379A mutation (UGT1A6 numbering) had an effect similar to that of the H371A mutations. A homology model of the putative UDPGA binding region of UGT1A6 was built using distant homologous protein structures from the “GT1 class.” The combined results of activity determinations, kinetic analyses, and modeling strongly suggest that His371 and Glu379 are directly involved in UDPGA binding but are not the general acid or general base.
In vertebrates, glucuronidation is an important detoxification and metabolic pathway for many endogenous compounds such as bile acids, bilirubin, and steroid hormones and for xenobiotics, including many drugs. Glucuronidation, catalyzed by a family of UDP-glucuronosyltransferases (UGTs; EC 2.4.1.17), increases the solubility of these compounds in water so that they are much more easily excreted from the body via bile or urine. The glucuronic acid moiety is donated by the cosubstrate UDP-glucuronic acid (UDPGA) and is conjugated to the aglycone substrate acceptor at a functional group, mostly hydroxyl, amino, or carboxyl groups. This process leads to the formation of β-d-glucuronides of the aglycones and, in most but not all cases, abolishes their biological activity (Radominska-Pandya et al., 1999; Tukey and Strassburg, 2000; Fisher et al., 2001; Ouzzine et al., 2003). Most UGTs can glucuronidate several substrates, often structurally unrelated, whereas one compound may be glucuronidated by several UGTs (Mackenzie et al., 2000).
UGTs are 529 to 534 amino acids in length, including an N-terminal signal sequence of approximately 25 residues that directs the nascent proteins to the endoplasmic reticulum (ER) and is later cleaved off (Kurkela et al., 2003). They are ER membrane proteins and are oriented such that the N terminus and most of the protein mass are located in the lumen. There is a single trans-membrane segment close to the C terminus, and the last 20 to 25 C-terminal residues are on the cytoplasmic side of the endoplasmic reticulum membrane (Radominska-Pandya et al., 1999). Many UGTs are expressed in the liver, the major site of glucuronidation, but a few are solely extrahepatic, and most human UGTs are expressed in more than one tissue (Tukey and Strassburg, 2001). The 19 human UGTs can be divided into two major families, UGT1 and UGT2, based on sequence similarities and gene organization (Mackenzie et al., 1997).
UGTs all contain two large and almost equally sized domains, the N-terminal and C-terminal halves (Radominska-Pandya et al., 1999). In the UGT1 subfamily, each isozyme has a variable N-terminal half encoded by different exons 1, whereas the C-terminal half is encoded by the shared exons 2 to 5 and so is identical (Ritter et al., 1992). The UGT2 proteins, with the exception of UGT2A1 and UGT2A2, are mostly encoded by separate genes (Mackenzie et al., 2005). Nonetheless, the C-terminal halves have approximately 60% identity with respect to the UGT1 proteins (data not shown). The very high sequence conservation in the C-terminal domain immediately suggests that it binds UDPGA, the cosubstrate that is used by all the UGTs. Consequently, the aglycone substrate is believed to bind within the more variable N-terminal domain of the UGTs (Mackenzie, 1990).
Thus far, there is no crystal structure of any UDP-glucuronosyltransferase. UGTs belong to the superfamily of glycosyltransferases (GT) and, so far, all the known structures of different members of this huge protein family have been found to adopt either the “GT-A” or “GT-B” folds (Bourne and Henrissat, 2001). The GT-B fold, to which the UGTs belong, consists of two rather similar Rossman fold domains separated by a linker region. This kind of fold was originally observed in the phage T4 β-glucosyltransferase (Vrielink et al., 1994). In the GT-B fold, the N-terminal domain binds the acceptor and the C-terminal binds the nucleotide-sugar (Bourne and Henrissat, 2001). Based on amino acid sequence similarity (Campbell et al., 1997) (available at http://afmb.cnrs-mrs.fr/CAZY/), UGTs belong to the GT1 family in the GT-B fold. Thus far, nine structures of GT1 family members are available of five different enzymes: the bacterial glycosyltransferases TDP-epi-vancosaminyltransferase GtfA, the UDP-glucosyltransferase GtfB, and the vancosaminyltransferase GtfD, all involved in vancomycin synthesis (Mulichak et al., 2001, 2003, 2004) and two plant glucosyltransferases, a triterpene/flavonoid glucosyltransferase (UGT71G1) and a flavonoid glucosyltransferase (VvGT) ((Shao et al., 2005; Offen et al., 2006). The level of sequence identity between each of these 5 GTs and any of the human UGTs is unfortunately below 20%, making it difficult to model a complete UGT. Nevertheless, there is a higher degree of identity within particular conserved regions, like the nucleotide-sugar binding site.

Relative expression levels (± S.E., n = 3) of control and mutant enzymes. Control UGT1A6 expression level was set to 1, and the expression levels of mutants 6H371A and 6E379A are presented relative to that. The same was carried out for UGT1A9 and the two mutants in this enzyme. See Materials and Methods for experimental details.
Which residues within the UGTs are directly involved in either UDPGA or aglycone binding is presently unknown. Photoaffinity labeling experiments suggest that the UDPGA binding site is located between amino acids 299 and 466 (Battaglia et al., 1998). In addition, there is a conserved, characteristic motif (Prosite PS00375) that can be used to identify distant homologues. The motif, located approximately between amino acids 369 and 407 in the C-terminal half of the UGTs, may function in UDPGA binding (Tukey and Strassburg, 2001). A much more specific suggestion was made about the role of one residue within this rather large segment. Based on mutation studies and protein modification with the reagent diethyl pyrocarbonate, His371 (UGT1A6 numbering; sometimes inaccurately referred to as His370, probably because of the presence of one UGT1A6 sequence in the gene bank that is short by one residue) was suggested to function as a catalytic base (Ouzzine et al., 2000).
Following on our earlier work (e.g., Kurkela et al., 2007), we were looking for a poorly active mutant of UGT1A9 for functional oligomerization studies and decided to mutate the “catalytic His” in UGT1A9 to Ala, based on this suggestion (Ouzzine et al., 2000). We expected that the equivalent UGT1A9 mutant, 9H369A, would have very low activity. However, the mutation had much a milder effect on scopoletin (7-hydroxy-6-methoxy-2H-1-benzopyran-2-one) glucuronidation than expected. We therefore re-examined the role of this strictly conserved His371 using mutagenesis, kinetic analyses, and modeling. We also examined the role of another highly conserved residue, Glu379 (UGT1A6 numbering). Our results suggest that His371, rather than being the previously proposed “catalytic residue,” plays an important role in UDPGA binding. The same also seems to be true for Glu379.
Materials and Methods
Recombinant Enzyme Preparation. The recombinant control UGTs UGT1A6 and UGT1A9 were expressed as C-terminal Histagged fusion proteins in baculovirus-infected insect cells, and membranes were isolated and stored as described previously (Kurkela et al., 2003). Point mutations were done by PCR, and the sequence of the entire DNA fragment that underwent PCR was verified by DNA sequencing. Mutant UGTs 6H371A, 6E379A, 9H369A, and 9E377A were expressed and produced as the control enzymes (A prefix of “6” indicates a UGT1A6 mutant; a prefix of “9” indicates a UGT1A9 mutant). Preparation of the control UGTs was optimized to achieve high normalized activity (activity/expression level) rather than specific activity (activity/protein amount). We have not observed stability differences between the control and the mutants studied here.
Normalization. The relative expression level of each recombinant UGT, either mutant or control, was determined by immunodetection on dot-blot using the monoclonal anti-His-tag IgG Tetra His (QIAGEN) as described previously (Kurkela et al., 2004, 2007). In brief, at least three replicates of each recombinant UGT membrane were made and blotted with the monoclonal antibody. The expression level of nonmutated UGT control membranes was set to 1, and the mutant expression levels were calculated relative to that. All the activity measurements were corrected by these relative expression levels (Fig. 1). The expression levels varied by less than a factor of 3, except for 9H369A.
HPLC Activity Measurements. Scopoletin, 4-methylumbellifer-one, umbelliferone (7-hydroxy-2H-1-benzopyran-2-one), and 1-naphthol were purchased from Sigma and UDPGA triammonium salt from Fluka Chemical Corp. (Ronkonkoma, NY). Activity assay measurements with at least three replicates were made in 100-μl reactions containing 50 mM phosphate buffer, pH 7.4, 10 mM MgCl2, 5 mM saccharolactone, and 0.036 to 1.0 mg/ml (protein concentration) of membranes containing recombinant protein of UGT1A6, UGT1A9, 6H371A, 6E379A, 9H369A, or 9E377A. The concentration of aglycone was 100 μM for 4-MU and 1-naphthol and 500 μM for scopoletin. The reaction was started by adding 5 mM UDPGA and performed in the linear range at 37°C for 20 to 30 min, depending on the substrate used. The reaction was stopped by adding 10 μl of 4 M ice-cold perchloric acid to the reaction mixture, after which the samples were centrifuged for 5 min at 16,000g and the cleared supernatant used for HPLC analysis. An Agilent model 1100 (Agilent Technologies, Waldbronn, Germany) was used to analyze the glucuronidation products formed.
Kinetic Measurements. Kinetic measurements were done using at least eight substrate concentrations. The protein concentrations in the assays were adjusted so that no more than 10% of the substrate was used during the glucuronidation reaction. Scopoletin (10–4000 μM), 1-naphthol (1–200 μM) or 4-MU (10–1500 μM) with either 5 or 20 mM UDPGA was used in the measurement of the aglycone kinetics, whereas the kinetics with respect to UDPGA was done in the presence of 2 mM scopoletin and 25 to 20,000 μM UDPGA. To obtain kinetic constants, we fit the experimental data to the Michaelis-Menten equation by nonlinear regression using Prism software (GraphPad Software, San Diego, CA). Two-substrate kinetics was done with eight UDPGA and seven scopoletin concentrations. Data from bisubstrate kinetics were fitted to eq. 1, taken from Luukkanen et al. (2005):  v = where Vmax is the maximum velocity of the reaction, Kd(UDPGA) is the dissociation constant for the enzyme-UDPGA complex and Km(AGLY) and Km(UDPGA) are Michaelis constants for aglycone and UDPGA, respectively. This equation is derived from the compulsory ordered bi bi mechanism (Fig. 2) assuming steady-state conditions. Up to three replicates were used in kinetic measurements.
v = where Vmax is the maximum velocity of the reaction, Kd(UDPGA) is the dissociation constant for the enzyme-UDPGA complex and Km(AGLY) and Km(UDPGA) are Michaelis constants for aglycone and UDPGA, respectively. This equation is derived from the compulsory ordered bi bi mechanism (Fig. 2) assuming steady-state conditions. Up to three replicates were used in kinetic measurements.
Modeling. We built a model of the UGT1A6 C-terminal domain residues 288 to 447 using the Homology module in Insight II (Accelrys, San Diego, CA). We used the following structures as templates: the triterpene/flavonoid glcyosyltransferase (UGT71G1) from the legume Medicago truncatula (PDB entry 2ACV with UDP and 2ACW with UDP-glucose) (Shao et al., 2005) and the UPD-glucose:flavonoid 3-O-glycosyltransferase from red grape (Vitis vinifera) (VvGT) (PDB entries 2C1X with UDP and 2C1Z with UDP-2-deoxy-2-fluoro glucose and 2C9Z with UDP and quercetin) (Offen et al., 2006). We initially aligned the sequences using ClustalW (http://www.ebi.ac.uk/clustalw/) and manually modified them using the information from the Jpred secondary structure prediction (http://www.compbio.dundee.ac.uk/~www-jpred/), and 3D-pssm (http://www.sbg.bio.ic.ac.uk/~3dpssm/) and 3d-jigsaw (http://www.bmm.icnet.uk/~3djigsaw/) fold servers. The sequence identity was 21.5% to UGT71G1 and 24.7% to VvGT over the entire modeled region. Minimization was done in Insight II using the Amber force field and steepest descent and conjugate gradient minimizations. UDPGA was docked manually to the partial model of the UGT1A6 C-terminal domain.
Results
In the beginning, we prepared the 9H369A mutant for functional oligomerization studies, expressed it in baculovirus-infected insect cells and examined its activity with scopoletin (Fig. 3) as the aglycone substrate. The normalized glucuronidation of scopoletin by the mutant, approximately 40% of the control UGT1A9 under the assay conditions, was much higher then expected, assuming that this His369 residue functions as a catalytic residue in all the UGTs (Fig. 4). Previous studies of the activity of the 6H371A mutant were performed using only 4-MU (Fig. 3) as the aglycone substrate (Ouzzine et al., 2000). The same substrate was therefore tested with UGT1A9 and the 9H369A mutant, revealing that the effect of this mutation is substrate-dependent (Fig. 4). These findings prompted us to re-examine the suggested role of this strictly conserved His residue in UGT1A9, as well as in UGT1A6, the isoenzyme that was mutated in the original studies (Ouzzine et al., 2000).
We therefore made the corresponding mutant in UGT1A6, 6H371A. When its activity was screened with three different aglycones (Fig. 3), it seemed that the mutation had a much more drastic effect on UGT1A6 (Fig. 5) than on UGT1A9 (Fig. 4). In both isoenzymes, however, the activities for scopoletin were much higher than for 4-MU (Figs. 4 and 5) and, even though the activities of 6H371A with scopoletin and 1-naphthol were low, they were nonetheless reduced by a factor of 30 or less. A key catalytic residue would be expected to lower the activity by a factor of at least 100.
The realization that His371 may not be the catalytic His prompted us to investigate the effect of a mutation of another highly conserved residue in its vicinity, Glu379. This residue, strictly conserved among the human UGTs, was mutated to Ala in both UGT1A6 and UGT1A9, yielding the 6E379A and 9E377A mutants. Like the H(369/371)A mutants, both E(377/379)A mutants exhibited very low 4-MU glucuronidation activity (Figs. 4 and 5). However, the scopoletin glucuronidation activity of the 9E377A mutant was almost 90% of the corresponding activity in the control UGT1A9 (Fig. 4). Even in UGT1A6, where the mutation seemed to affect activity more, the activity of the 6E379A mutant, particularly toward 1-naphthol, was higher than the corresponding activity of the 6H371A mutant (Fig. 5). When the two isoenzymes are compared, it is clear that both mutations had a very similar and dramatic effect on glucuronidation activity for all three substrates in UGT1A6 (Fig. 5), whereas the effects varied in UGT1A9 (Fig. 4)

The compulsory ordered bi bi mechanism for UGTs (see also Luukkanen et al., 2005). E, UGT; Agly, aglycone; GA, glucuronic acid.
We continued to reinvestigate the role of both His371 and Glu379 in UGTs by kinetic analyses, starting with UGT1A9. The Km values were highly affected (Table 1), and so we could not determine reliable Vmax for the 9E377A mutant, although it seemed to be more active than control (data not shown). The 4 mM scopoletin (close to the solubility limit) and 20 mM UDPGA concentrations we used were still far from saturating conditions. The changes were not so dramatic for the UGT1A6 mutants, so we decided to focus on UGT1A6. The results indicated that both mutations increased the Km for aglycone substrates substantially (Tables 2, 3, 4). The Km for the cosubstrate, UDPGA, increased much more than the Km for the aglycones (Tables 2 and 3). The same was also true for the full bisubstrate kinetic analysis for the control and for the mutants 6H371A and 6E379A (Table 4, Fig. 6). The dissociation constant Kd(UDPGA) increased approximately 9-fold in the H371A mutant and over 30-fold in the E379A mutant, indicating severely impaired UDPGA binding (Table 4). The mutations have a bigger effect on UDPGA than on the aglycone binding, particularly for the E379A mutant.
Kinetic analysis of UGT1A9 control and 9H369A and 9E377A mutant forms (n = 3)
The scopoletin kinetics was measured with nine concentrations between 25 and 4000 μM and 20 mM UDPGA. UDPGA kinetics was measured using nine concentrations between 25 and 20,000 μM and 4000 μM scopoletin. Numbers in parentheses indicate the ratio Km(mutant)/Km(control) (i.e., to indicate how much the mutant Km value increased). Values are presented as mean ± S.E.
Kinetic analysis of UGT1A6 control and 6H371A and 6E379A mutant forms
Constant UDPGA concentration (5 or 20 mM). 1-Naphthol and 4-MU kinetics was measured with eight aglycone concentrations (1-naphthol between 1 and 200 μM and 4-MU between 10 and 1500 μM and 5 mM UDPGA). The scopoletin kinetics was measured with nine different concentrations between 25 and 4000 μM and 20 mM UDPGA. Numbers in parentheses indicate the ratio Km(mutant)/Km(control) (i.e., to indicate how much the mutant Km value increased).
Kinetic analysis of UGT1A6 control and 6H371A and 6E379A mutant forms
Constant scopoletin concentration (2 mM). UDPGA kinetics was measured using nine UDPGA concentrations between 25 and 20,000 μM and 2000 μM scopoletin (n = 3). Numbers in parentheses indicate the ratio Km(mutant)/Km(control) (i.e., to indicate how much the mutant Km value increased).
Kinetic analysis of UGT1A6 control and 6H371A and 6E379A mutant forms
Bisubstrate kinetics was measured using 7 scopoletin (25-4000 μM) and 8 (25-20000 μM) UDPGA concentrations. Numbers in parentheses indicate the ratio Km(mutant)/Km(control) (i.e., to indicate how much the mutant Km value increased).
Structures of the three substrates used in activity and kinetic studies.

A comparison of the scopoletin and 4-MU glucuronidation activities of mutants 9H369A and 9E377A with UGT1A9, normalized according to expression levels, shown as percentages (± S.E., n = 3). The substrate concentrations were either 500 μM scopoletin or 100 μM 4-MU for the aglycone and 5 mM UDPGA.

A comparison of the normalized glucuronidation rates of mutants 6H371A and 6E379A mutant with UGT1A6 control, measured in the presence of either 500 μM scopoletin, 100 μM 1-naphthol or 4-MU, and 5 mM UDPGA. Activities are represented as percentages (± S.E., n = 3).
To obtain further insight into the UDPGA binding site of the UGTs, we modeled the C-terminal domain of UGT1A6 up to the membrane anchor. A structural model for the C-terminal UDPGA binding site of UGT1A6 was built using Insight II, and the structures of UGT71G1 and VvGT, two GT1 superfamily plant homologues, were used as templates (Shao et al., 2005; Offen et al., 2006) (see Materials and Methods). Both of these glycosyltransferases catalyze the addition of glucose from UDP-glucose to acceptor molecules such as saponin (UGT71G1) or cyanidin (VvGT) (Shao et al., 2005; Offen et al., 2006). Hence, these two plant enzymes catalyze a kind of reaction similar to that of human UGTs, but they employ UDP-glucose, not UDP-glucuronic acid. The sequence around His371 is quite conserved in the GT1 superfamily, and the percentage identity of the UGT1A6 sequence to the template structures is higher in this region than over the entire C-terminal domain (Fig. 7), thus increasing the reliability of our model. The model was minimized in Insight II using the Amber force field. The overall structural geometry was good according to Procheck analysis (Laskowski et al., 1993). UDP-glucose adopts the same conformation in both UGT71G1 and VvGT, and so UDPGA was manually docked into the model in the same conformation as the UDP-glucose in VvGT.
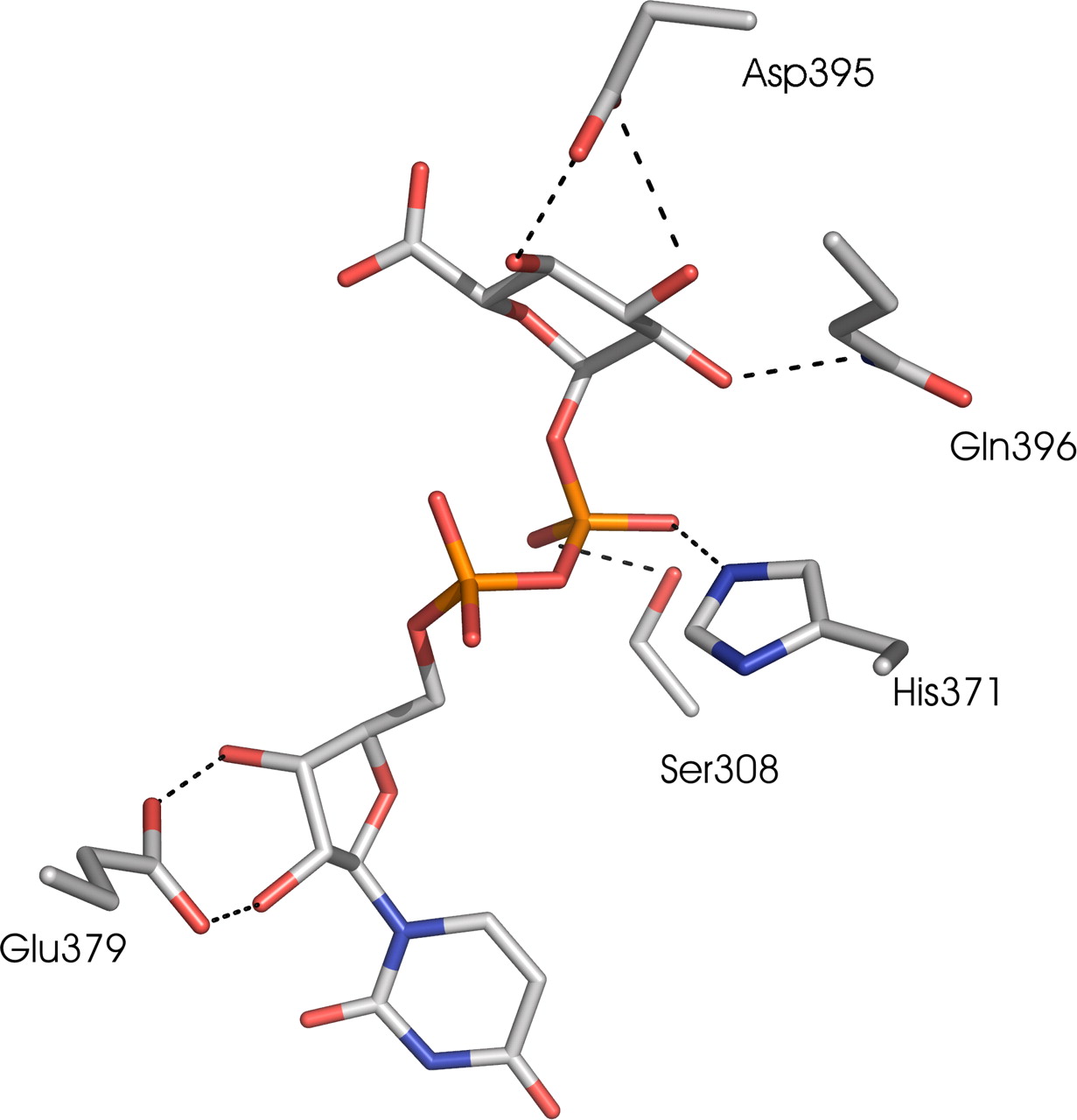
In our model (Fig. 8), His371 hydrogen-bound to the β-phosphate group of UDPGA and thus the model supported the suggestion that His371 of UGT1A6 is directly involved in UDPGA binding. The model also suggests that Glu379 forms hydrogen bonds to the ribose ring. However, the reason that the mutations lead to changes in substrate specificity is currently unclear. For instance, the main difference between the E→ A and the H→ A mutants, particularly for UGT1A9, was that the 9E377A mutation seemed to lower the scopoletin glucuronidation rate at most only slightly, leading to an even larger substrate-dependence of its effect than for 9H369A (Fig. 4). Nonetheless, there is good agreement between the model and our experimental results, thus strengthening the inferences we draw below.
What can be concluded from the results? We confirmed earlier results (Ouzzine et al., 2000), that the 6H371A mutant has very low 4-MU activity. This naively would suggest that H371A is a catalytic residue. However, the fact that 9H369A has 40% of the activity of the control UGT1A9 with scopoletin (Fig. 4) clearly indicated that this is not the case. Mutation of a key catalytic residue should have a much more severe impact on Vmax with all substrates and for both isoenzymes tested.

Bisubstrate kinetic curves of control UGT1A6 and mutants 6H371A and 6E378A. Seven scopoletin (25, 100, 250, 500, 1000, 2000 and 4000 μM) and eight UDPGA concentrations (25, 100, 250, 500, 1000, 2500, 10,000, and 20,000 μM) were used. All the activities were normalized for expression levels. The derived kinetic constants are listed in Table 4.
Discussion
Much is currently known about the gene structure and different genetic elements that affect the expression of different human UGTs in our tissues. However, with respect to the structure of the UGTs, all we know is that there are two domains and a C-terminal transmembrane helix and that the overall fold they adopt is GT-B (see http://afmb.cnrs-mrs.fr/CAZY/) (Campbell et al., 1997). Studies on the roles of individual residues have yielded little detailed information. When a specific residue, His371 in UGT1A6 (then assumed to be His370), was reported to serve as the general base by Ouzzine et al. (2000), it was regarded as a major development. In their model, His371 deprotonates the nucleophilic group on the aglycone, which then attacks the UDPGA, with subsequent release of the UDP group. The conclusion was based on site-directed mutagenesis, 4-MU glucuronidation activity of the UGT1A6 mutants and on the interpretation of chemical modification studies, taken to indicate the presence of a “catalytic His” in the UGTs (Ouzzine et al., 2000, 2003). Our new results, however, do not support this suggestion. Based on our activity, kinetic, and modeling studies, neither His371 nor Glu379 is in contact with the aglycone, but both are involved in UDPGA binding. His371 cannot be a general base and Glu379 cannot be a general acid.
The kinetic analyses revealed that changing either His371 or Glu379 to Ala clearly increases the Km values for three different aglycones (Tables 1, 2, 3, 4). Why should this be so if both residues play important roles in UDPGA binding? The answer is that both mutations increase the Km values for UDPGA significantly more than they increase the aglycone Km values. Moreover, we have recently suggested that the UGTs (at least those of UGT1 subfamily) bind the two substrates sequentially with UDPGA binding first (Luukkanen et al., 2005). If so, the effect of either H371A or E379A mutations on the Km for the different aglycone substrates is “secondary”; the “primary” effect is on the Km for UDPGA. The mutation affects how UDPGA binds, and thus affects aglycone binding (see below).
In UGT1A9, the Km value for both UDPGA and aglycone increased, suggesting that substrate binding is impaired. However, Vmax doubled for 9H369A (Tables 2, 3, 4) and also seemed to increase substantially for 9HE377A, although the increase in Km for this mutant meant that we could not determine the value reliably (data not shown). One possible explanation is that the reduction in UDPGA binding affinity allows aglycone to bind in a catalytically more productive manner, thus reducing the effect of the substrate inhibition typically observed in UGTs (Luukkanen et al., 2005). The existence of such an effect is consistent with the changes we see in UGT1A6 (see above) and is not consistent with a key catalytic role for either His371 or Glu377.
His-tag antibody detection cannot differentiate between properly folded and misfolded protein. Could the changes in activity we observe (see Figs. 4 and 5) therefore be due to differences in the proportion of active protein in the control and mutant preparations? The answer to this is clearly no. The Km and Kd values are insensitive to the amount of active enzyme, and so are any differences in the mutant responses to various substrates. The only thing that might be affected is Vmax. Nonetheless, we maximize the specific activity of the control UGTs during protein preparation; the relative amount of the active control enzyme is thus as high as possible. Furthermore, we have not detected any stability differences between the control and the mutants studied here and, if the mutants were less stable, their real Vmax would be higher than the one we have measured (Figs. 4 and 5) (i.e., in most cases, more similar to control). Finally, any differences between the activity UGT1A6 and UGT1A9 toward different substrates upon mutation are independent of the ratio of active enzyme, because that ratio does not change from substrate to substrate (Figs. 4 and 5).

Sequence alignment of UGT1A6 (1A6) with the plant proteins UGT71G1 (2ACW) and VvGT (2C1Z) used in the modeling (Fig. 8). The original alignment was made by ClustalW and manually modified according to structural information from the Jpred, 3d-pssm and 3d-jigsaw servers. The most conserved areas are highlighted with gray, and the mutated residues His371 and Glu379 are bold. The figure was made using GeneDoc.
The central role of His371 in UDPGA binding is supported by its conservation over a wide range of different glycosyltransferases, including GtfA (His293) and GtfD (His309), where it makes contacts with the β-phosphate in the TDP sugar donor (Mulichak et al., 2003, 2004). It is noteworthy that the equivalent residue is Arg261 in the bacterial UDP-N-acetylglucosaminyltransferase MurG, which has the GT-B fold but does not belong to the same class. Arg261 is located close to the β-phosphate of UDP-N-acetylglucosamine and is proposed to stabilize the leaving group during catalysis (Ha et al., 2001). It has been speculated that His371 in human UGTs may have a similar kind of role (Radominska-Pandya et al., 2005).
The activity and the kinetic results strongly suggest that, in addition to His371, Glu379 is one of the key residues involved in UDPGA binding (Fig. 8). Consistent with this, even very distant homologues of the UGTs use the equivalent Glu to bind the nucleotide. For instance, Glu269 in MurG has been suggested to have a role in discriminating between UDP and dTDP (Hu et al., 2003). Likewise, in Escherichia coli T4 bacteriophage β-glucosyltransferase, which catalyzes the transfer of glucose from UDP-glucose to hydroxymethylated cytosines, Glu272 makes the equivalent contacts to the ribose moiety (Moréra et al., 1999). However, in three of the five GT1 structures, UDP-glucosyltransferase GtfB (PDB entry 1IIR), vancosaminyltransferase GtfD (PDB entry 1RRV), and TDP-epi-vancosaminyltransferase GtfA (PDB entries 1PN3, 1PNV), there is a hydrophobic residue in the position equivalent to Glu379. It has been proposed that a hydrophobic residue at this position lowers the affinity of the enzymes for their respective sugar donors (Hu et al., 2003). In GtfB and GtfD, the Km values for the nucleotide-sugar donors are 1 to 2 mM (Mulichak et al., 2001, 2004), whereas it is in the micromolar range in MurG (Hu et al., 2003).
The new model, available on request from us, also has predictive power. For example, it suggests that Ser308 is hydrogen-bonded to the β-phosphate group in UDPGA, much like Ser285 in the structure of UGT71G1. Likewise, GtfA, GtfB, and GtfD all have serines in this position [Ser230, Ser247, and Ser246, respectively (Mulichak et al., 2001, 2003, 2004)], whereas VvGT has a threonine (Thr280). In MurG, Ser192 can make a contact to the β-phosphate of UDP-N-acetylglucosamine; replacing this Ser with Ala affects all the kinetic parameters (Hu et al., 2003). The role of this Ser was also examined in UGT71G1, and the Ser→ Ala mutant was inactive (He et al., 2006). The highly conserved residues Asp395 and Qln396 seemed to form hydrogen bonds to the sugar moiety of UDPGA (Fig. 8). The initial results from mutation studies of Asp395 and Qln396 support the model (Y. Xiong, S. Bratton, A. Zielinska, M. Finel, A. Radominska-Pandya, submitted). The equivalent residues in VvGT, Asp374 and Qln375, as well as Glu381 and Qln382 in UGT71G1, make corresponding contacts and were suggested to play a role in sugar recognition (Offen et al., 2006). The mutants D374A of VvGT and E381A of UGT71G1 were catalytically inactive, whereas mutants Q375N and Q375H of VvGT1 exhibited seriously impaired activities (Offen et al., 2006) (Shao et al., 2005).
In conclusion, the results of this study suggest that both His371 and Glu379 of UGT1A6 are involved in UDPGA binding, but not as a general base or acid. Our study also identifies other possible UDP-binding residues. The identity of the “real” catalytic residues remains open.

A ball-and-stick representation of the UDPGA binding site of the UGT1A6 C-terminal model (carbon, gray; oxygen, red; nitrogen, blue; phosphate, orange). Dashed lines represent hydrogen bonds. The figure was produced in PyMOL (http://www.pymol.org). The model suggests that both His371 and Glu379 make hydrogen bonds to UDPGA. In our model, His371 and Ser308 make hydrogen bonds to the β-phosphate, and Glu379 makes hydrogen bonds to the 2′ and 3′ hydroxyls of the ribose moiety of UDPGA. Asp395 and Gln396 hydrogen bond to the glucopyranose ring of UDPGA. See text for details.
Acknowledgments
We are grateful for the excellent technical assistance of Johanna Mosorin.
Footnotes
-
This work was supported by the Academy of Finland (A.G. projects 1105157 and 1114752, M.F. projects 207535 and 210933) and the Sigrid Juselius Foundation (A.G. and M.F.). A.G. is a member of Biocentrum Helsinki, which partly supported this work.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.107.036871.
-
ABBREVIATIONS: UGT, UDP-glucuronosyltransferase; UDPGA, UDP-glucuronic acid; ER, endoplasmic reticulum; 4-MU, 4-methylumbelliferone; GT, glycosyltransferase; CAZY, carbohydrate active enzymes; PDB, Protein Data Bank; UGT71G1, triterpene/flavonoid glycosyltransferase from legume Medicago truncatula; VvGT, UDP-glucose:flavonoid 3-O-glycosyltransferase from red grape (Vitis vinifera); GtfA, TDP-epi-vancosaminyl-transferase from Amycolatopsis orientalis; GtfB, UDP-glucosyltransferase from Amycolatopsis orientalis; GtfD, vancosaminyltransferase from Amycolatopsis orientalis; MurG UDP-N-acetylglucosaminyltransferase from Escherichia coli; UGT1A6, human UDP-glucuronosyltransferase isoenzyme 1A6; UGT1A9, human UDP-glucuronosyltransferase isoenzyme UGT1A9.
- Received April 5, 2007.
- Accepted June 19, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}