


Abstract
Human cytochrome P450 (CYP) 2C enzymes metabolize ∼30% of clinically prescribed drugs and various environmental chemicals. CYP2C8, an important member of this subfamily, metabolizes the anticancer drug paclitaxel, certain antidiabetic drugs, and endogenous substrates, including arachidonic acid, to physiologically active epoxyeicosatrienoic acids. Previous studies from our laboratory showed that microRNA 107 (miR107) and microRNA 103 downregulate CYP2C8 post-transcriptionally. miR107 is located in intron 5 of the pantothenate kinase 1 (PANK1) gene. p53 has been reported to coregulate the induction of PANK1 and miR107. Here, we examine the possible downregulation of CYP2C8 by drugs capable of inducing miR107. Hypolipidemic drugs, such as bezafibrate, known activators of the peroxisome proliferator-activated receptor α (PPARα), induce both the PANK1 gene and miR107 (∼2.5-fold) in primary human hepatocytes. Surprisingly, CYP2C8 mRNA and protein levels were induced by bezafibrate. CYP2C8 promoter activity was increased by ectopic expression of PPARα in HepG2 cells, with a further increase after bezafibrate (∼18-fold), 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio acetic acid (∼10-fold) treatment, or the antidiabetic drug rosiglitazone, all known PPAR activators. Promoter sequence analyses, deletion studies, mutagenesis studies, and electrophoretic mobility shift assays identified a PPARα response element located at position −2109 base pair relative to the translation start site of CYP2C8. Chromatin immunopreciptation assay analysis confirmed recruitment of PPARα to this PPARα response element after bezafibrate treatment of human hepatocytes. Thus, we show for the first time that CYP2C8 is transcriptionally regulated by PPARα, suggesting the potential for drug-drug interactions due to upregulation of CYP2C8 by PPAR activators.
Introduction
Cytochrome P450 (CYP) 2C8 is the second most abundant CYP2C enzyme in the human liver after CYP2C9 (Lai et al., 2009). It metabolizes the antidiabetic drugs rosiglitazone and troglitazone, anticancer drug paclitaxel, cholesterol-lowering drug cerivastatin, antiarrhythmic drug amiodarone, calcium channel blocker verapamil, and antimalarials amodiaquine and chloroquine (Totah and Rettie, 2005). CYP2C8 also metabolizes the endogenous molecule arachidonic acid to 11,12- and 14,15-epoxyeicosatrienoic acids (EETs) (Fisslthaler et al., 1999). It is highly expressed in human liver, but is also expressed in extrahepatic tissues, such as the kidney, lung, nasal mucosa, arteries, endothelial mucosa, and heart (Fisslthaler et al., 1999; Klose et al., 1999; Ding and Kaminsky, 2003; Delozier et al., 2007). Because CYP2C8 is expressed in endothelial cells, arteries, and the heart and metabolizes arachidonic acid to physiologically active EETs, CYP2C8 has been proposed as an endothelial-derived hyperpolarizing factor synthetase (Fisslthaler et al., 1999; Zeldin, 2001).
Human CYP2C8 is the most inducible of the CYP2C genes in human hepatocytes in response to microsomal inducers, such as rifampicin, phenobarbital, and CITCO (Gerbal-Chaloin et al., 2001; Ferguson et al., 2005; Chen and Goldstein, 2009; Lai et al., 2009). CYP2C8 is also induced by phenytoin, hyperforin, paclitaxel (a CYP2C8 substrate), and the synthetic glucocorticoid dexamethasone (Synold et al., 2001; Raucy et al., 2002; Garcia-Martin et al., 2006). Induction of CYP2C8 by xenobiotics contributes to the interindividual variability in drug metabolism in human populations, which can lead to a change in the half-life of drugs and result in drug tolerance or therapeutic failure. The induction of the CYP2C8 gene by drugs and xenobiotics is mediated by the constitutive androstane receptor (CAR), pregnane X receptor (PXR), and glucocorticoid receptor, whereas hepatocyte nuclear factor 4α (HNF4α) appears to play a role in basal expression (Ferguson et al., 2005).
Recently, our laboratory demonstrated that CYP2C8 protein levels were downregulated post-transcriptionally by microRNA 107 (miR107) and microRNA 103 (miR103) in human liver (Zhang et al., 2012). MicroRNAs (miRNAs) play important roles in the regulation of target genes by binding to the 3′-untranslated region and promote mRNA degradation or repress mRNA translation (Bartel, 2004). miR107 and miR103 (paralogs) are encoded within the introns of three pantothenate kinase (PANK) genes located on separate chromosomes (Wilfred et al., 2007). PANK genes catalyze the rate limiting step in coenzyme A biosynthesis and are involved in the regulation of acetyl-CoA levels and lipid metabolism (Wilfred et al., 2007; Trajkovski et al., 2011). Although miR103 is not completely coregulated with the corresponding PANK genes (Wilfred et al., 2007), previous studies have shown that p53 coregulates PANK1 and miR107 in different cellular systems (Yamakuchi et al., 2010; Bohlig et al., 2011). Pank1 expression is upregulated by the peroxisome proliferator-activated receptor (PPAR) α agonist bezafibrate (BF) in HepG2 cells, resulting in elevated CoA levels (Ramaswamy et al., 2004).
PPARs act as lipid sensors to control the expression of gene networks involved in lipid homeostasis and inflammatory responses (Lalloyer and Staels, 2010). There are three functional PPARs: PPARα, PPARβ, and PPARγ. PPARα is highly expressed in the liver and functions primarily to regulate the expression of genes involved in peroxisomal and mitochondrial β-oxidation and microsomal ω-hydroxylation (Gulick et al., 1994; Schoonjans et al., 1996). The activated PPARα heterodimerizes with the retinoic acid X receptor (RXR), and this complex binds to specific DNA sequences called peroxisome proliferator response elements (PPREs), which are located in the promoter regions of target genes to upregulate their expression (Kliewer et al., 1992; Wahli and Michalik, 2012).
Until recently, CYP4 family members, which function as microsomal fatty acid ω-hydroxylases, were the only cytochromes P450 reported to be directly regulated by PPARα (Waxman, 1999; Hsu et al., 2007). However, the drug-metabolizing cytochromes P450 CYP3A4, CYP2B6, CYP2C8, CYP1A1, and CYP1A2 were recently reported to be induced by fibrates (Thomas et al., 2013). Studies using primary human hepatocytes and a CYP3A4/3A7-humanized mouse model showed that CYP3A4 is directly regulated by PPARα (Thomas et al., 2013). However, the mechanism of regulation of CYP2C8 by PPARα has not been investigated. It is not known whether the regulation of CYP2C8 by PPARα agonists/ligands is modulated by transcriptional activation by PPARα or CAR/PXR or indirectly by changes in miR107 expression.
The purpose of this study was to examine the regulation of CYP2C8 expression by xenobiotics capable of inducing PANK1/miR107 in cultured primary human hepatocytes and whether PPARα affects CYP2C8 transcription directly or indirectly. Surprisingly, the hypolipidemic fibrate BF induced both Pank1 and miR107 expression in primary human hepatocytes and also induced CYP2C8 expression. Here, we provide evidence to support the hypothesis that the CYP2C8 gene is directly upregulated by PPARα in human hepatocytes.
Materials and Methods
Chemicals and Reagents.
BF, 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio acetic acid (WY14643), and rosiglitazone were purchased from Sigma-Aldrich Company, Inc. (St Louis, MO). Anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (clone 6C5, MAB374) was purchased from Millipore (Temecula, CA). Rabbit polyclonal antibodies against PPARα (sc-9000), RXRα (sc-553), and RNA Pol II (sc-899) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The expression plasmids for human PPARα (pcDNA3-hPPARα) and human RXRα (pGEM-3T-RXRα) were kindly provided by Dr. Masahiko Negishi (Laboratory of Reproductive and Developmental Toxicology, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC). The expression plasmids pSG5-PPARα (identity 22751) and pcDNA-Flag-PPARγ (identity 8895) and the luciferase construct PPRE-X3-TK-Luc (identity 1015), containing three copies of the PPRE from rat acyl-CoA oxidase (ACOX) cloned upstream of the TK gene promoter, were obtained from Addgene (Cambridge, MA). The luciferase reporter constructs 2C8-3k, 2C9-3k, 2C19-2.7k, 2C8-3k/−8.9–8.5 [−8.9 to −8.5 kilobase (kb) region of the CYP2C8 promoter containing the CAR site cloned upstream of the 2C8-3k], 2C8-2.5k, 2C8-2k, 2C8-1.5k, 2C8-500, 2C8-300, 2C8-3k/ΔBgl II, and 2C8-2.5k/ΔBgl II (generated by digestion and ligation of Bgl II restriction sites at positions −2342 and −698) were previously described (Ferguson et al., 2005; Chen et al., 2009).
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction.
Total RNA containing small RNA were isolated from human primary hepatocytes and HepG2 cells using the miRNeasy mini kit (Qiagen, Valencia, CA), according to the manufacturer’s instructions, and reverse transcribed to cDNA using the SuperScript III First Strand Synthesis System for RT-PCR kit (Invitrogen, Carlsbad, CA) with Oligo (dT) primers. Quantitative polymerase chain reaction (qPCR) was performed using the ABI Prism 7900 Sequence Detector System (Applied Biosystems, Foster City, CA), with the following primer and probe sets purchased from Applied Biosystems: CYP2C8 (Hs00258314_m1), CYP2C9 (Hs00426397_m1), CYP2C19 (Hs00426380_m1), ACOX1 (Hs01074241_m1), carnithine palmitoyl transferase 1A (Hs00912671_m1), PANK1 (Hs00332073_s1), and GAPDH (Hs03929097_g1). Each cDNA (100 ng) was mixed with 1X Taqman Universal PCR Master Mix (Applied Biosystems). mRNA levels were normalized with GAPDH as the endogenous control. To analyze miRNA expression, TaqMan microRNA assays (Applied Biosystems) were used to quantify levels of mature miRNAs following the manufacturer’s instructions. Briefly, total RNA, including small RNA isolated using the miRNeasy mini kit, was reverse transcribed using the TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems) and the expression of mature miRNA-107 and miRNA-103 was quantitated using the individual TaqMan microRNA assays. RNU44 was used as the endogenous control to normalize miRNA expression. The primers/probes for qPCR were purchased from Applied Biosystems as follows: Hsa-miR107 (000443), Hsa-miR103 (000439), and RNU44 (001094). All qPCR experiments were performed in triplicate with cDNA samples from independent samples, as previously described.
Preparation of Microsomes.
Cultured human primary hepatocytes in six-well plates were obtained from Triangle Research Laboratories (Research Triangle Park, NC). The information of the human donors is shown in Table 2. Hepatocytes were cultured in hepatocyte maintenance media containing supplements (MM250) according to instructions provided by Triangle Research Laboratories. Immunoblotting experiments were performed in microsomes to detect CYP2C proteins before and after treatment with BF or dimethylsulfoxide (DMSO) controls. Microsomes were prepared from cultured human hepatocytes as previously described (Makia et al., 2014). Briefly, cells were suspended in ice-cold buffer (0.1 M potassium phosphate, pH 7.4, containing 0.25 M sucrose and 1 mM EDTA) and homogenized using a Potter-Elvehjem homogenizer. The 10,000g supernatant was subject to ultracentrifugation for 2 hours at 112,000g using a TLA-55 rotor (Beckman Coulter, Palo Alto, CA). The microsomal pellet was resuspended in 50 mM potassium phosphate buffer, pH 7.4, containing 2% glycerol and 1 mM EDTA and stored at −80°C.
Western Blotting.
Total cell and nuclear extracts were prepared from HepG2 cells as previously described (Makia et al., 2012). The extracts were separated on 4–20% SDS-PAGE gels and transferred onto nitrocellulose membranes. Membranes were probed with antibodies against GAPDH (1:5000) as the endogenous control or 1:1000 dilutions of all other primary antibodies. Horseradish peroxidase–conjugated goat anti-rabbit or anti-mouse secondary (1:10,000) antibodies were used, and the proteins were visualized using SuperSignal West Pico or Dura Western blotting detection system (Thermo Scientific, Rockford, IL). Microsomal proteins were separated using a Protean II xi cell (Bio-Rad, Hercules, CA), and Western blots were performed with the following rabbit antibodies: 1590 (raised to recombinant purified CYP2C9 expressed in Escherichia coli, which recognizes CYP2C9 > CYP2C19 >> CYP2C8), 1592 (raised to recombinant purified CYP2C9 expressed in E. coli, which recognizes CYP2C9 >> CYP2C19 and CYP2C8), and 1937 (a specific anti-CYP2C8 peptide antibody that recognizes only CYP2C8) (Zhang et al., 2012). The following CYP2C standards were also used: human liver microsomes (Gentest Corp., Woburn, MA), and recombinant yeast CYP2C9, CYP2C8, and CYP2C19 proteins.
Transcription Factor–Binding Site Analysis.
The Genomatrix MatInspector software was used to analyze the human CYP2C8 (−3077/+1) promoter for putative PPREs. The canonical PPRE is a direct repeat with the distance of one base (DR1) with the sequence AGGTCAAAGGTCA.
Site-Directed Mutagenesis.
A QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used to mutate the DR1-B site located at positions −2109 of the human CYP2C8 promoter according to the manufacturer’s instructions. The primers for mutagenesis of the DR1-B site at position −2109 base pair (bp) to generate the luciferase reporter constructs 2C8-3k/DR1BM and 2C8-2.5k/DR1BM were as follows: forward, 5′- CAGCAAATTACTACTTCGGTTTGCGGTGGATAAAGGGTTCA-3′, and reverse, 5′- TGAACCCTTTATCCACCGCAAACCGAAGTAGTAATTTGCTG-3′. The mutated sites are underlined. The mutation of the DR1-B site was confirmed by sequencing to eliminate spurious mutations.
Transfection of HepG2 Cells and Luciferase Reporter Assays.
The human hepatocellular carcinoma cell line HepG2 (HB8065; American Type Culture Collection, Rockville, MD) was maintained in minimal essential medium supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 1 mM sodium pyruvate, 2 mM glutamine, and penicillin-streptomycin (Life Technologies, Carlsbad, CA). Cells were seeded in 24-well plates and transfected with 200 ng/well of the luciferase reporter construct and 20 ng of Renilla luciferase plasmid (pRL-TK) using Lipofectamine 2000 (Life Technologies, Carlsbad, CA). The cells were cotransfected with pcDNA3.1 (control vector), pcDNA3-hPPARα (100 ng), pSG5-mPPARα (100 ng), and pcDNA-Flag-hPPARγ (100 ng) plasmids. Twenty-four hours after transfection, cells were treated with BF (0.5 mM), WY14643 (50 µM), or rosiglitazone (5 µM) for 24 hours. The cells were resuspended in passive lysis buffer (Promega, Madison, WI), and luciferase activity was assayed with a Dual-Glo luciferase reporter assay system. The data were expressed relative to Renilla luciferase activity to normalize for transfection efficiency. Transfection experiments were performed in triplicate and repeated at least twice for confirmation.
In Vitro Transcription/Translation of PPARα and RXRα proteins.
Human PPARα and RXRα proteins were synthesized in vitro from 1 μg of pcDNA3-hPPARα or pGEM-3T-RXRα plasmids using a TNT Quick Coupled Transcription/Translation System (Promega) in the presence of unlabeled methionine following the manufacturer’s instructions. Additional controls containing the TNT Quick master mix with no template DNA were also performed. The proteins were separated on a 4–20% Tris–glycine polyacrylamide gel and verified by Western blotting using antibodies against PPARα and RXRα.
Electrophoretic Mobility Shift Assays.
Electrophoretic mobility shift assays (EMSAs) with the in vitro transcribed proteins were performed essentially as described (Makia et al., 2014) by incubating 2 μl of the in vitro translated PPARα and RXRα proteins with labeled double-stranded oligonucleotides containing the PPRE control and various DR1s. The sequences of the complementary oligonucleotides were as follows: PPRE control: forward, 5′-CAGGGGACCAGGACAAAGGTCACGTTCGGGA-3′, and reverse, 5′-TCCCGAACGTGACCTTTGTCCTGGTCCCCTG-3′; DR1-A: forward, 5′-ACCCTATGTGAACTTCGAACTTTGGTTGATG-3′, and reverse, 5′- CATCAACCAAAGTTCGAAGTTCACATAGGGT-3′; DR1-B: forward, 5′-AATTACTACTTCCCTTTGCCCTGGATAAAGG-3′, and reverse, 5′- CCTTTATCCAGGGCAAAGGGAAGTAGTAATT-3′; DR1BMut: forward, 5′-AATTACTACTTCGGTTTGCGGTGGATAAAGG-3′, and reverse, 5′- CCTTTATCCACCGCAAACCGAAGTAGTAATT-3′; DR1-C: forward, TAAAACCAAACACGTCTGACCCACATTTTAC-3′, and reverse, 5′- GTAAAATGTGGGTCAGACGTGTTTGGTTTTA-3′; and DR1-D: forward, 5′-TAAAAAGAAAGGTCAAGGCAGGAGCCTCAGC-3′, and reverse, 5′-GCTGAGGCTCCTGCCTTGACCTTTCTTTTTA-3′. The complementary oligonucleotides (1 pmol/μl) were annealed by incubating at 95°C for 5 minutes with annealing buffer (10 mM Tris-HCl, 1 mM EDTA, and 50 mM NaCl, pH 8.0). Double-stranded oligonucleotides were labeled with [γ-32P]ATP using a T4 polynucleotide kinase kit following the supplier’s instructions (Promega). Unincorporated nucleotides were removed by chromatography on microspin G-25 columns (GE Healthcare, Piscataway, NJ). Protein-DNA complexes were formed by incubating 2 μl of in vitro translated proteins and 106 cpm of the 32P-labeled oligonucleotide probe for 30 minutes at room temperature in a total volume of 20 μl with binding buffer [10 mM Tris-HCl, 1 mM EDTA, 1 mM MgCl2, 5% glycerol, 1 mM DTT, and 50 ng/μl poly(dI-dC)]. Binding specificity was assessed by the addition of 100-fold excess of unlabeled double-stranded oligonucleotides. For super shift analyses, antibodies (4 μg) were added to the binding reactions after the initial 20-minute incubation and incubation was continued for 2 hours at 4°C. Loading buffer was added to the reactions, and 10 μl of the binding reactions were resolved by electrophoresis on 5% polyacrylamide gels using 0.5X TBE buffer (50 mM Tris, pH 8.3, 50 mM sodium borate, and 1 mM EDTA) at 200 V for 2 hours. The gel was transferred to Whatman 3MM filter paper, dried, and exposed to film overnight at −80°C.
Chromatin Immunoprecipitation Assay.
Chromatin immunoprecipitation (ChIP) assays were performed as previously described (Makia et al., 2012) using the MAGnify Chromatin-Immunoprecipitation System (Life Technologies) according to the manufacturer’s protocol, with minor modifications. Briefly, human primary hepatocytes in 10-cm dishes treated with either DMSO (control) or BF (0.5 mM) were fixed in 1% formaldehyde at room temperature for 10 minutes to crosslink the nuclear proteins to DNA, and the reaction was stopped by incubation with 125 mM glycine for 10 minutes. Cells were resuspended in lysis buffer (10 mM Tris-HCl, 10 mM NaCl, and 0.5% NP-40) containing 1× complete protease inhibitor (Roche Diagnostics, Indianapolis, IN) and incubated on ice for 30 minutes. The nuclei were harvested and solubilized in buffer containing 50 mM Tris-Cl, pH 8.0, 1% SDS, 10 mM EDTA, and 0.5 mM phenylmethanesulfonyl fluoride with 1× complete protease inhibitor. The homogenate was sonicated on ice at a 40% setting (Branson Sonicator, North Olmsted, OH) to shear the chromosomal DNA into fragments of ∼200–500 bp in size. Immunoprecipitation of the sonicated DNA fragments was performed overnight at 4°C with 8-μg antibodies against IgG (negative control), PPARα, RXRα, or RNA Pol II conjugated to Dynabeads protein A/G. The crosslinked protein-DNA complexes were uncrosslinked in the presence of proteinase K, and the purified DNA was analyzed by polymerase chain reaction (PCR) using PCR SuperMix High Fidelity (Life Technologies), with primers spanning DR1-B (position −2109), the negative control region (position −1500 in the promoter of CYP2C8), the PPRE of human 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR) (positive control for PPARα and RXRα), and α-actin (negative control for all antibodies). The sequences of the primers were as follows: 2C8-DR1B: forward, 5′-ATTGCTCTAAAGAGAGAAAG-3′, and reverse, 5′-AAT TCT AGC ACC AGT TGA GT-3′; 2C8 negative control: forward, 5′-AGGAGTAGGACAAAAGAACA-3′, and reverse, 5′-TAAGACAGCTGTGAGCTTGC-3′; human HMGCR promoter: forward, 5′-ACGCTGATTTGGGTCTATGG-3′, and reverse, 5′-GTGTAAATGGCTCCGGTCAC-3′; and human α-actin coding region: forward, 5′-CTTCTGCCCTCCGCAGCTGA-3′, and reverse, 5′-GTGAATGCCCGCCGACTCCA-3′. PCR products were resolved on a 1.5% agarose gel and visualized by ethidium bromide staining. For qPCR analysis, the purified DNA (10 µl), immunoprecipitated with antibodies against PPARα and IgG, were mixed with 1× Power SYBR Green PCR Master Mix (Applied Biosystems) containing 290 nM of the forward and reverse primers. All PCR results were normalized to input control and presented as fold enrichment over IgG.
Statistical Analysis.
All error bars indicate the mean ± S.E.M. Data were analyzed with Student’s t tests or one-way analysis of variance followed by Tukey’s test using Sigma Stat software (Sigma Stat 3.5; SPSS, San Jose, CA). Differences were considered to be statistically significant at a P level of 0.05.
Results
BF Induces PANK1/miR107 Expression in Primary Human Hepatocytes.
MicroRNA 107 is located in intron 5 of the PANK1 gene (Fig. 1A). Previous studies showed that the PANK1 gene is a target of PPARα in HepG2 cells (Ramaswamy et al., 2004). We examined whether miR107 expression is also induced by activation of PPARα in primary human hepatocytes at 24 and 48 hours after BF (0.5 mM) treatment. BF induced miR107 expression, whereas miR103 expression was not increased (Fig. 1B). Consistent with possible coregulation of the PANK1 gene and miR107 expression, we also observed BF concurrently induced expression of PANK1 (pri-miR107) (Fig. 1C). Treatment of HepG2 cells with BF also resulted in a 1.5-fold increase in miR107 expression. Ectopic expression of PPARα in HepG2 cells and treatment with BF resulted in a 3-fold increase in miR107 expression (Fig. 1D).
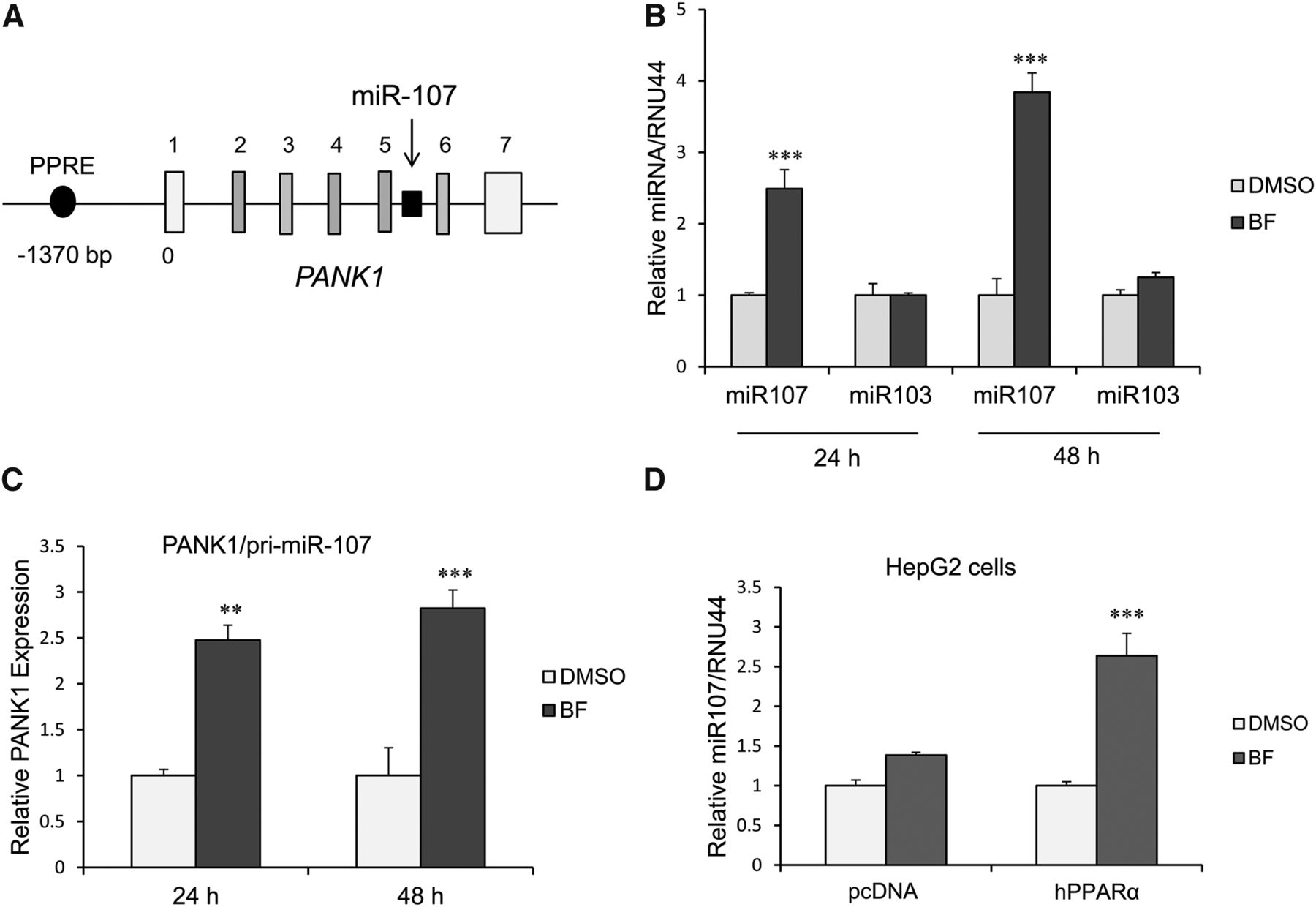
miR107 expression is induced by BF in human primary hepatocytes and HepG2 cells. (A) Human PANK1 gene structure [adapted from Yamakuchi et al. (2010)]. miR107 is intronic to the PANK1 gene and coregulated with the PANK1 gene. The gene encoding miR107 is shown in black. PANK1 gene untranslated and translated exons are shown in white and gray, respectively. (B) miR107, but not miR103 expression, is induced by BF in primary human hepatocytes. Human primary hepatocytes were treated with BF (0.5 mM) for 24 or 48 hours, and miRNA expression was measured by quantitative PCR normalized to RNU44. Data represent the human hepatocytes isolated from at least three different donors, as shown in Table 2. Each donor sample was analyzed in triplicate, and data indicate the mean ± S.E.M. from at least three different donors. ***P < 0.001 compared with DMSO control (Student’s t test). (C) PANK1/pri-miR107 expression is upregulated by exposure of human primary hepatocytes with BF at 24 and 48 hours. Data represent human hepatocytes isolated from four different donors, as shown in Table 2. Each donor sample was analyzed in triplicate, and data indicate the mean ± S.E.M. from at least three different donor hepatocyte preparations. **P < 0.01 compared with DMSO control; ***P < 0.001 compared with DMSO control (Student’s t test). (D) miR107 is induced by BF in HepG2 cells, and the induction is depended on activation of PPARα. ***P < 0.001 compared with vector control (one-way analysis of variance followed by Tukey’s test).
BF Induces CYP2C8 Expression in Primary Human Hepatocytes but Had Minimal Effects on CYP2C9 or CYP2C19 Expression.
BF (a PPARα ligand/agonist) had only a marginal effect on the expression of two other CYP2C genes (CYP2C9 and CYP2C19 mRNA) (Fig. 2, A and B). ACOX1 and carnithine palmitoyl transferase 1A (CPT1A) are positive controls for PPARα activation. We examined the effect of varying concentrations of BF on the expression of PANK1 and CYP2C genes at 48 hours. There was a concentration-dependent increase in CYP2C8 mRNA to a maximum of a 3.5-fold increase at 0.5 mM (Fig. 2C) and a 2.5-fold increase at 0.75 mM. PANK1 mRNA was induced ∼2.5-fold at 0.125 mM BF, which remained constant through a concentration of 0.75 mM BF. CYP2C9 and CYP2C19 mRNA were increased only slightly (1.5-fold) at 0.75 mM BF. Treatment of primary hepatocytes with BF also induced expression of CYP2C8 protein (∼6.5-fold) after 48 hours, but did not appear to induce CYP2C9 or CYP2C19 protein (Fig. 3).

Human CYP2C8 mRNA levels were induced by BF in primary human hepatocytes. (A–C) represent human hepatocytes isolated from at least three different donors, as shown in Table 2. CYP2C8, CYP2C9, and CYP2C19 mRNA expression in cultured human hepatocytes following treatment with 0.5 mM BF for (A) 24 and (B) 48 hours were analyzed by quantitative PCR. CYP2C8 mRNA was significantly increased 24 and 48 hours after BF (P < 0.001) compared with minimal increases. *P < 0.05 in CYP2C9 and CYP2C19 mRNA 48 hours after BF compared with DMSO control. CPT1A and ACOX, known PPARα target genes, were dramatically increased by BF. ***P < 0.001 compared with DMSO control (Student’s t test). (C) Human primary hepatocytes were treated with 0.125, 0.25, 0.5, and 0.75 mM BF for 48 hours. Total RNA was extracted, and gene expression was examined by quantitative PCR. Each donor sample was analyzed in triplicate, and data indicate the mean ± S.E.M. of at least three different donor preparations. *P < 0.05 compared with DMSO control; **P < 0.01 compared with DMSO control; ***P < 0.001 compared with DMSO control (one-way analysis of variance followed by Tukey’s test).

CYP2C8, but not CYP2C9, protein levels were induced by BF in human primary hepatocytes. Human primary hepatocytes isolated from between three to five different donors were treated with either DMSO control or 0.5 mM BF for 48 hours. Microsomes prepared from DMSO control or BF-treated cells were analyzed by Western blot for CYP2C proteins. The figure is a representative blot. HLM, human liver microsomes (Gentest, Woburn, MA). The following recombinant yeast CYP2C protein controls were used: CYP2C9 (2C9), CYP2C8 (2C8), and CYP2C19 (2C19). 1590 is an anti-CYP2C antibody, whereas 1937 is a specific antibody for a CYP2C8 peptide that does not crossreact with other human CYP2C proteins. The specificity of the antibodies is presented in Materials and Methods. (B) Densitometry analysis of Western blot using Image J software. Data indicate the mean ± S.E.M. calculations of a minimum of three independent donor preparations. ***P < 0.001 compared with DMSO control (Student’s t test).
CYP2C8 Promoter Luciferase Activity Is Transactivated by Ectopic Expression of PPARα and Treatment with BF in HepG2 Cells.
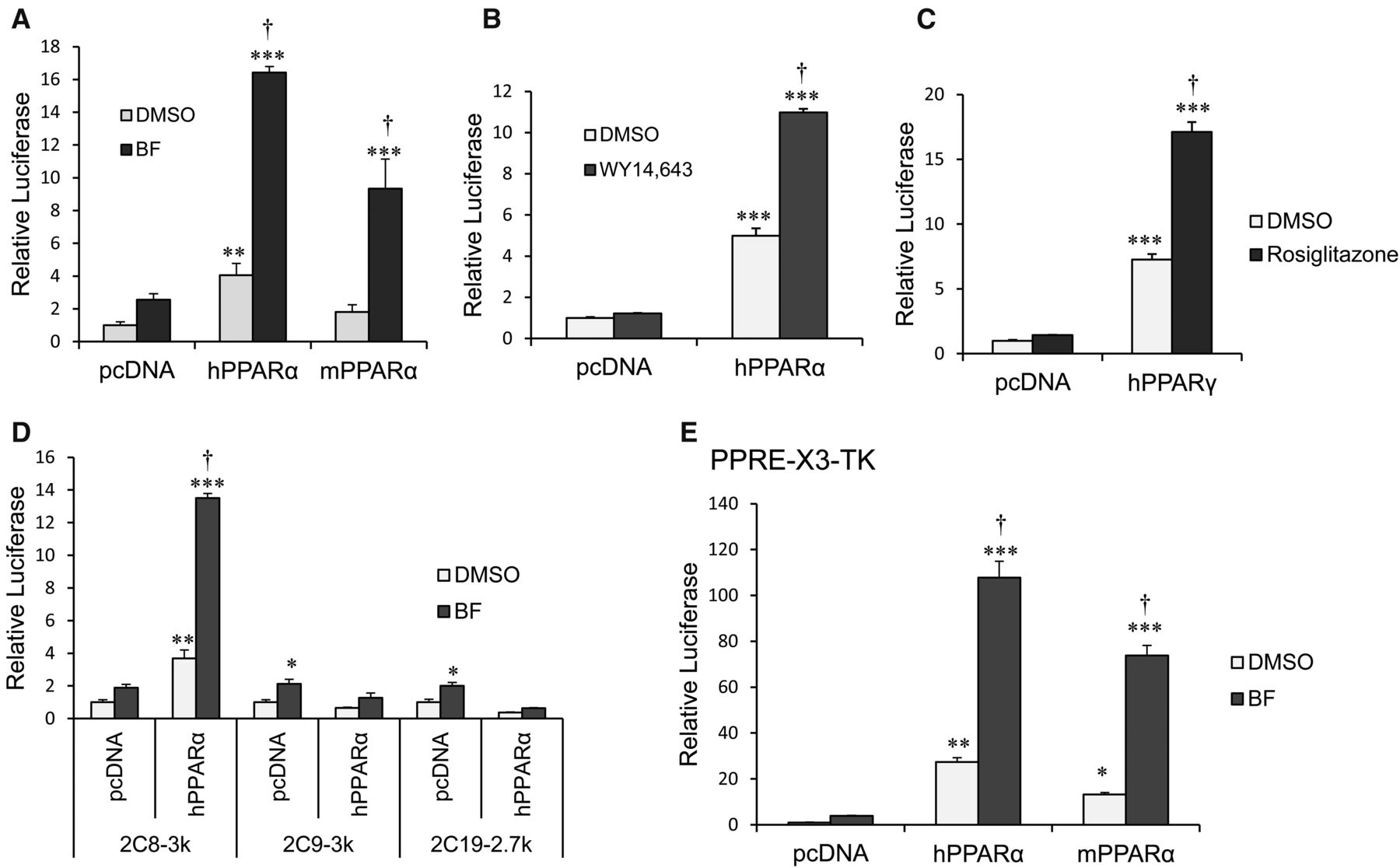
We examined whether CYP2C8 transcription is induced by PPARα activation in HepG2 cells. HepG2 cells were cotransfected with a 2C8-3k luciferase reporter construct (containing −2966 to +1 of the CYP2C8 promoter upstream in a luciferase reporter) and vector control or with human or mouse PPARα expression plasmids. Cells were treated with either DMSO control or BF (0.5 mM) for 24 hours. We observed ∼16- and ∼10-fold increases in CYP2C8 promoter luciferase activity after transfection with human and mouse PPARα, respectively, and treatment with BF (Fig. 4A). CYP2C8 promoter activity was increased ∼12-fold by activation of human PPARα using a more specific PPARα ligand, 50 µM WY14643 (Fig. 4B), and was ∼18-fold after treatment with rosiglitazone (5 μM) and cotransfection with PPARγ (Fig. 4C). In contrast, neither the CYP2C9 (2C9-3k) nor the CYP2C19 (2C19-2.7k) promoter was induced by treatment of ectopically expressed human PPARα with BF (Fig. 4D). As a control for PPARα transcriptional activation, the PPRE-X3-TK luciferase construct that contains three copies of the rat ACOX1-PPRE upstream of the TK promoter was transactivated by activation of human (120-fold) and mouse (80-fold) PPARα in HepG2 cells (Fig. 4E).

CYP2C8 promoter luciferase activity is transactivated by ectopic expression of PPARα and treatment with BF in HepG2 cells. (A) Activation of ectopic expressed PPARα by BF transcriptionally activates CYP2C8 luciferase activity. HepG2 cells were cotransfected with 2C8-3k luciferase reporter construct (spanning −2966 to +1 of the CYP2C8 promoter) together with the control vector (pcDNA3.1), hPPARα, or mouse PPARα (mPPARα) expression plasmids, as indicated. Transfected cells were subsequently exposed to either DMSO (0.1%) or 0.5 mM BF. (B) Transfected cells were treated with either DMSO (0.1%) or 50 µM WY14643. Cells were harvested, and luciferase activity was normalized to Renilla luciferase activity. The results were expressed as fold induction from at least three independent experiments compared with that of vector control. (C) HepG2 cells were cotransfected with 2C8-3k luc together with control vector (pcDNA3.1) or human PPARγ (hPPARγ) expression plasmids. Transfected cells were treated with either DMSO (0.1%) or 5 µM rosiglitazone. (D) HepG2 cells were cotransfected with 2C8-3k, 2C9-3k, or 2C19-2.7k luciferase reporter constructs together with either control vector (pcDNA3.1) or hPPARα expression plasmids. Transfected cells were treated with either DMSO (0.1%) or 0.5 mM BF for 24 hours. (E) HepG2 cells were transfected with PPRE-X3-TK luciferase constructs (contains three copies of ACOX1-PPRE upstream of the TK gene promoter) and either control vector (pcDNA3.1), hPPARα, or mouse PPARα expression plasmids. Transfected cells were treated with either DMSO (0.1%) or 0.5 mM BF for 24 hours. The PPRE-X3-TK is a positive control for PPARα transcription regulation. Luciferase activity was normalized to Renilla luciferase, and the results were expressed as fold induction from at least three independent experiments compared with that of control transfected cells. *P < 0.05 compared with pcDNA3/DMSO-treated cells; **P < 0.01 compared with pcDNA3/DMSO-treated cells; ***P < 0.001 compared with pcDNA3/DMSO-treated cells (one-way analysis of variance followed by Tukey’s test); †P < 0.05 compared with PPARα/DMSO-treated cells (one-way analysis of variance followed by Tukey’s test).
Functional Characterization of DR-1 in the CYP2C8 Promoter in HepG2 Cells.
Promoter sequence analysis of the CYP2C8 gene promoter revealed five putative PPRE sites located at positions −2772 (DR1-A), −2109 (DR1-B), −2039 (DR1-C), −1501 (DR1-D), and −152 (DR1-E) relative to the translation start site. The sequences of the DR1s and their 5′-flanking regions and the strand orientation are shown in Table 1. The consensus sequence of PPRE (AGGTCAAAGGTCA) is also shown. Figure 5A is a schematic diagram of the CYP2C8 promoter showing the position of the various DR1s. HepG2 cells were cotransfected with various deletion luciferase constructs and the human PPARα expression plasmid. Cells were treated with either DMSO control or BF for 24 hours. We observed about a 20-fold increase in CYP2C8 luciferase activity with the 2C8-3k construct. Similar levels of CYP2C8 luciferase activity were observed with the 2C8-3k/−8.9–8.5 (containing the upstream CAR/PXR site) and 2C8-2.5k constructs. No significant effect on CYP2C8 luciferase activity was observed with 2C8-2k, 2C8-1.5k, 2C8-500, and 2C8-300. This indicates that DR1-B rather than DR1-A might be essential for transactivation of the CYP2C8 promoter activity by PPARα. We then used the 2C8-3k/ΔBgl II constructs generated by digestion and ligation of Bgl II restriction sites at positions −2342 and −698. This construct contains DR1-A, but not the DR1-B site. When HepG2 cells were transfected with this construct, no significant difference in CYP2C8 luciferase activity was observed, suggesting again that the DR1-A site might not be important for the increased CYP2C8 promoter activation by PPARα. Site-directed mutagenesis was used to investigate the relative importance of DR1-B in the transactivation of the CYP2C8 promoter by PPARα activation with BF. Mutation of DR1-B in the 2C8-3k or 2C8-2.5k constructs completely abolished CYP2C8 reporter activation by BF (Fig. 5B). These results confirm that DR1-B is required for maximal transactivation of the CYP2C8 luciferase promoter construct by PPARα.
DR1 identified in the CYP2C8 promoter
Clinical histories of human donors

PPARα-regulated transcription of CYP2C8 is mediated by a PPRE (DR1-B) located at −2109 bp upstream of the translation start site. (A) Computer analysis of the CYP2C8 promoter showed five putative PPRE sites located at −2772 (DR1-A), −2109 (DR1-B), −2039 (DR1-C), −1501 (DR1-D), and −152 (DR1-E) bp relative to the translation start site. (B) HepG2 cells were transiently transfected with various luciferase constructs containing the indicated sequences of the CYP2C8 promoter cotransfected with hPPARα expression plasmids. Twenty-four hours after transfection, cells were treated with either DMSO (0.1%) or BF (0.5 mM) for 24 hours. Cells were harvested, and luciferase and Renilla luciferase activity were measured. The results were expressed as a fold induction compared with DMSO-treated cells. ***P < 0.001 compared with DMSO-treated cells (one-way analysis of variance followed by Tukey’s test).
EMSA Analysis of CYP2C8 DR1 and PPRE Control Using In Vitro Translated PPARα or RXRα Proteins.
Human PPARα and RXRα synthesized using a TNT Quick Coupled Transcription/Translation System in the presence of unlabeled methionine were verified by Western blot using antibodies to human PPARα and RXRα (Supplemental Fig. 1). EMSA was performed to determine whether PPARα and RXRα bind to the various DR1s of CYP2C8 and PPRE control (rat ACOX1). The sequences of the oligonucleotide used for the EMSA are shown in Fig. 6A. 32P-labeled double-stranded oligonucleotides containing CYP2C8 DR1-A, DR1-B, DR1-C, DR1-D, DR1-BM (mutation of GG to CC in DR1-B), and a positive control for PPARα binding (PPRE from the rat ACOX1 gene) were incubated for 30 minutes with in vitro transcribed/translated RXRα or both hPPARα and RXRα proteins at 4°C. We observed the formation of a DNA-protein complex with the labeled PPRE consensus and DR1-B oligonucleotides when incubated with both PPARα and RXRα. No complex was formed with incubation of DR1-A, DR1-C, or DR1-D oligonucleotides with PPARα and RXRα (Fig. 6A). The DNA-protein complex with DR1-B and PPRE were effectively inhibited by the addition of excess unlabeled oligonucleotides (cold), but not excess unlabeled mutant oligonucleotides, indicating specificity of binding to DR1-B (Fig. 6B). Supershift experiments showed that the protein complexes formed with the CYP2C8 DR1-B and PPRE control oligonucleotides were shifted by antibodies against PPARα and RXRα, but not with IgG. These results indicate specific binding of PPARα and RXRα to the DR1-B site at −2109 of the CYP2C8 promoter.

Specific binding of PPARα to DR1-B at −2109 bp of the CYP2C8 promoter by gel shift assay. (A) Sequences of the oligonucleotides used for the binding assays. Putative and known PPREs are written in red, and the DR1 half-sites are underlined. GG in DR1-B half-sites were mutated to CC to generate DR1BM. (A) 32P-labeled double-stranded oligonucleotides containing CYP2C8 DR1-A, DR1-B, DR1-C, DR1-D, and a positive control for PPARα binding (PPRE from the rat ACOX1 gene) were incubated for 30 minutes with in vitro transcribed/translated RXRα or hPPARα and RXRα proteins at 4°C. (B) 32P-labeled double-stranded oligonucleotides containing CYP2C8 DR1-B and the known PPRE (rat ACOX1) were incubated for 30 minutes with in vitro translated hPPARα and RXRα at 4°C. In competition experiments, 100-fold excess of unlabeled double-stranded oligonucleotides inhibited formation of the complex. Supershift experiments were performed by incubating the binding reactions with 4 μg of antibodies to PPARα, RXRα, or IgG for 2 hours at 4°C.
ChIP Analysis of Transcription Factor Binding to DR1-B in Primary Human Hepatocytes before and after Treatment with BF.
ChIP experiments were used to assess in vivo recruitment of PPARα and RXRα to DR1-B before and after treatment of primary human hepatocytes with BF. Chromatin was prepared from cultured human hepatocytes treated with DMSO control or BF for 48 hours. The chromatin was sheared by sonication into fragments approximately 200–500 bp in size and immunoprecipitated with IgG, PPARα, RXRα, or RNA Pol II (positive control for transcriptionally active genes) antibodies conjugated to Dynabeads. Conventional PCR was performed with primers spanning the CYP2C8 DR1-B site, CYP2C8 promoter region that does not contain DR1 (negative control), PPRE of the human HMGCR promoter (positive control for PPARα and RXRα binding), and human α-actin coding region (a negative control for all antibodies). The positions of the primers used for ChIP analyses are shown in Fig. 7A. As seen in Fig. 7B, we observed enhanced recruitment of PPARα, RXRα, and Pol II to DR1-B and HMGCR after treatment with BF compared with DMSO control treatment. These factors were not recruited to the CYP2C8 negative control site or α-actin promoter. SYBR green quantitative PCR was performed using primers flanking the CYP2C8 DR1-B, a CYP2C8 promoter region that does not contain DR1 (negative control), the PPRE of human HMGCR, and α-actin, with chromatin extracted from control or BF-treated primary human hepatocytes immunoprecipitated with antibodies specific to PPARα or IgG. Again, we observed enhanced enrichment of PPARα to the DR1-B and HMGCR after treatment with BF compared with DMSO control treatment, but not the 2C8 negative control site or α-actin. Consistent with the EMSA, these results indicate the in vivo binding of PPARα to the CYP2C8 DR1-B site.

ChIP analysis showed in vivo binding of PPARα and RXRα to the CYP2C8 DR1-B sites. (A) CYP2C8 promoter with position of primers used for ChIP PCR analysis. (B) Chromatin was prepared from cultured primary human hepatocytes treated with DMSO control or BF (0.5 mM) for 48 hours, sheared, immunoprecipitated with IgG (negative control), PPARα, RXRα, or RNA Pol II (positive control for transcriptional active genes) antibodies conjugated to Dynabeads protein A/G. PCR was performed with primers spanning the CYP2C8 DR1-B, CYP2C8 promoter region that does not contain DR1 (negative control), PPRE of human HMGCR promoter (positive control for PPARα and RXRα binding), and human α-actin coding region (a negative control for all antibodies). (C) Chromatin extracted from control and BF-treated primary human hepatocytes was immunoprecipitated with antibodies specific to PPARα and IgG. SYBR green qPCR was performed using primers flanking the CYP2C8 DR1-B, CYP2C8 promoter region that does not contain DR1 (negative control), PPRE of human HMGCR, and α-actin. Data represent the human hepatocytes isolated from at least three different donors, as shown in Table 2. Each treatment was analyzed in triplicate, and data indicate the mean ± S.E.M. from at least three different donor preparations. ***P < 0.001 compared with DMSO control (Student’s t test).
Discussion
In the present study, we show for the first time that CYP2C8, but not CYP2C9 or CYP2C19, is transcriptionally upregulated by PPARα activation in primary human hepatocytes. We demonstrate that this induction is independent of activation of CAR/PXR or changes in miR107 by hypolipidemic drugs via PPARα activation. Until recently, the microsomal fatty acid ω-hydroxylase CYP members, the CYP4 family, were the only known PPARα-regulated CYP genes (Aldridge et al., 1995). A recent study investigated the possible involvement of PPARα in the regulation of drug biotransformation genes using gene silencing experiments in primary human hepatocytes (Thomas et al., 2013). Their data showed that CYP1A1, CYP1A2, CYP2B6, CYP2C8, CYP3A4, and CYP7A1, but not CYP2C9, CYP2C19, CYP2D6, or CYP2E1, were selectively regulated by PPARα agonists, such as WY14643 or fibrates, in primary human hepatocytes (Thomas et al., 2013). These data are consistent with other previous studies using mouse and human hepatocytes (Prueksaritanont et al., 2005; Rakhshandehroo et al., 2009). These studies imply an involvement of PPARα in modulating the constitutive and xenobiotic-induced expression of the CYP2C8 gene. However, except for CYP3A4 and CYP1A1, no other studies have formally addressed direct regulation of other CYP genes by PPARα (Seree et al., 2004; Villard et al., 2011; Thomas et al., 2013). It is now known that CYP1A1 is directly regulated by PPARα via two PPREs within its proximal promoter (Seree et al., 2004; Villard et al., 2011). Our data are consistent with these previous studies showing induction of CYP2C8, but not CYP2C9 or CYP2C19, by PPARα ligand/agonists in primary human hepatocytes. Moreover, for the first time, we identified a PPRE (DR1-B) at position −2109 bp in the promoter of CYP2C8 and showed that in vitro translated PPARα binds to this site in the presence of RXRα. We also observed enhanced recruitment of PPARα and RXRα to the DR1-B site, but not a negative control region in the CYP2C8 promoter by bezafibrate. Apart from CYPs, PPARα has also been shown to directly regulate other drug biotransformation enzymes, including UDP-glucuronosyltransferases (Barbier et al., 2003a,b), sulfotransferase (Fang et al., 2005), and transporters (Moffit et al., 2006; Hoque et al., 2012; Ghonem et al., 2014).
The CYP2C8 gene is regulated post-transcriptionally by miRNA107 and miRNA103 in primary human hepatocytes (Zhang et al., 2012). miR107 had little or no effect on CYP2C8 mRNA levels, but decreases CYP2C8, CYP2C9, and CYP2C19 proteins in primary human hepatocytes. miR107 is transcribed from the intron of the PANK1 gene, which catalyzes the rate-limiting step of CoA biosynthesis and is involved in the regulation of acetyl-CoA levels, cell stress, insulin sensitivity, and lipid metabolism (Wilfred et al., 2007; Trajkovski et al., 2011). Previous studies demonstrate that PANK1 and miR107 are coregulated by p53 in different cellular systems through a p53-binding site on the PANK1 promoter (Yamakuchi et al., 2010; Bohlig et al., 2011). PANK1 transcription is regulated by PPARα in HepG2 cells, leading to increased CoA levels in cells (Ramaswamy et al., 2004). The induction of the PANK1 gene by PPARα was mediated by the PPREs located within its proximal promoter (Ramaswamy et al., 2004). We showed that miR107 was induced by PPARα activation using BF in primary human hepatocytes, whereas miR103 expression was not upregulated. Consistent with the hypothesis of coregulation between the PANK1 gene and miR107 expression by PPARα, miR107 and PANK1 gene expression were induced by activated PPARα in primary human hepatocytes and HepG2 cells. But contrary to the expectation that CYP2C genes might be post-transcriptionally decreased by miR107 upregulation after BF treatment, CYP2C8 protein expression was significantly increased in primary human hepatocytes by BF treatment. However, neither CYP2C9 nor CYP2C19 proteins were induced by activation of PPARα. Therefore, we hypothesize that the regulation of CYP2C8 by PPARα is transcriptional rather than indirect by enhancing miR107 expression in human liver. If increases in miR107 affect CYP2C8 transcription, the effects are not substantial enough to override the direct induction by PPARα. CYP2C8 luciferase activity was induced by activated ectopically expressed human and mouse PPARα in HepG2 cells. Interestingly, CYP2C8 promoter activity was also induced by the CYP2C8 substrate rosiglitazone, and the induction was dependent on human PPARγ activation. Previous studies showed that fibrates and other hypolipidemic drugs regulate gene expression by activation of the nuclear receptor CAR and PXR (Prueksaritanont et al., 2005; Aouabdi et al., 2006). However, the absence of transactivation of CYP2C9 and CYP2C19 promoter luciferase activity by BF, which are known CAR/PXR-regulated genes, indicates that the regulation of the CYP2C8 gene is independent of CAR/PXR. Furthermore, there was no significant difference in the transactivation of CYP2C8 luciferase by activated PPARα between the CYP2C8 luciferase construct that contains (2C8-3k/−8.9–8.5) or lacks (2C8-3k) a distal CAR/PXR-binding site located at position −8.8 kb. This CAR/PXR-binding site was shown to be essential for induction of the CYP2C8 reporter by rifampicin (PXR agonist) and CITCO (CAR ligand) in primary human hepatocytes (Ferguson et al., 2005).
A unique feature of the CYP2C8 promoter compared with other CYP2C subfamily members is that no CAR/PXR-responsive elements were identified within the proximal 3 kb of the CYP2C8 promoter but rather one element at −8.8 kb (Ferguson et al., 2005). Thus, the observed induction of CYP2C8 luciferase reporter activity by BF is likely not due to activation of CAR/PXR. Promoter sequence analysis of the CYP2C8 gene promoter revealed five putative PPRE sites within the proximal 3-kb region relative to the translation start site located at positions −2772 (DR1-A), −2109 (DR1-B), −2039 (DR1-C), −1501 (DR1-D), and −152 (DR1-E). PPARα regulates gene expression by binding to PPRE on the target gene promoters. The consensus sequence of the PPRE is AGGTCAAAGGTCA, which consists of a direct repeat of AGGTCA half-sites with one base-pair spacing (DR1) (Kliewer et al., 1992; Wahli and Michalik, 2012). However, imperfect half-sites of the canonical DR1 have been identified in PPARα-regulated genes, such as CYP4A1 (AGGGTAAAGTTCA), very low density lipoprotein receptor (AGGTCAGATGGCA), cyclic AMP response element binding (AGGTCAAAGGACA), the mouse hypoxia-inducible lipid droplet associated (AGGGGAAAGGTCA), and the rat ACOX (AGGACAAAGGTCA) (Juge-Aubry et al., 1997; Roy et al., 2013; Gao et al., 2014; Mattijssen et al., 2014). It is widely accepted that the sequence of the 5′-flanking region (6- or 7-bp segment) in addition to the consensus DR1 sequence is important for binding of PPAR to various PPREs (Palmer et al., 1995; Juge-Aubry et al., 1997; Chandra et al., 2013). This is due to the fact that the hinge region of PPARα and PPARγ recognizes additional 6-bp segments located upstream to the DR1 core element. The half-site of the CYP2C8 DR1-C site at position −2039 was previously shown to bind retinoid-related orphan receptorα (RORα) and RORγ and is important for transcriptional regulation of CYP2C8 by ROR in the human liver and other tissues, such as the colon and intestine (Chen et al., 2009). Both the HNF4α homodimer and the PPAR/RXRα heterodimer are known to recognize the same DR1 sequence (Pineda Torra et al., 2002; Chandra et al., 2013). An in vitro binding assay showed that DR1-E is an HNF4α-binding site located within the CYP2C8 basal promoter (Ferguson et al., 2005). This site was shown to be necessary for constitutive activation of the CYP2C8 reporter by cotransfected HNF4α in Hela cells and primary human hepatocytes (Rana et al., 2010). We observed about a 20-fold increase in CYP2C8 luciferase activity with the 2C8-3k and 2C8-2.5k constructs. The CYP2C8 promoter contains several potential PPREs, but site-directed mutagenesis and deletion analyses demonstrate that DR1-B at −2109 bp with the sequence AGGGCAAAGGGAA was solely required for transactivation of the CYP2C8 luciferase by PPARα activation. The DR1-B sequence is absent in the promoter of CYP2C9 and CYP2C19, which might explain the specific regulation of CYP2C8 by PPARα. Since both PPARα and PPARγ are known to bind identical PPREs, we anticipate that the DR1-B site is also essential for PPARγ-mediated regulation of CYP2C8 by rosiglitazone.
In conclusion, we showed for the first time that CYP2C8 is directly transcriptionally regulated by activated PPARα in human primary hepatocytes. PPARα-dependent regulation of CYP2C8 is mediated by the DR1-B site at position −2109 bp in the CYP2C8 promoter. This regulation is specific for CYP2C8, but not CYP2C9 or CYP2C19. Because CYP2C8 is involved in the metabolism of fatty acids, it is reasonable to suggest that, like CYP4A, induction of endogenous CYP2C8 by PPARα may serve a role in the oxidative metabolism of arachidonic acid to EETs. EETs have been shown to have potent vasodilatory and anti-inflammatory functions (Wray and Bishop-Bailey, 2008). Because CYP2C8 has also been detected in endothelial cells and arteries, oxidation of arachidonic acid to produce 11,12- and 14,15-EETs may play vasodilatory and anti-inflammatory roles (Delozier et al., 2007; Wray and Bishop-Bailey, 2008). This study also indicates the possibility of a previously underrepresented drug-drug interaction due to upregulation of CYP2C8 by hypolipidemic (fibrates and WY14643) and antidiabetic (rosiglitazone) drugs. The discovery that CYP2C8 is transactivated by PPARα and PPARγ in hepatocytes suggests the possibility that the clearance of CYP2C8 substrates might be modulated by ligands of PPARα/PPARγ.
Acknowledgments
We would like to thank Ms. Joyce Blaisdell and Dr. Sailesh Surapureddi, National Institute of Environmental Health Sciences, for assistance with this work. We also wish to thank Dr. Stephen Ferguson of the National Toxicology Program and Dr. Masahiko Negishi of the Laboratory of Reproductive and Developmental Toxicology, National Institute of Environmental Health Sciences, for reviewing the manuscript.
Authorship Contributions
Participated in research design: Makia, Goldstein.
Conducted experiments: Makia.
Performed data analysis: Makia, Goldstein.
Wrote or contributed to the writing of the manuscript: Makia, Goldstein.
Footnotes
- Received June 5, 2015.
- Accepted October 13, 2015.
This work was supported by the Intramural Research Program of the National Institutes of Health and the National Institute of Environmental Health Sciences [Grant Z01 ES021024-32].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ACOX
- acyl-CoA oxidase 1
- BF
- bezafibrate
- bp
- base pair
- CAR
- constitutive androstane receptor
- ChIP
- chromatin immunopreciptation assay
- CYP
- cytochrome P450
- DMSO
- dimethylsulfoxide
- DR1
- direct repeat with the distance of one base
- EET
- epoxyeicosatrienoic acid
- EMSA
- electrophoretic mobility shift assay
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- HMGCR
- 3-hydroxy-3-methyl-glutaryl-CoA reductase
- HNF4α
- hepatocyte nuclear factor 4α
- hPPARα
- human peroxisome proliferator-activated receptor α
- kb
- kilobase
- miR103
- microRNA 103
- miR107
- microRNA 107
- miRNA
- microRNA
- PANK
- pantothenate kinase
- PCR
- polymerase chain reaction
- PPAR
- peroxisome proliferator-activated receptor
- PPRE
- peroxisome proliferator-activated receptor response element
- PXR
- pregnane X receptor
- qPCR
- quantitative polymerase chain reaction
- RXR
- retinoid X receptor
- WY14643
- 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio acetic acid
- U.S. Government work not protected by U.S. copyright





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}