


Abstract
The 190-kDa phosphoglycoprotein multidrug resistance protein 1 (MRP1) (ABCC1) confers resistance to a broad spectrum of anticancer drugs and also actively transports certain xenobiotics with reduced glutathione (GSH) (cotransport) as well as conjugated organic anions such as leukotriene C4 (LTC4). In the present study, we have investigated a series of bioflavonoids for their ability to influence different aspects of MRP1 function. Most flavonoids inhibited MRP1-mediated LTC4 transport in membrane vesicles and inhibition by several flavonoids was enhanced by GSH. Five of the flavonoids were competitive inhibitors of LTC4 transport (K i, 2.4–21 μM) in the following rank order of potency: kaempferol > apigenin (+ GSH) > quercetin > myricetin > naringenin (+ GSH). These flavonoids were less effective inhibitors of 17β-estradiol 17β-(d-glucuronide) transport. Moreover, their rank order of inhibitory potency for this substrate differed from that for LTC4 transport inhibition but correlated with their relative lipophilicity. Several flavonoids, especially naringenin and apigenin, markedly stimulated GSH transport by MRP1, suggesting they may be cotransported with this tripeptide. Quercetin inhibited the ATPase activity of purified reconstituted MRP1 but stimulated vanadate-induced trapping of 8-azido-α-[32P]ADP by MRP1. In contrast, kaempferol and naringenin stimulated both MRP1 ATPase activity and trapping of ADP. In intact MRP1-overexpressing cells, quercetin reduced vincristine resistance from 8.9- to 2.2-fold, whereas kaempferol and naringenin had no effect. We conclude that dietary flavonoids may modulate the organic anion and GSH transport, ATPase, and/or drug resistance-conferring properties of MRP1. However, the activity profile of the flavonoids tested differed from one another, suggesting that at least some of these compounds may interact with different sites on the MRP1 molecule.
Resistance to multiple anticancer drugs is a major obstacle to the successful treatment outcome of many human malignancies. In tumor cell lines, multidrug resistance is often associated with an ATP-dependent decrease in cellular drug accumulation attributed to the overexpression of one or other of the plasma membrane drug transporters: the 170-kDa P-glycoprotein (Juliano and Ling, 1976; Sharom, 1997) or the 190-kDa multidrug resistance protein 1 (MRP1) (Cole et al., 1992; Hipfner et al., 1999a). Increased expression of MRP1 and P-glycoprotein has also been reported in a variety of hematological and solid tumors, suggesting an important clinical role for these transport proteins.
MRP1 and P-glycoprotein belong to the ATP-binding cassette (ABC) superfamily of transport proteins but share very limited amino acid identity (Cole et al., 1992). Moreover, P-glycoprotein (ABCB1) has a four domain structure typical of most ABC transporters with two nucleotide binding domains (NBDs) each preceded by a hydrophobic membrane spanning domain. In contrast, MRP1 (ABCC1) has a five domain structure with a third NH2-proximal membrane spanning domain with five transmembrane segments and an extracytosolic NH2 terminus (Cole et al., 1992; Bakos et al., 1996; Hipfner et al., 1997, 1999a). Nevertheless, despite these structural differences, both proteins confer resistance to a similar spectrum of natural product type drugs as well as the folic acid antimetabolite methotrexate (Cole et al., 1994; Hipfner et al., 1999a;Hooijberg et al., 1999a). On the other hand, only MRP1 is an active ATP-dependent transporter of various conjugated organic anions and its substrates range from potential physiological substrates such as the cysteinyl leukotriene LTC4, and 17β-estradiol 17-(β-d-glucuronide) (E217βG), to substrates of toxicological relevance such as the exo andendo GSH conjugates of the mycotoxin aflatoxin B1 (Hipfner et al., 1999a). The mechanisms by which MRP1 and P-glycoprotein transport unconjugated xenobiotics and thereby decrease cellular drug accumulation in human tumor cells are very different. P-glycoprotein has been shown to directly bind and transport drugs to which it confers resistance, whereas MRP1 does not (Loe et al., 1996; Sharom, 1997). Instead, MRP1 seems to efflux some xenobiotics (e.g., vincristine, daunorubicin) through a cotransport mechanism with reduced glutathione (GSH) (Loe et al., 1998; Rappa et al., 1999; Renes et al., 1999). In addition to enhancing direct transport of some MRP1 substrates, GSH has been shown to increase the potency of certain compounds to inhibit conjugated organic anion transport activity by MRP1. For example, transport of LTC4 is poorly inhibited by vincristine or verapamil alone but in the presence of GSH, inhibition by these drugs is enhanced more than 20-fold (Loe et al., 1996, 1998, 2000).
The flavonoids are a large group of polyphenolic antioxidants found in vegetables, fruits, and beverages such as tea and wine. In addition to their well known antioxidant effects, flavonoids have a broad range of biological activities that includes inhibition of tyrosine kinases and serine/threonine kinases (Akiyama et al., 1987), topoisomerase II (Yamashita et al., 1990), and the drug-metabolizing cytochromes P450 1A1/2 (Edenharder et al., 1997), as well as activation of p53 (Plaumann et al., 1996) and the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel (Illek et al., 1999). Furthermore, flavopiridol, a synthetic flavone currently in phase I clinical trials as an antineoplastic agent, has been shown to be a potent inhibitor of several cyclin-dependent kinases, including CDK2 and CDK4 (Carlson et al., 1996). Natural flavonoids are abundant in a normal diet and are also available commercially, marketed as antioxidant tablets. Flavonoids are generally thought to have many beneficial health effects, including antiproliferative and anticarcinogenic effects (Le Marchand et al., 2000) but recently, it has been suggested that maternal ingestion of these compounds may contribute to infant leukemia through a mechanism proposed to involve topoisomerase II (Strick et al., 2000).
Mega-dose quantities of certain flavonoids are frequently ingested by healthy individuals as antioxidant supplements and by cancer patients as a form of alternative or complementary therapy. Consequently, it is important to know whether these compounds affect the function of proteins involved in drug metabolism and/or transport because this may have a significant impact on the physiological functions carried out by these proteins and possibly, the pharmacokinetics and thus efficacy of chemotherapy. In the present study, we have investigated several bioflavonoids for their ability to modulate the transport, ATPase, and drug resistance-conferring activities of MRP1. We found that the effects of flavonoids on these activities were variable, and that the ability of a flavonoid to modulate one activity of MRP1 was not necessarily linked to its ability to modulate another activity of the protein. Our results also indicate that the flavonoids should be considered individually rather than as a class of compounds with respect to their interactions with MRP1 and probably with other related ABC proteins.
Experimental Procedures
Materials and Cell Lines.
[14,15,19,20-3H]LTC4(115.3 Ci/mmol), [6,7-3H]E217βG (55 Ci/mmol), and [glycine 2-3H]GSH (50 Ci/mmol) were purchased from PerkinElmer Life Science Products (Guelph, ON, Canada), and 8-azido-α-[32P]ATP (20 Ci/mmol) was from ICN Biomedicals (Irvine, CA). LTC4 was obtained from Calbiochem (San Diego, CA), and DTT and 2-mercaptoethanol from ICN (Aurora, OH). ATP, AMP, E217βG, GSH, GSSG, sodium orthovanadate, ouabain, vincristine, genistin, genistein, naringin, naringenin, quercetin, kaempferol, apigenin, and myricetin were from Sigma Chemical (Oakville, ON, Canada). Creatine kinase and creatine phosphate were from Roche Molecular Biochemicals (Laval, PQ, Canada). The HeLa cell line T5 is stably transfected with a pRc/CMV vector containing the complete coding sequence of MRP1 and has been described previously (Cole et al., 1994; Grant et al., 1994). The C1 HeLa cell line is stably transfected with the pRc/CMV vector alone. HeLa cells were maintained in RPMI 1640 medium supplemented with 4 mM l-glutamine and 5% defined bovine calf serum and 400 μg/ml geneticin (G418) (Life Technologies, Burlington, ON, Canada).
High-Performance Liquid Chromatography (HPLC) Analysis of Flavonoids.
To determine the relative hydrophilicity of the flavonoids, HPLC was carried out using a Waters 717 HPLC system equipped with a multisolvent delivery system and dual wavelength absorbance detector based on a method described by Oliveira and Watson (2000). Flavonoids were prepared at a concentration of 500 μM in DMSO and 10 μl was injected onto a 3.9 × 150-mm Nova Pak C18 column (Waters, Mississauga, ON, Canada). The isocratic mobile phase consisted of 0.1% trifluoroacetic acid and methanol (6:4, v/v) and was used with a flow rate of 1 ml/min. Absorbance was measured at 254 nm and peaks were analyzed using Waters Millenium 32 software.
Membrane Vesicle Preparation.
Plasma membrane vesicles were prepared as described with modifications (Loe et al., 1996). Briefly, transfected HeLa cells were homogenized in buffer containing 250 mM sucrose/50 mM Tris pH 7.5/0.25 mM CaCl2 and protease inhibitor cocktail tablets (Complete, mini EDTA free) (Roche Molecular Biochemicals, Indianapolis, IN). Cells were disrupted by N2 cavitation (5-min equilibration at 200 psi), and then released to atmospheric pressure and EDTA added to 1 mM. The suspension was centrifuged at 800g at 4°C for 15 min and the supernatant was layered onto a 10-ml 35% (w/w) sucrose/1 mM EDTA/50 mM Tris, pH 7.4 cushion after centrifugation at 100,000g at 4°C for 1 h, the interface was removed and placed in a 25 mM sucrose/50 mM Tris, pH 7.4 solution and centrifuged at 100,000g at 4°C for 30 min. The membranes were washed with buffer (250 mM sucrose, 50 mM Tris pH 7.4) and then resuspended by vigorous syringing with a 27-gauge needle. Protein concentration was determined using a Bradford assay (Bio-Rad, Mississauga, ON, Canada) and aliquots of membranes were stored at −70°C. Relative levels of MRP1 protein in membrane vesicles were determined by immunoblot analysis as before with the human MRP1-specific monoclonal antibody QCRL-1 (Hipfner et al., 1994).
[3H]LTC4 Transport Studies.
LTC4 transport inhibition assays were carried out by a rapid filtration method as described previously (Loe et al., 1996). Assays were carried out at 23°C in a 50-μl volume containing vesicle protein (2 μg), ATP, or AMP (4 mM), MgCl2 (10 mM), GSH (0, 1 or 3 mM), DTT (10 mM), creatine phosphate (10 mM), creatine kinase (100 μg/ml), [3H]LTC4 (50 nM; 40 nCi), and flavonoid dissolved in DMSO (0.7%). After incubations for 60 s, the entire reaction mixture was removed and added to 800 μl of ice-cold Tris sucrose buffer. The solution was then filtered through glass fiber filters (type A/E), washed twice with Tris sucrose buffer, and radioactivity quantitated by liquid scintillation counting. Transport in the presence of AMP was subtracted from transport in the presence of ATP and reported as ATP-dependent [3H]LTC4 uptake. Results are expressed as percentage of control (ATP-dependent [3H]LTC4 transport with vehicle alone) and all compounds were tested in at least three independent experiments.
The mode of LTC4 transport inhibition by selected flavonoids was examined by determining theirK i values. At least two concentrations of flavonoid were used and transport was carried out as described above except LTC4 was added at eight different concentrations (10–500 nM). Reactions were incubated over a time period during which uptake was linear (30 or 45 s depending on the activity of the individual T5 membrane vesicle preparation).
[3H]E217βG Transport Studies.
T5 membrane vesicles (8–10 μg of protein) were incubated at 37°C for 90 s in a total reaction volume of 50 μl containing [3H]E217βG (400 nM, 40 nCi), DTT (10 mM), GSH (±3 mM), and components as described above for LTC4 transport. Flavonoids were added at a final DMSO concentration of ≤1%.
[3H]GSH Transport Studies.
ATP-dependent transport of [3H]GSH into membrane vesicles was measured as described above in a total reaction volume of 60 μl containing 100 μM [3H]GSH (120 nCi/reaction) and DTT (10 mM). To minimize GSH catabolism by γ-glutamyl transpeptidase during transport, membranes (20 μg/reaction) were preincubated with 500 μM acivicin at 37°C for 10 min (Loe et al., 1998). Uptake assays were carried out for 20 min at 37°C in the presence of verapamil (VRP) (100 μM), previously demonstrated to stimulate MRP1-specific GSH transport (Loe et al., 2000), or flavonoid (30 μM) dissolved in DMSO (final concentration 0.7%). [3H]GSH uptake was also measured with membrane vesicles prepared from control transfected C1 cells.
ATPase Activity of Purified Reconstituted MRP1.
To measure the effect of quercetin, kaempferol, and naringenin (± GSH) on the ATPase activity of MRP1, MRP1 was immunoaffinity purified from drug-resistant H69AR lung cancer cells and reconstituted as described previously (Mao et al., 1999) with modifications (Mao et al., 2000). Purified reconstituted MRP1 (1 μg) was incubated at 37°C in 0.1 ml of buffer containing 50 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 2.5 mM ATP, and flavonoids (10 and 50 μM) dissolved in DMSO for 4 h. The effect of naringenin was measured in the presence of 3 mM GSH and 3 mM DTT. As a positive control, proteoliposomes were incubated with GSSG (500 μM), a known stimulator of MRP1 ATPase activity (Chang et al., 1997; Mao et al., 1999). After terminating the reactions, the amount of inorganic phosphate released was determined immediately as described previously (Mao et al., 1999). The final concentration of DMSO in the reactions was 5%, which was determined to have no significant effect on MRP1 ATPase activity.
Orthovanadate-Induced Trapping of 8-Azido-α-[32P]ADP by MRP1.
Sodium orthovanadate (10 mM) was prepared in Tris sucrose buffer (50 mM Tris, 250 mM sucrose, pH 7.5) and the pH adjusted to 7.5. This solution was boiled for approximately 10 min until its yellow color disappeared and then stored at 4°C until use. MRP1-enriched membrane vesicles (20 μg) were incubated in Tris sucrose buffer containing MgCl2 (5 mM), sodium orthovanadate (1 mM), 8-azido-α-[32P]ATP (5 μM, 2 μCi), EGTA (0.1 mM), ouabain (1 mM), and sodium azide (0.02% w/v) for 15 min at 37°C, in the presence of flavonoid (50 μM) (5% v/v DMSO) and freshly prepared GSH (3 mM) as indicated. Preliminary experiments showed that DMSO had little or no effect on vanadate-induced trapping. Trapping in the presence of LTC4 (1 μM) was included as a positive control and membrane vesicles prepared from empty vector transfected C1 HeLa cells were included as a negative control. Reactions were terminated by the addition of 500 μl of ice-cold Tris-EGTA buffer (50 mM Tris HCl, pH 7.4, 0.1 mM EGTA, 5 mM MgCl2) and then centrifuged at 21,000gfor 15 min at 4°C. Resuspended membrane proteins were washed once with 500 μl of Tris-EGTA buffer, centrifuged, and resuspended in 20 μl of the same buffer. Samples were then transferred to an open, flexible 96-well plate and exposed to UV light at 302 nm for 8 min at a distance of 8 cm. After addition of Laemmli buffer, samples were transferred to Microfuge tubes, vortexed, and centrifuged at 21,000g for 5 min at 4°C. The supernatants were subjected to electrophoresis on a 10% SDS-polyacrylamide running gel. After drying, 32P-labeled proteins in the gel were detected using a STORM 800 PhosphorImager and quantitated using ImageQuaNT software (Molecular Dynamics, Sunnyvale, CA). Two independent experiments were carried out and modulation of orthovanadate-induced trapping of 8-azido-α-[32P]ADP was expressed as a mean value relative to the DMSO control.
Chemosensitivity Testing.
The effect of quercetin, naringenin, and kaempferol on the sensitivity of the MRP1-transfected HeLa T5 cells to vincristine was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide microtiter plate assay as described previously (Cole et al., 1994). Dose-response curves were first obtained for quercetin, naringenin, and kaempferol alone, and these compounds were then tested at a concentration (10 μM) that produced <10% cytotoxicity in combination with several concentrations of vincristine. Within each experiment, determinations were carried out in quadruplicate. Relative resistance factors were calculated as the ratio of the IC50 value of the MRP1-transfected T5 cells to the IC50 value of the C1 cells transfected with empty pRc/CMV vector alone.
Results
HPLC Analysis and Hydrophilicity of Flavonoids.
The bioflavonoids examined in the present study were analyzed by HPLC to provide an estimate of their relative hydrophilicity. Their retention times on a reverse phase column are shown in Fig.1 and their structures are illustrated in Fig. 2. The glycosides naringin and genistin with retention times of <4 min were the most hydrophilic compounds, presumably due to their attached sugar groups. Myricetin, a flavon-3-ol with six hydroxyl groups, had the next shortest retention time (5 min) followed by quercetin (10.2 min), another flavon-3-ol with five hydroxyl groups. The flavanone naringenin and the isoflavone genistein (each with three hydroxyl groups) were relatively lipophilic with retention times of 10.9 and 15 min, respectively. Kaempferol (a flavon-3-ol with four hydroxyl groups) and apigenin (a flavone with three hydroxyl groups) were the most lipophilic compounds with retention times of 20 and 23 min, respectively.

HPLC retention times of dietary flavonoids. The flavonoids were analyzed by reverse phase HPLC as described underExperimental Procedures and their retention times plotted in rank order (left to right) from the shortest (most hydrophilic) to the longest (most hydrophobic).

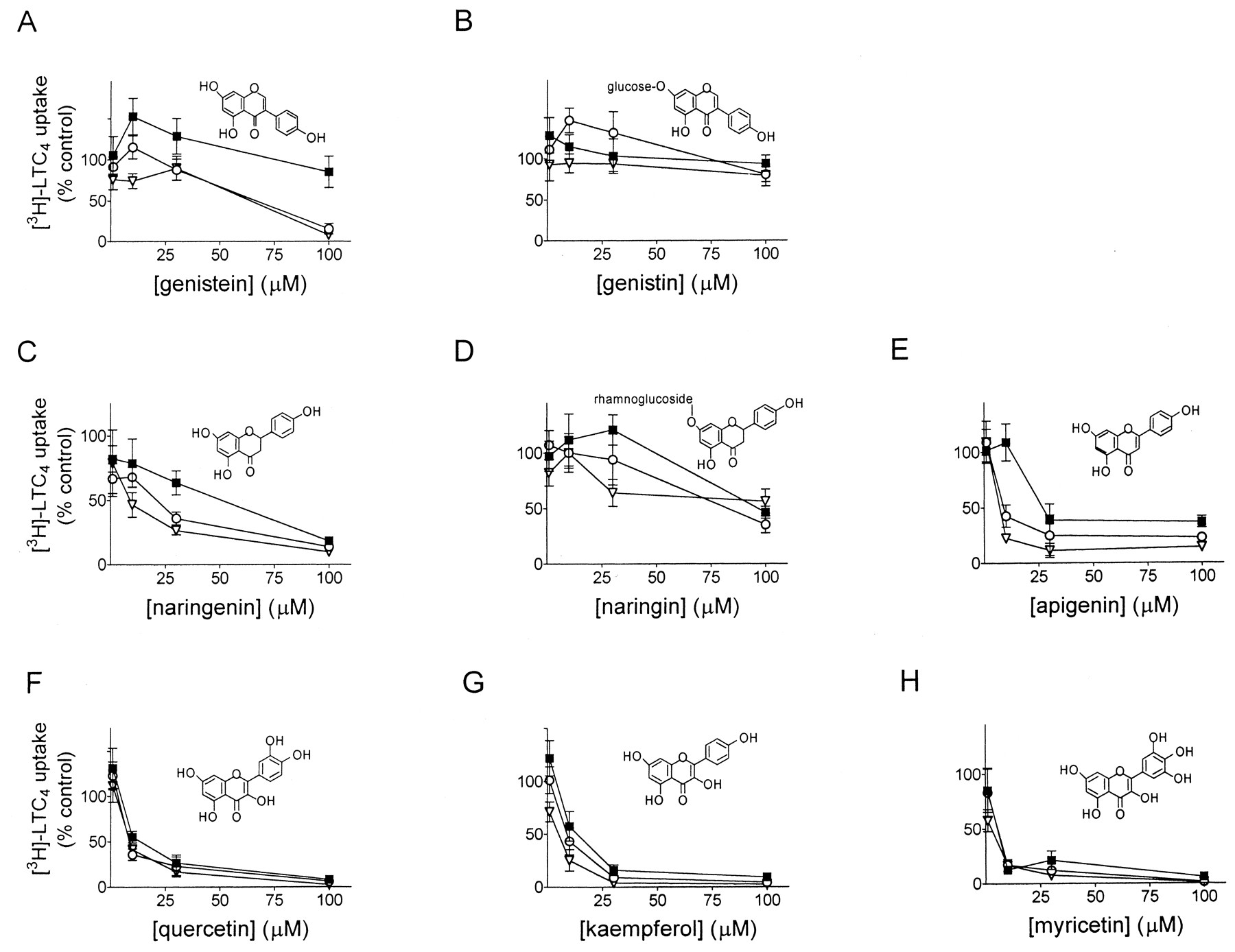
Effect of dietary flavonoids on [3H]LTC4 uptake by membrane vesicles from MRP1-transfected HeLa cells. MRP1-enriched membrane vesicles were incubated for 60 s at 23°C with 50 nM [3H]LTC4 in the absence (▪) or presence (○, 1 mM; ▿, 3 mM) of GSH and flavonoid. A, genistein; B, genistin; C, naringenin; D, naringin; E, apigenin; F, quercetin; G, kaempferol; H, myricetin. Data were calculated as percentage of control uptake in the absence of GSH and flavonoid. The graphs shown represent results from a single experiment and the symbols are means of triplicate determinations (±S.D.). Each compound was tested in one to three additional independent experiments and similar results were obtained.
Inhibition of LTC4 Transport Activity by Bioflavonoids.
ATP-dependent LTC4 uptake by MRP1-enriched T5 membrane vesicles was measured at several different concentrations of flavonoid in the presence and absence of GSH (Fig.2). In the absence of GSH, the aglycosides genistein and naringenin were relatively weak inhibitors of LTC4 uptake (IC50 values >100 and 66 μM) (Fig. 2, A and C) and their glycoside counterparts, genistin and naringin, were even less potent (IC50 values >200 μM) (Fig. 2, B and D). GSH enhanced inhibition of LTC4 transport by genistein and naringenin by 2- and 7-fold (IC50values of 65 and 9.5 μM, respectively, with 3 mM GSH), but had little effect on the corresponding glycosides genistin and naringin (IC50 values >200 and 98 μM, respectively, with 3 mM GSH). The flavone apigenin was a potent inhibitor of LTC4 transport (IC50, 25 μM) and inhibition by this compound was enhanced approximately 3.4-fold by GSH (IC50, 7.4 μM) (Fig. 2E). Alone, the flavon-3-ols myricetin, quercetin, and kaempferol were even more potent inhibitors of LTC4 transport (IC50 values, 7–12 μM) than apigenin but the addition of GSH had little effect on these compounds (Fig. 2, F–H).
Determination of Ki Values for Selected Flavonoids.
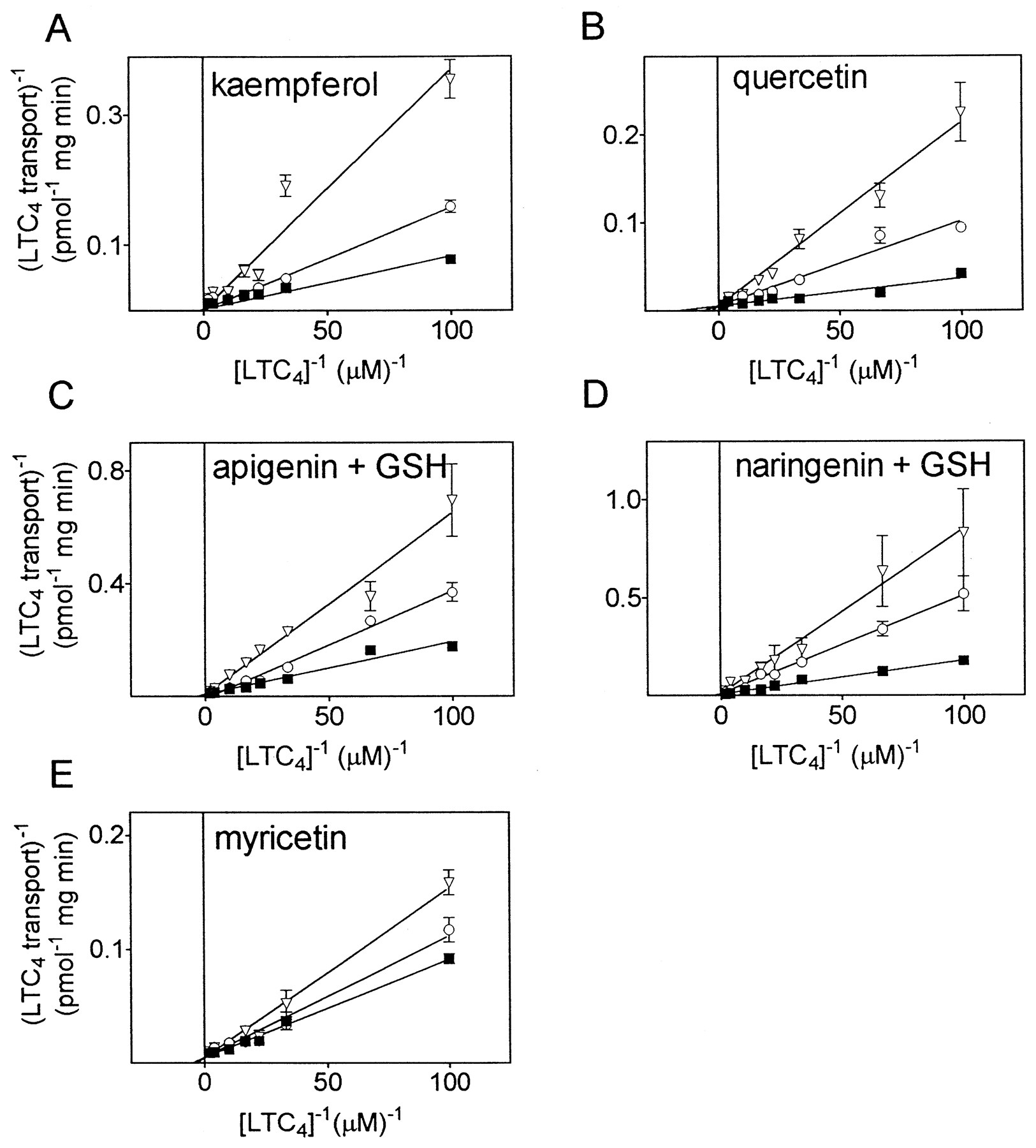
Those flavonoids found to be relatively potent inhibitors of LTC4 transport [kaempferol, quercetin, myricetin, apigenin (+ GSH), and naringenin (+ GSH)] were further characterized by determining the mode of inhibition (Fig.3). Lineweaver-Burk plots showed that kaempferol (Fig. 3A), quercetin (Fig. 3B), apigenin (+ GSH) (Fig. 3C), naringenin (+ GSH) (Fig. 3D), and myricetin (Fig. 3E) behaved as competitive inhibitors of MRP1-mediated LTC4transport. The K i values for these compounds are summarized in Table 1 and range from 2.4 ± 1.6 μM for kaempferol to 20.8 ± 6.4 μM for naringenin in the presence of 3 mM GSH. Their rank order for potency of LTC4 transport inhibition was kaempferol > apigenin (+ GSH) > quercetin > myricetin > naringenin (+ GSH), which correlated poorly with the relative lipophilicity of these compounds as inferred from their HPLC retention times.

Kinetic analysis of the LTC4 transport inhibition by flavonoids in MRP1-enriched HeLa membrane vesicles. Uptake of [3H]LTC4 (10–500 nM) at 23°C was measured in the presence of various concentrations of each flavonoid. A, kaempferol (0 μM, ▪; 5 μM, ○; 10 μM, ▿); B, quercetin (0 μM, ▪; 20 μM, ○; 40 μM, ▿); C, apigenin (+ 3 mM GSH) (0 μM, ▪; 5 μM, ○; 10 μM, ▿); D, naringenin (+ 3 mM GSH) (0 μM, ▪; 50 μM, ○; 100 μM, ▿); and E, myricetin (0 μM, ▪; 5 μM, ○; 10 μM, ▿). Double-reciprocal plots were generated andK i values were calculated from the apparentK m value.. In all cases, data points represent means (±S.D.) of triplicate determinations in a typical experiment. Each flavonoid was tested at two different concentrations in at least two independent experiments. A summary of the K i values determined in multiple experiments is presented in Table 1.
Inhibitory constants of selected flavonoids for [3H]LTC4 transport
Inhibition of [3H]E217βG Transport by Bioflavonoids
The same five flavonoid inhibitors of LTC4 transport [kaempferol, quercetin, myricetin, apigenin (+ GSH), and naringenin (+ GSH)] were also tested for their ability to inhibit [3H]E217βG uptake by MRP1-enriched membrane vesicles (Fig.4A). Overall, these compounds were less potent with respect to inhibition of E217βG transport than observed with LTC4 as a substrate. For example, 60 μM myricetin did not inhibit [3H]E217βG uptake, and quercetin caused only 30% inhibition at this concentration, whereas LTC4 transport was inhibited more than 90% at the same concentrations of these flavonoids. Kaempferol was also a less potent inhibitor of E217βG transport (40 and 70% inhibition at 10 and 30 μM, respectively) compared with its effect on LTC4 transport (50 and 90% inhibition at 10 and 30 μM, respectively) as was apigenin (+ 3 mM GSH) (E217βG transport was inhibited 55 and 80%, whereas LTC4 transport was inhibited 70 and 90% at 10 and 30 μM, respectively). In contrast, naringenin (+ GSH) inhibited E217βG and LTC4 transport with a comparable potency (approximately 50 and 60% inhibition at 10 and 30 μM, respectively). Thus, the rank order of potency for E217βG transport inhibition by these flavonoids was apigenin (+ GSH) > kaempferol > naringenin (+ GSH) > quercetin ≫ myricetin. In contrast to the inhibition of LTC4 transport, this rank order potency is in very good agreement with the relative lipophilicity of these compounds, as indicated by their HPLC retention times.

Effect of flavonoids on [3H]E217βG and [3H]GSH uptake by membrane vesicles from MRP1-transfected HeLa cells. A, MRP1-enriched membrane vesicles were incubated for 90 s at 37°C with transport buffer and 400 nM [3H]E217βG, 10 mM dithiothreitol, ± 3 mM GSH (as indicated) and flavonoid [0 μM (open columns), 10 μM (hatched columns), 30 μM (light solid columns), and 60 μM (dark solid columns)]. Columns are means of triplicate determinations (±S.D.). The graph shown represents results obtained in a single experiment and similar results were obtained in two additional independent experiments. B, membrane vesicles prepared from MRP1-transfected HeLa cells (T5) (open columns) and vector control-transfected HeLa cells (C1) (solid bars) were incubated with 100 μM [3H]GSH (120 nCi), DTT (10 mM), in transport buffer at 37°C for 20 min. Control assays contained vehicle alone (0.7% DMSO); VRP (100 μM) was added as a positive control and each flavonoid was added at 30 μM. Columns are means of triplicate determinations (±S.D.) in a single experiment. [3H]GSH uptake by T5 HeLa membrane vesicles in the absence of VRP or flavonoid was 0.27 ± 0.9 nmol/mg.
Stimulation of [3H]GSH Transport by Bioflavonoids
Kaempferol, quercetin, myricetin, apigenin, and naringenin were tested at a concentration of 30 μM for their ability to stimulate transport of [3H]GSH (Fig.4B). VRP (100 μM) was included as a positive control and stimulated transport 4-fold, consistent with our previous results (Loe et al., 2000). Kaempferol had no effect on [3H]GSH transport, whereas myricetin and quercetin caused a moderate stimulation of transport (1.4- and 1.7-fold, respectively). Apigenin and naringenin strongly stimulated GSH uptake of GSH by 4.3- and 2.3-fold, respectively. Thus, apigenin at 30 μM stimulated GSH uptake to the same degree as 100 μM VRP. The rank order of stimulation of GSH transport by the flavonoids was apigenin > naringenin > quercetin > myricetin > kaempferol, which did not correlate with their rank order of either LTC4 or E217βG transport inhibition, nor with their relative lipophilicity as inferred from their HPLC retention times.
Effect of Flavonoids on the ATPase Activity of Purified Reconstituted MRP1.
As two of the most abundant dietary flavonoids and potent inhibitors of MRP1-mediated LTC4transport, the flavon-3-ols kaempferol and quercetin were investigated for their ability to modulate the ATPase activity of purified reconstituted MRP1. The flavanone naringenin was also examined. We have shown previously that H69AR cells from which MRP1 was originally cloned express approximately 8-fold more MRP1 than the transfected HeLa T5 cells and thus, for practical reasons, the protein was purified from H69AR cells. Quercetin inhibited MRP1 ATPase activity by 22 and 37% at 10 and 50 μM, respectively (Table 2). In contrast, kaempferol and naringenin at 50 μM both stimulated MRP1 ATPase activity by 9%, whereas a lower concentration of these compounds (10 μM) had no effect. GSH (3 mM) by itself had no effect on basal ATPase activity of purified MRP1 as reported previously by us (Mao et al., 1999), in contrast to observations by other laboratories that reported stimulation by this tripeptide (Chang et al., 1997;Hooijberg et al., 2000). On the other hand, GSH increased stimulation by 50 μM naringenin to 116% of control values. Finally, GSSG (500 μM) stimulated basal ATPase activity of purified MRP1 by approximately 20%, consistent with previous findings (Chang et al., 1997; Mao et al., 1999).
Effect of flavonoids on the ATPase activity of purified reconstituted MRP1
Effect of Flavonoids on Orthovanadate-Induced Trapping of 8-Azido-α-[32P]ADP by MRP1.
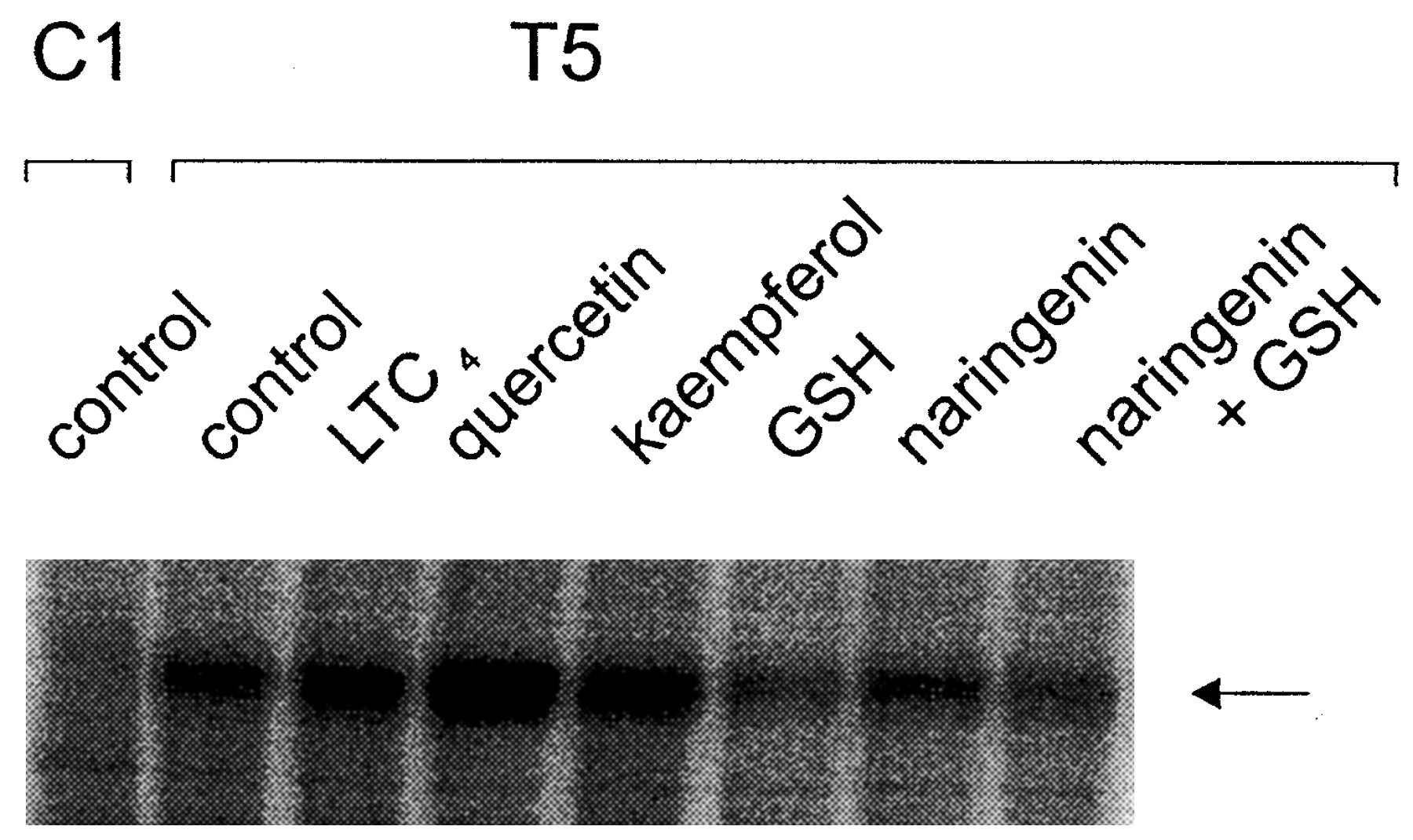
We and others have previously shown that vanadate-induced trapping of 8-azido-α-[32P]ADP by MRP1 can be stimulated by LTC4 (Bakos et al., 2000; Gao et al., 2000). To examine whether flavonoids might have a similar effect, the ability of quercetin, kaempferol, and naringenin (± GSH) to stimulate orthovanadate-induced trapping of 8-azido-α-[32P]ADP by MRP1 was investigated. In agreement with our previous studies in MRP1-enriched insect cell membranes (Gao et al., 2000), LTC4 (1 μM) stimulated the vanadate-induced trapping of 8-azido-α-[32P]ADP by MRP1 in T5 membrane vesicles by 1.4-fold compared with vehicle control (Fig.5). Quercetin (50 μM) stimulated labeling to an even greater extent than LTC4(approximately 2-fold), whereas kaempferol (50 μM) stimulated trapping by approximately 1.2-fold. When tested alone, naringenin also stimulated vanadate-induced trapping approximately 1.2-fold. In contrast, naringenin in the presence of 3 mM GSH inhibited trapping by approximately 25%. However, this inhibition appears attributable to GSH rather than the naringenin, because vanadate-induced trapping was decreased by approximately 50% by 3 mM GSH alone.

Effect of flavonoids on the orthovanadate-induced nucleotide trapping by MRP1. Membrane proteins prepared from MRP1-transfected T5 HeLa cells were incubated with 8-azido-α-[32P]ATP, 1 mM sodium orthovanadate in the presence or absence of 50 μM of the indicated flavonoids or 1 μM LTC4 at 37°C, following the trapping method described under Experimental Procedures. Proteins were resolved by SDS-polyacrylamide electrophoresis and 32P-labeled proteins detected and quantitated by analysis on a PhosphorImager. The labeled MRP1 is indicated. The autoradiograph shown is the result of a typical experiment and similar results were obtained in a second experiment.
Our observation that GSH significantly decreases vanadate-induced 8-azido-α-[32P]ADP trapping contrasts with the findings of other investigators who reported an increase in trapping with this tripeptide (Taguchi et al., 1997). Consequently, we examined the effect of two additional sulfhydryl-reducing agents for their ability to modulate trapping; GSSG, which has been previously shown to stimulate trapping was included in these experiments as a positive control (Taguchi et al., 1997). 2-Mercaptoethanol (3 mM) had no significant effect on trapping, whereas DTT at the same concentration abolished MRP1-specific labeling altogether (data not shown). GSSG stimulated trapping (1.2-fold) as expected (Taguchi et al., 1997).
Effect of Kaempferol, Naringenin, and Quercetin on Vincristine Sensitivity of Transfected HeLa Cells.
Kaempferol, quercetin, and naringenin were tested for their ability to sensitize intact MRP1-transfected HeLa T5 cells to the cytotoxic effects of vincristine. In initial experiments with the flavonoids alone, it was noted that the T5 cells were not resistant to these compounds relative to control C1 cells, consistent with the reports of others (Hooijberg et al., 1999b). Moreover, the IC10 for kaempferol, naringenin, or quercetin was approximately 10 μM and consequently, this concentration of bioflavonoid was used in subsequent combination experiments with vincristine. Neither kaempferol nor naringenin had any effect on the vincristine sensitivity of the MRP1 T5 cells or the control C1 cells (Fig. 6, A and B). In contrast, quercetin (10 μM) decreased the relative resistance of T5 cells to vincristine from 8.9- to 2.2-fold (Fig. 6C). None of the bioflavonoids at a concentration of 10 μM had statistically significant effect on the vincristine sensitivity of the control transfected C1 cells.
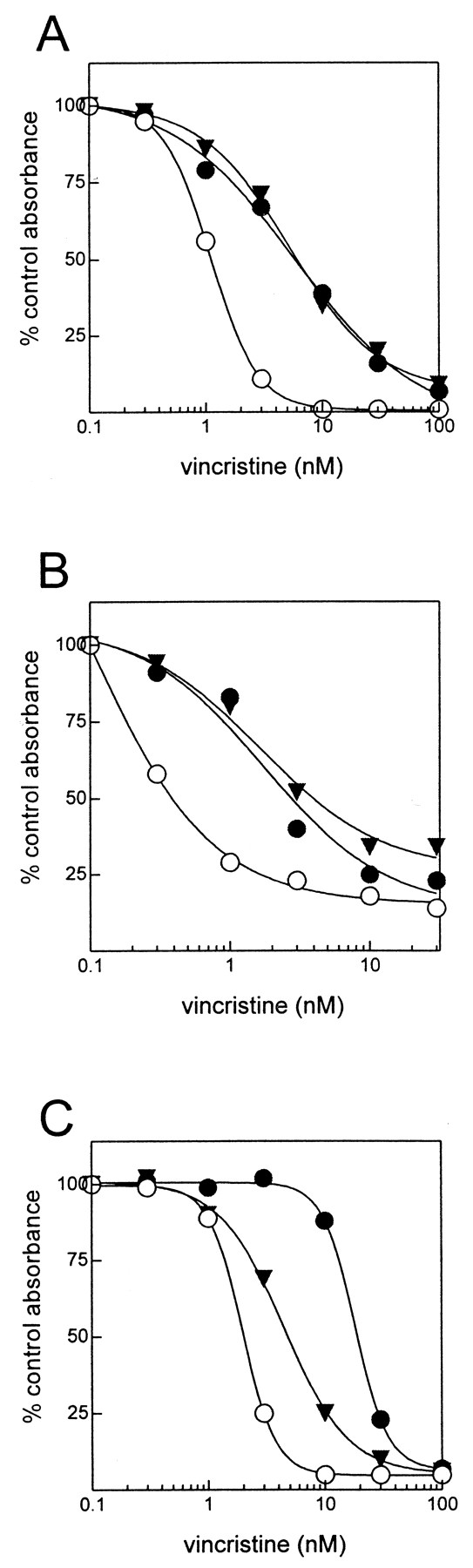
Effect of flavonoids on vincristine resistance in MRP1-transfected HeLa cells. Vector control transfected C1 HeLa cells (○) and MRP1-transfected HeLa cells (●, ▾) were incubated in the presence (▾) and absence (○, ●) of 10 μM flavonoid. A, kaempferol; B, naringenin; and C, quercetin. None of the flavonoids had a statistically significant effect on vincristine sensitivity of the control C1 cells and these data have been omitted for clarity. Data points represent the mean (±S.D.) of quadruplicate determinations in a single experiment. Similar results were obtained in one to two additional independent experiments.
Discussion
In the present study, we have examined the ability of several naturally occurring flavonoids to modulate conjugated organic anion and GSH transport and ATPase activity associated with MRP1. We also investigated the effects of these compounds on MRP1-mediated vanadate-induced trapping of ADP and to a limited extent, drug resistance. For these studies, we used inside-out membrane vesicles from HeLa cells stably transfected with MRP1 cDNA, as well as native MRP1 purified to >85% homogeneity and reconstituted into phospholipid vesicles, both systems that allow any observed effects of the flavonoids to be attributed directly to the properties of MRP1.
Initially, the effect of a series of flavonoids and several glycoside derivatives on the uptake of the well characterized MRP1-conjugated organic anion substrate [3H]LTC4 into inside-out membrane vesicles was measured as an indicator of their interaction with MRP1. The glycosylated flavonoids have been reported to have generally lower biological activity than their aglycoside partners (Williamson et al., 1996; Conseil et al., 1998). However, most substrates and inhibitors of MRP1 are relatively hydrophilic and consequently, it was not necessarily expected that naringin and genistin would be poorer inhibitors of LTC4transport than their more lipophilic aglycoside derivatives naringenin and genistein. Thus, although the low inhibitory potency of the glycosides we observed suggests that their interaction with MRP1 in inside-out membrane vesicles is significantly less than the aglycosides, it is not clear why this is so.
We and others have shown previously that the ability of certain unconjugated xenobiotics to inhibit MRP1 transport of organic anions in inside-out membrane vesicles is markedly enhanced by the addition of GSH (Loe et al., 1996, 1998). We found that inhibition of LTC4 transport by apigenin and naringenin was also enhanced by GSH, whereas other flavonoids (notably the flavon-3-ols myricetin, quercetin, and kaempferol) were relatively potent inhibitors without the addition of this reducing tripeptide. LTC4 transport inhibition by genistein (an isoflavone) was enhanced by GSH, but this occurred only at high concentrations of flavonoid that are unlikely to be biologically relevant. The mechanism by which GSH enhances the inhibitory potency of certain compounds on MRP1-mediated conjugated organic anion transport appears to result from increasing their affinity for the protein (Loe et al., 1998, 2000; Qian et al., 2001). However, the structural and/or physical properties of a molecule that determine whether its inhibitory action on LTC4 will be increased by GSH have yet to be well defined. In the case of the polyphenolic flavonoids, our results indicate that again, it is not simply related to the relative hydrophilicity of the compound as reflected by their HPLC retention times.
As mentioned above, the flavone apigenin and the flavanone naringenin had the greatest inhibitory effect on [3H]LTC4 transport by MRP1 when GSH was present. These two compounds were also the most potent stimulators of [3H]GSH transport. Stimulation of GSH transport by the flavonoids was unexpected and may have implications for cellular redox homeostasis. Whether these flavonoids are cotransported with GSH as demonstrated previously for vincristine (Loe et al., 1998) is not known. It may be that these compounds interact with MRP1 to stimulate [3H]GSH transport without being transported themselves as shown recently for verapamil (Loe et al., 2000). The lack of cross-resistance of the T5 cells to the flavonoids does not seem consistent with a cotransport mechanism but direct transport studies with the flavonoids are required before any conclusions can be drawn. There is good evidence in other model systems, including human colonic carcinoma Caco-2 cells, that certain flavonoids and their glucoside-, glucuronide-, and sulfate-conjugated metabolites are transported by MRP2, a highly related ABC protein with 67% amino acid similarity to MRP1 (Walle et al., 1999; Walgren et al., 2000). However, despite the similarity of their primary structures, MRP1 and MRP2 differ in a number of important respects. For example, MRP1 is localized to basolateral membranes, whereas MRP2 is localized on apical membranes of polarized cells, including gastrointestinal epithelia, and whereas the two transporters have many substrates in common, their substrate specificities are not identical. These differences are certain to have important consequences with respect to the relative influence these two transporters might have on the bioavailability and elimination of dietary flavonoids.
The differences in rank order potency of flavonoid-mediated inhibition of E217βG and LTC4transport, and stimulation of GSH transport are of interest because they support the idea that these organic anion substrates bind to different or mutually exclusive sites on MRP1. This conclusion is in agreement with our recent findings that substitution of a conserved tryptophan residue in the last predicted transmembrane segment of MRP1 eliminates E217βG transport activity but leaves LTC4- and verapamil-stimulated GSH transport intact (Ito et al., 2001).
Five of the flavonoids that inhibited MRP1-mediated LTC4 transport at biologically relevant concentrations were determined to be competitive inhibitors. TheK i values of the flavonoids tested [which included the flavon-3-ols myricetin, quercetin, and kaempferol alone as well as the flavanone naringenin (+ GSH) and the flavone apigenin (+ GSH)] were in the low micromolar range, which is within a biologically achievable plasma level (Manach et al., 1998). The competitive nature of LTC4 transport inhibition by these flavonoids indicates that they bind to the same or mutually exclusive binding site(s) on MRP1 as LTC4 but differences in their effects on other MRP1 activities suggest the latter possibility is more likely. These findings also imply that LTC4transport and associated physiological processes mediated by this cysteinyl leukotriene might be affected in individuals who ingest significant amounts of these flavonoids.
It has been reported that genistein, kaempferol, apigenin, and the synthetic flavonoid flavopiridol increase daunorubicin accumulation in a drug-selected MRP1 overexpressing lung cancer cell line exposed to a 50 μM concentration of these compounds (Hooijberg et al., 1999b). We were unable to achieve this high a concentration of flavonoids in our transfected HeLa cells without significant toxicity. However, at a 5-fold lower concentration (10 μM), we found that only quercetin, but not kaempferol or naringenin, increased vincristine cytotoxicity in the T5 cells. Thus, although these three flavonoids are relatively good inhibitors of organic anion transport by MRP1, they seem to be poor candidates for reversing MRP1-mediated resistance to anticancer drugs.
The mechanistic relationship between the ATPase activity of ABC transporters and transport of their substrates is still not fully understood. However, it is generally assumed that the ability of a compound to stimulate the ATPase activity of an ABC protein is a reflection of its interaction with the protein and in the case of P-glycoprotein, this has often been the sole criterion for concluding that a molecule is transported by this ABC protein (Rao et al., 1994;Hooijberg et al., 1997). However, there are substrates (e.g., vinblastine and daunorubicin) that inhibit rather than stimulate P-glycoprotein ATPase activity (Sharom, 1997). Moreover, verapamil is one of the most effective stimulators of P-glycoprotein ATPase activity and yet is poorly transported by this protein. Finally, although not widely considered, many P-glycoprotein substrates have a biphasic effect, stimulating ATPase activity at low concentrations and inhibiting at high concentrations (Sharom, 1997). At present there is no satisfactory mechanistic explanation for these observations. In the case of MRP1, kaempferol and naringenin stimulated its ATPase activity, whereas quercetin was inhibitory. Inhibition of MRP1 ATPase activity by quercetin has not been reported previously but is not completely unexpected given that this flavon-3-ol is a well known inhibitor of the ATPase activity of several other ABC proteins, including purified reconstituted P-glycoprotein (Shapiro and Ling, 1997). It has been previously reported that other flavonoids (genistein, kaempferol, flavopiridol, and apigenin) modestly stimulate MRP1 ATPase in membranes prepared from a drug-selected lung cancer cell line pretreated with various inhibitors to suppress the activity of other membrane ATPases (Hooijberg et al., 1999b, 1997, 2000). Although it is difficult to compare these prior studies with our current data obtained using purified reconstituted MRP1 because of methodological differences, it is worth noting that kaempferol modestly stimulated the ATPase activity in both systems. Nevertheless, our findings support the assertion ofSharom (1997) that it is not wise to presume that drug-stimulated ATPase activity is always representative of the transport function of an ABC transporter.
Another method commonly used to measure interaction of compounds with ABC transporters is their ability to stimulate orthovanadate-induced nucleotide trapping (Taguchi et al., 1997). Naringenin and kaempferol caused a significant stimulation of MRP1-mediated orthovanadate-induced trapping of ADP consistent with their ability to stimulate MRP1 ATPase activity. On the other hand, quercetin also caused a large stimulation of trapping by MRP1, which appears inconsistent with its inhibitory effect on MRP1 ATPase activity. Thus, although both assays reflect ATP binding and hydrolysis, our results indicate that the interaction of quercetin with MRP1 differs from that of the other flavonoids.
Studies of purified recombinant polypeptides encoding the NBD2 of murine P-glycoprotein (Conseil et al., 1998) and the P-glycoprotein-like Leishmania tropica transporter (Perez-Victoria et al., 1999) have suggested that quercetin and perhaps other flavonoids interact with the ATP binding site and a vicinal region of these proteins that binds certain steroids. These same flavonoids have also been reported to interact with NBD1 of murine P-glycoprotein by a similar mechanism but with apparently lower affinity (Conseil et al., 1998). The chloride channel activity of CFTR (which is more closely related to MRP1 than P-glycoprotein) is also stimulated by flavonoids such as genistein, kaempferol, apigenin, and quercetin (Wang et al., 1998; Illek et al., 1999). Initially, it was proposed that genistein acted in an indirect manner by inhibiting protein tyrosine kinases (Akiyama et al., 1987). However, more recent evidence suggests that genistein interacts directly with NBD2 of CFTR to prevent ATP hydrolysis, resulting in a prolonged open channel conformation of the protein (Wang et al., 1998; Randak et al., 1999). In support of a direct and relatively specific interaction of flavonoids with NBD2 of CFTR, genistein and quercetin were observed to have no effect on the ATPase activity of a fusion protein encoding NBD1 and the regulatory R domain of this protein (Howell et al., 2000). Results from the present study suggest that quercetin in particular interacts with at least one NBD of MRP1 causing a significant stimulation of vanadate-dependent trapping and inhibition of ATPase activity. We and others have shown that essentially all ADP trapping occurs at NBD2 of MRP1 and thus quercetin might interact specifically with this domain to increase the efficiency of ADP trapping (Gao et al., 2000; Nagata et al., 2000). Although it remains possible that at least some flavonoids might interact with the drug- or substrate-binding sites found within one or more of the polytopic membrane spanning domains of these ABC proteins, the studies described above provide compelling evidence that NBD2 is a common site of quercetin action.
In summary, our results indicate that dietary flavonoids have the potential to influence the conjugated organic anion and GSH transport, drug resistance-conferring, as well as the ATPase properties of MRP1. However, it is clear that a flavonoid's ability to modulate one activity is not necessarily linked to its ability to modulate another activity of the protein. Our data also indicate that the flavonoids should be considered individually rather than as a class of compounds, because their effects on different MRP1 activities are variable. Finally, our results and those of others (Hooijberg et al., 1997,1999b) indicate that the flavonoids probably bind to more than one site on MRP1 but in the case of quercetin at least, these sites may be localized to the NBDs. We have recently reported the expression of correctly folded soluble polypeptides corresponding to NBD1 and NBD2 of MRP1 (Hipfner et al., 1999b). The availability of these MRP1 domains should facilitate future mechanistic studies of flavonoid interactions with MRP1.
Acknowledgments
We thank Drs. Doug Loe and Mian Gao for expert advice, Kathy Sparks and Libby Eastman for excellent technical assistance, and Maureen Rogers for expert word processing and assistance with the preparation of the figures.
Footnotes
- Received October 12, 2000.
- Accepted January 12, 2001.
-
Send reprint requests to: Dr. Susan P. C. Cole, Cancer Research Laboratories, Room 328, Botterell Hall, Queen's University, Kingston, Ontario, Canada, K7L 3N6. E-mail:coles{at}post.queensu.ca
-
This work was supported by a grant (MT-10519) from the Medical Research Council of Canada. E.M.L. and C.J.O. are recipients of Medical Research Council of Canada Doctoral Awards, and E. M. L. is the past recipient of an Ontario Graduate Scholarship. R.G.D. is the Stauffer Cancer Research Professor of Queen's University and S.P.C.C. is a Senior Scientist of Cancer Care Ontario.
Abbreviations
- MRP1
- human multidrug resistance protein 1
- ABC
- ATP-binding cassette
- NBD
- nucleotide binding domain
- LTC4
- leukotriene C4
- E217βG
- 17β-estradiol 17-(β-d-glucuronide)
- GSH
- reduced glutathione
- CFTR
- cystic fibrosis transmembrane conductance regulator
- DTT
- dithiothreitol
- GSSG
- glutathione disulfide (oxidized glutathione)
- HPLC
- high-performance liquid chromatography
- DMSO
- dimethyl sulfoxide
- VRP
- verapamil
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}