


Abstract
CCAAT/enhancer-binding proteins (C/EBPs) are key transcription factors involved in the constitutive expression of several cytochrome P450 genes in the liver. Their concentration and activity change in several pathophysiological conditions. For instance, during inflammation, released cytokines induce repressive C/EBPβ-liver inhibitory protein (LIP), which antagonizes constitutive C/EBP transactivators [C/EBPα and C/EBPβ-liver activating protein (LAP)], down-regulating genes such as CYP3A4. However, the mechanism by which hepatic C/EBP factors modulate transcription of the CYP3A4 gene is not known. To elucidate the mechanism of action, we cotransfected luciferase reporter vectors, containing 5′-flanking deletions of the CYP3A4 gene, along with expression vectors for C/EBPβ-LAP, C/EBPβ-LIP, and C/EBPα, in hepatic (HepG2) and nonhepatic (HeLa) cells. Analysis of the –3557 to –6954 base pair (bp) region demonstrated the existence of a 288-bp sequence at –5.95 kilobases (kb), which showed maximal response to C/EBPβ-LAP (∼30-fold increase in HepG2 cells). Coexpression of LAP with increasing amounts of LIP reduced the activating effect by ∼70%. Site-directed mutagenesis of predicted C/EBPβ binding sites demonstrated the presence of four functional C/EBPβ-responsive motifs within this distal flanking region. Further experiments using chromatin immunoprecipitation proved the binding of endogenous C/EBPβ to the –5.95-kilobase enhancer of the CYP3A4 gene in human hepatocytes. Expression of recombinant LAP and LIP by means of adenoviral vectors resulted in their binding to this region, which was followed by activation/repression of CYP3A4. Together, our results uncover a new distal enhancer site in the CYP3A4 gene where C/EBPβ-LAP binds and activates transcription, whereas the truncated form, C/EBPβ-LIP, antagonizes LAP activity and causes gene repression.
Cytochrome P450 enzymes are key players in the oxidative metabolism of xenobiotics, including the majority of clinically used drugs, environmental procarcinogens and toxins (Nelson et al., 1996). CYP3A4 is the most abundant human cytochrome P450 isoform in the liver and small intestine and plays a major role in the biotransformation of many drugs because of its broad substrate specificity. Indeed, CYP3A4 is involved in the metabolism of approximately half of the drugs in use today (Guengerich, 1999).
CYP3A4 activity shows a wide interindividual variability, which represents a basis for clinically significant drug interactions and toxicities (Lehmann et al., 1998). Part of this variability can result from the induction of CYP3A4 transcription by xenobiotics. The molecular mechanism that underlies this phenomenon is complex, with several nuclear receptors, including PXR (Lehmann et al., 1998), CAR (Goodwin et al., 2002), glucocorticoid receptor (Pascussi et al., 2003), and vitamin D receptor (Drocourt et al., 2002), playing a decisive role.
However, CYP3A4 gene transcription relies not only on xenobiotic-activated nuclear receptors. We have recently shown that CYP3A4 is transactivated in human hepatocytes by several liver-enriched factors (LETFs), such as hepatocyte nuclear factor (HNF) 4α and 3γ, and CCAAT/enhancer-binding protein (C/EBP) α and β (Jover et al., 2001, 2002; Rodriguez-Antona et al., 2003). These LETFs are coordinately involved in the constitutive transcription of CYP3A4 gene, resulting in one of the highest enzyme mRNA productions in hepatocytes (Yamashita et al., 2000).
Some of these transcription factors show a complex regulation and variable expression in the liver, and it is likely they play a role in the well recognized CYP3A4 phenotypic variability (Schuetz, 2004). One example of variably expressed LETF is the family of C/EBP proteins, which show tissue- and stage-specific expression, leaky ribosomal reading, post-transcriptional modifications, and irregular DNA binding specificity (Lekstrom-Himes and Xanthopoulos, 1998).
C/EBP transcription factors are critical for normal cellular differentiation and function in a variety of tissues. The prototypic C/EBP contains of a basic region involved in DNA binding followed by a leucine zipper motif involved in homo- and heterodimerization with other family members.
C/EBPα and C/EBPβ are the predominant C/EBP isoforms expressed by adult hepatocytes in healthy liver (Diehl, 1998; Lekstrom-Himes and Xanthopoulos, 1998). C/EBPβ (originally isolated as LAP, nuclear factor for IL-6 expression, IL-6DBP, α1-acid glycoprotein/enhancer-binding protein, and CRP2) was identified as a factor binding to regulatory elements of albumin, interleukin-6, and several acute-phase genes (Akira et al., 1990; Descombes et al., 1990; Poli et al., 1990). Transcription of C/EBPβ results in a single mRNA that can be translated into full-length and amino-terminally truncated proteins. The liver isoform C/EBPβ-LIP (20 kDa), a shorter translation product initiated at the third AUG codon of the C/EBPβ mRNA, behaves as a transcription antagonist. It shares with C/EBPβ-LAP (35 kDa) the DNA binding and leucine zipper domains but lacks the transactivation domain (Descombes and Schibler, 1991). Changes in the LAP:LIP isoform ratio are likely to affect hepatocyte proliferation and differentiation (Descombes and Schibler, 1991; Rana et al., 1995). This ratio increases significantly during liver development with a transient perinatal peak of LAP expression (Descombes and Schibler, 1991). On the other hand, partial hepatectomy or administration of LPS, results in an increase in the nuclear expression of the transcriptional repressor LIP (Rana et al., 1995; An et al., 1996; Hsieh et al., 1998; Welm et al., 2000).
The physiopathological conditions leading to changes in the LAP:LIP ratio also frequently lead to an altered expression of cytochrome P450 genes. For example, partial hepatectomy of rats results in the loss of several cytochrome P450 activities, with a pronounced reduction of CYP3As expression (Tamasi et al., 2001). Likewise, LPS injection to mice results in reduced expression of CYP3A in the liver (Sachdeva et al., 2003). The mechanisms of down-regulation of hepatic CYP3A in response to these stimuli (e.g., hepatectomy and inflammation) are not completely understood, but changes in the expression of C/EBP factors seem to be involved. Recent experimental evidence obtained in our laboratory supports this idea (Jover et al., 2002). We found that the down-regulation of CYP3A4 by the proinflammatory cytokine interleukin-6 was associated with a translational induction of C/EBPβ-LIP, that caused a sharp decrease in the LAP:LIP ratio (Jover et al., 2002). Despite the strong experimental evidence, the molecular mechanism explaining how these C/EBP factors activate/repress the CYP3A4 gene is not known. In the present study we demonstrate the existence of a distal enhancer site at –5.95 kb in the CYP3A4 gene, where C/EBPβ-LAP binds and activates transcription, whereas the truncated form, C/EBPβ-LIP, antagonizes LAP activity and causes gene repression. Our results also reveal that changes in the LAP:LIP isoform ratio can largely influence the regulation of CYP3A4 by other well characterized mechanisms such as rifampicin induction.
Materials and Methods
Plasmid Constructs. Chimeric luciferase reporter constructs with different lengths of the 5′-flanking region of the human CYP3A4 gene were prepared as follows.
First, pGL3-B-9388, pGL3-B-6954, and pGL3-B-3557 were obtained by cloning CYP3A4 5′-flanking sequences into the enhancer-less, promoterless pGL3-Basic vector (Promega, Madison, WI). Three DNA fragments encompassing bases +30 to –9388, –6954, and –3557 bp of the CYP3A4 5′-flanking region were PCR-cloned from human genomic DNA (Roche Diagnostics, Barcelona, Spain) using the Expand 20kbPLUS PCR System (Roche Diagnostics). The PCR primers used are described in Table 1. To facilitate the posterior cloning of the PCR fragments, restriction enzyme sites were added to the 5′ end of the different primers. After the PCR reaction, fragments were double-digested with MluI and XhoI, and ligated to pGL3-Basic vector previously digested with the same enzymes. All pGL3-B-3A4 constructs were confirmed by restriction digestion and double-strand DNA sequencing.
Oligonucleotides used for cloning
Second, 11 DNA deletion fragments within the –3557 to –6954 bp 5′-flanking region of the CYP3A4 gene were prepared according to standard techniques using various restriction enzymes as illustrated in Fig. 3. DNA fragments were cloned into the pGL3-Promoter vector (Promega), which contains a simian virus 40 promoter upstream of the luciferase gene.
Third, pGL3-P-5950/-5663 and pGL3-P-5692/-5329 were constructed to analyze in more detail the 590-bp region comprised between EcoRI and PvuII sites at approximately –6 kb upstream of the CYP3A4 start site. Fragments encompassing bases –5950 to –5663 bp, and –5692 to –5329 bp were PCR-cloned from the plasmid pGL3-P-MluI-SacI (150 ng) using the Expand high-fidelity PCR system (Roche Diagnostics). The PCR primers used and the restriction enzyme sites added are described in Table 1. After the PCR reaction, fragments were digested and ligated into the pGL3-Promoter vector, and constructs were confirmed by restriction digestion and DNA sequencing.
Fourth, deletion mutants lacking predicted C/EBPβ binding sites in the –5950/–5663-bp fragment were obtained by means of PCR-based site-directed deletions (Weiner et al., 1994). The PCR primers flanking each deleted site are described in Table 2. Increased template concentration and reduced cycling number were selected to avoid potential second-site mutations during the PCR. Amplified DNA was treated with DpnI to digest parental DNA and to select for mutation-containing amplified DNA. Cloned Pfu DNA polymerase was used before end-to-end ligation of the linear template to remove any bases extended onto the 3′ ends of the product by the Expand high-fidelity PCR system polymerase blend (Roche Diagnostics). The recircularized ligated vector DNA incorporating the desired deletions was then transformed into competent DH5α Escherichia coli.
Oligonucleotides used for PCR-based site-directed deletions
Fifth, base-substitution mutants were also generated to investigate the relevance of the four C/EBPβ binding sites separately or in combination. Mutants were obtained by means of two independent PCR reactions with primers (a-5′ or b-3′) flanking the –5950/–5663-bp fragment, and primers (Mut-a or Mut-b), which overlap C/EBPβ binding sites and contain base-substitutions changing the target sequence into a Kpn2I restriction site (Table 3). Amplified DNA was digested with SacI/Kpn2I (fragment a) or with Kpn2I/XhoI (fragment b), and ligated into the pGL3-Promoter vector (Promega) previously digested with SacI/XhoI. Mutation 4 was constructed in a single PCR reaction using the pGL3-P-5950/-5663 plasmid as a template, with the –5950/–5663-FP-a forward primer and the Mut-4-FP-b reverse primer, which contains both the mutant sequence (Kpn2I) and the 3′ cloning site (XhoI) (Table 3). Double mutants 1 + 4, 2 + 4, and 3 + 4 were obtained with the same pair of primers but using the plasmids Mut-2, Mut-3, and Mut-4 as templates. A construct containing four-site mutations was obtained with the “two independent PCR reactions” strategy described above but with the primers Mut-2 + 3-RP-a and -b (Table 3) and the plasmid Mut-1 + 4 as a template. Constructs were confirmed by restriction digestion analysis and DNA sequencing.
Oligonucleotides used for base-substitution mutagenesis
Cell Culture. HepG2 cells were plated in Ham's F-12/Leibovitz L-15 [1:1 (v/v)] supplemented with 6% newborn calf serum and cultured to 70 to 80% confluence. HeLa and human embryonic kidney 293 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% newborn calf serum and maintained as monolayer cultures. Culture medium for 293 cells was also supplemented with 3.5 g/l glucose. Human hepatocytes were isolated from liver biopsies (1–3 g) of patients undergoing liver surgery after informed consent. None of the patients were habitual consumers of alcohol or other drugs. A total of eight liver biopsies (three male and five female, ages ranging from 25 to 70 years) were used. Hepatocytes were isolated using a two-step perfusion technique (Gomez-Lechon and Castell, 2000) and seeded on plates coated with fibronectin (3.6 μg/cm2) at a density of 8 × 104 cells/cm2. The culture medium was Ham's F-12/Leibovitz L-15 [1:1 (v/v)] supplemented with 2% newborn calf serum, 5 mM glucose, 0.2% bovine serum albumin, and 10–8 M insulin. The medium was changed 1 h later to remove unattached hepatocytes. After 24 h, the culture medium was changed to serum-free medium containing 10–8 M dexamethasone. Cultures were routinely supplemented with 50 U/ml penicillin and 50 μg/ml streptomycin.
Adenoviral Vectors. Recombinant adenoviruses encoding the C/EBPα, C/EBPβ-LAP, and C/EBPβ-LIP were prepared as described previously (Jover et al., 2002; Rodriguez-Antona et al., 2003). The resulting viruses (called Ad-C/EBPα, Ad-LAP, and Ad-LIP) were plaque-purified, expanded into a high-concentration stock, and titrated by plaque assay as described previously (Castell et al., 1997). Cell lines and primary hepatocytes were infected with recombinant adenoviruses for 120 min at a multiplicity of infection (m.o.i.) ranging from 2.5 to 75 plaque-forming units per cell. Thereafter, cells were washed and fresh medium was added. Forty-eight hours after transfection, cells were analyzed or harvested and frozen in liquid N2.
Transfection Assays. Plasmid DNAs were purified on QIAGEN Maxiprep kit columns (QIAGEN, Valencia, CA) and quantified by optical density260 and fluorescence using PicoGreen (Molecular Probes, Leiden, The Netherlands). The day before transfection, cells were plated in 35-mm dishes with 1.5 ml of DMEM/Nut F12 (Invitrogen, Barcelona, Spain) supplemented with 6% newborn calf serum, 50 U/ml penicillin, and 50 mg/ml streptomycin. Firefly luciferase expression constructs (CYP3A4-LUC) (0.65 μg) were transfected with varying amounts of pAC-C/EBP plasmids (0–0.25 μg), as indicated in the figures, by calcium phosphate precipitation. The total amount of expression vector was kept constant by adding empty expression vector (pAC-CMV). In parallel, 0.08 μg of pRL-CMV, a plasmid expressing Renilla reniformis luciferase under the CMV immediate early enhancer/promoter, was cotransfected to correct variations in transfection efficiency. Calcium phosphate/DNA coprecipitates were added directly to each culture, and cells were incubated for an additional 48 h. Luciferase activities were assayed using the dual-luciferase reporter kit (Promega).
Quantification of mRNA Levels. Total cellular RNA was extracted with the RNeasy total RNA kit (QIAGEN), and contaminating genomic DNA was removed by incubation with DNase I amplification grade (Invitrogen). RNA (1 μg) was reverse transcribed as described previously (Rodriguez-Antona et al., 2000; Perez et al., 2003). Diluted cDNA (3 μl) was amplified with a rapid thermal cycler (LightCycler instrument; Roche Diagnostics) in 15 μl of LightCycler DNA Master SYBR Green I (Roche Diagnostics), 5 mM MgCl2, and 0.3 μM concentrations of each oligonucleotide (Table 4). In parallel, we analyzed the mRNA concentration of the human housekeeping porphobilinogen deaminase (hydroxymethylbilane synthase) as an internal control for normalization (Table 4). PCR amplicons were confirmed to be specific by size and melting curve analysis. After denaturing for 30 s at 95°C, amplification was performed by 40 cycles of 1 s at 94°C, 5 s at 62°C, and 20 s at 72°C. The real-time monitoring of the PCR reaction and precise quantification of the products in the exponential phase of the amplification was done by using the SYBR Green I format as described previously (Rodriguez-Antona et al., 2000; Perez et al., 2003). To ensure that specificity was high enough to discriminate between CYP3A4 and CYP3A7, we performed PCR and digested amplified DNA with HindIII, which recognizes only CYP3A4 amplicons. Agarose gel analysis revealed that almost 100% of the PCR product was HindIII digested.
Oligonucleotides used for quantitative RT-PCR
Extraction of Nuclear Proteins and Immunoblotting. Nuclear extracts from cultured hepatocytes were prepared as described previously (Andrews and Faller, 1991) and electrophoresed in an SDS-polyacrylamide gel (20 μg of protein/lane). Proteins were transferred to Immobilon membranes (Millipore Corporation, Billerica, MA), and sheets were incubated with a rabbit polyclonal antibody raised against the carboxyl terminus of C/EBPβ (Santa Cruz Biotechnology Inc., Santa Cruz, CA). After washing, blots were developed with horseradish peroxidase-labeled IgG, using an enhanced chemiluminescence kit (Amersham Biosciences UK, Ltd., Little Chalfont, Buckinghamshire, UK).
Chromatin Immunoprecipitation Assay. Cells were treated with 1% formaldehyde in phosphate-buffered saline buffer under gentle agitation for 10 min at room temperature to cross-link transcription factors to DNA. Thereafter, cells were collected by centrifugation, washed, resuspended in lysis buffer as described previously (Borras et al., 2003), and sonicated on ice for 10 steps of 10 s at 30% output in a Branson sonicator. Samples were centrifuged to clear the supernatants. DNA content was carefully measured by fluorescence with PicoGreen dye (Molecular Probes) and properly diluted to have an equivalent amount of DNA in all samples (input DNA). The immunofractionation of C/EBPβ-DNA complexes was performed by addition of 10 μg/ml C/EBPβ antibody and incubation at 4°C overnight on a 360° rotator (antibody-bound DNA fraction). For each cell preparation, an additional mock immunoprecipitation with no antibody or with a human β-actin antibody was performed (background DNA fraction). The inmunocomplexes were affinity-absorbed with 10 mg of protein A-Sepharose (prewashed with lysis buffer for 4 h at 4°C under gentle rotation) and collected by centrifugation (6500g, 1 min). The antibody-bound and background DNA fractions were washed as described previously (Borras et al., 2003). The cross-links were reversed by heating the samples at 65°C overnight. The DNA from bound, background, and input fractions was purified, diluted (1/10, bound and background fractions; 1/100, input fraction) and subjected to quantitative real-time PCR with a LightCycler instrument. Amplification was real-time monitored and allowed to proceed in the exponential phase, until fluorescent signals reached a significant value. Then, amplified DNA was analyzed by agarose gel electrophoresis. To check that the immunoprecipitation contains the –5953/–5667-bp fragment of the CYP3A4 5′-flanking region among the pool of DNA, DNA samples were amplified with specific primers flanking this region (Table 5). As positive and negative controls, the binding of C/EBPβ to the D element in the albumin promoter and to the CYP3A4 exon 2 was analyzed with primers specific to these targets (Table 5).
Oligonucleotides used for ChIP assay
Statistical Analysis. Each experiment was performed in several independent cell cultures as indicated. Each quantitative determination was done in triplicate. The results are expressed as the mean value ± S.D. Statistical significance was calculated by Student's t test.
Results
Transactivation of CYP3A4 by C/EBPβ-LAP through a Far Upstream 5′-Flanking Region (–5950 to –5663 bp). CYP3A4 is modulated by C/EBP factors (i.e., C/EBPβ-LAP, C/EBPβ-LIP, and C/EBPα) (Jover et al., 2002). Therefore, the presence of C/EBP regulatory element(s) within CYP3A4 promoter/enhancer sequences is expected. The occurrence of such element(s) was first investigated by reporter gene assays using deletions of the CYP3A4 5′-flanking region.
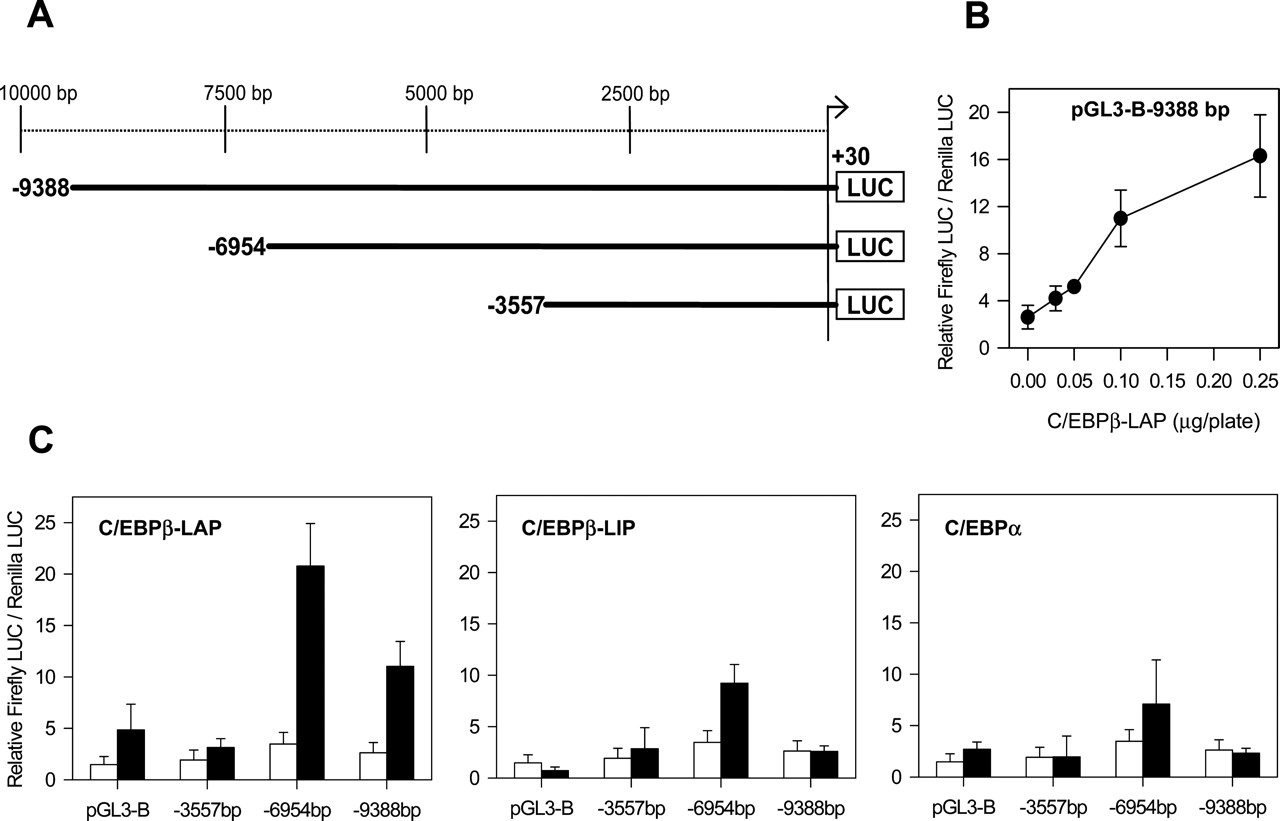
Three large fragments of the CYP3A4 5′-flanking region (+30 to –3557, –6954, and –9388 bp) were fused in frame to the firefly luciferase gene in the pGL3-Basic plasmid (Fig. 1A). Thereafter, the effect of C/EBP factors was investigated by cotransfection of reporter constructs with expression plasmids encoding for LAP, LIP, and C/EBPα, in the human cervix carcinoma cell line HeLa.
Reporter assays using the largest fragment (–9388 to +30 bp) showed a dose-dependent transactivation of CYP3A4 by C/EBPβ-LAP (Fig. 1B). A concentration of 0.1 μg of pAC-CEBPβ-LAP/plate caused already a significant 4.2-fold transactivation. Subsequent reporter analysis showed that although pGL3-B-9388 (–9388 to +30 bp) and pGL3-B-6954 (–6954 to +30 bp) responded to CEBPβ-LAP, the construct pGL3-B-3557 (–3557 to +30 bp) did not, suggesting that a C/EBPβ-responsive sequence is contained within the –3557 to –6954-bp region (Fig. 1C). Comparative analysis of the three C/EBP factors showed that C/EBPβ-LAP induces a more prominent response than C/EBPβ-LIP and C/EBPα (Fig. 1C).
To extend our observations to liver-derived cells, we conducted similar experiments in the human hepatic cell line HepG2. As Fig. 2A shows, C/EBPβ-LAP elicited again the most potent transactivating effect in the +30 to –6954-bp fragment. The average response in the hepatic cell line was higher than in HeLa cells (10.6-versus 6.0-fold increase, respectively). C/EBPβ-LAP caused again a more important transactivation than C/EBPβ-LIP and C/EBPα on the +30 to –6954-bp fragment (Fig. 2B).

Identification of a far upstream CYP3A4 5′-flanking region responsive to C/EBP factors. A, schematic chimeric CYP3A4-luciferase reporter gene constructs. The indicated positions are relative to the transcription start site. B, dose-response of pGL3-B-9388 bp to increasing concentrations of recombinant C/EBPβ-LAP in transfected HeLa cells. C, basal activity of CYP3A4-LUC constructs (□) and transactivation by C/EBP factors (▪) in HeLa cells. Chimeric CYP3A4-LUC reporter vectors or the promoterless pGL3-Basic (0.65 μg) were cotransfected with pAC-C/EBP expression vectors (0.1 μg), and with the plasmid pRL-CMV (0.08 μg) to normalize transfection efficiency. The insertless plasmid pAC/CMVpLpA was added to have a constant amount of expression vector. Cells were harvested 48 h after DNA transfection, and luciferase activity was assayed. Values represent firefly luciferase/Renilla-luciferase enzymatic activity ratios. Data represent the mean ± S.D. from at least four independent experiments performed in triplicate.
Transactivation of the smaller +30 to –3557-bp fragment by C/EBPβ-LAP was marked in HepG2 cells but insignificant in HeLa cells (4.9-versus 1.6-fold increase, respectively; Figs. 1C and 2A), suggesting that additional hepatic-specific C/EBP elements exist in this more proximal region. This effect could be caused through binding of C/EBPβ-LAP to previously characterized hepatic-specific C/EBPα sites located in the proximal CYP3A4 5′-flanking region (Rodriguez-Antona et al., 2003).
To more precisely locate the C/EBPβ-responsive element in the –3557 to –6954-bp region, sequential deletion fragments were generated and cloned into the pGL3-Promoter vector, which contains a simian virus 40 promoter upstream of the luciferase gene (Fig. 3). Reporter assays with 11 different chimeric constructs revealed an EcoRI-PvuII 590-bp fragment, which showed maximal response to C/EBPβ-LAP (∼17-fold increase upon transfection in HepG2 cells with 0.1 μg of C/EBPβ-LAP expression vector) (Fig. 3). After this observation, the response to C/EBPβ-LAP was investigated in two PCR-cloned fragments overlapping the EcoRI-PvuII sequence. We found that the C/EBPβ-LAP response sequence was located between bases –5950 and –5663 of the CYP3A4 5′-flanking region. This 288-bp enhancer sequence showed a 23.1-fold activation upon transfection of 0.1 μg of C/EBPβ-LAP (Fig. 3).
C/EBPβ-LIP Antagonizes C/EBPβ-LAP-Mediated Activation at the –5950/–5663 bp CYP3A4 5′-Flanking Region. C/EBPβ mRNA can produce at least three isoforms: 38-kDa LAP*, 35-kDa LAP, and 20-kDa LIP (Descombes and Schibler, 1991). The 20-kDa protein LIP is believed to function as a dominant negative regulator of full-length C/EBP proteins, because it lacks most of the transactivation domain but contains the DNA binding and dimerization domains. To investigate the possible role of LIP in CYP3A4 transcription, we performed gene reporter assays in HepG2 cells. We co-transfected pGL3-P-5950/-5663 with a constant amount of C/EBPβ-LAP and increasing doses of C/EBPβ-LIP expression vectors. The results depicted in Fig. 4 show a repressive effect of LIP on the transactivation exerted by LAP. An equivalent concentration of LIP was sufficient to attenuate the action of LAP by more than 50%, evidencing a potent competitive effect between LAP and LIP on the CYP3A4 distal sequence.

Transactivation of chimeric CYP3A4-LUC reporter gene constructs by C/EBPβ-LAP in human hepatoma HepG2 cells. A, chimeric CYP3A4-LUC reporter vectors or the promoterless pGL3-Basic (0.65 μg) were cotransfected with (▪) or without (□) pAC-C/EBPβ-LAP expression vector (0.1 μg) in HepG2 cells. B, transactivation of pGL3-B-6954 bp reporter plasmid by recombinant C/EBP factors in transfected HepG2 cells. Normalized luciferase activities were obtained as described in legend to Fig. 1. Data represent the mean ± S.D. from at least three independent experiments performed in triplicate.
Site-Directed Mutagenesis of Putative C/EBPβ Binding Sites in the –5950/–5663 5′-Flanking Region Reveals the Existence of Functional C/EBPβ-LAP-Responsive Elements. The –5950/–5663 5′-flanking region was analyzed for consensus C/EBPβ motifs by using the MatInspector software and TRANSFAC database. Four putative C/EBPβ binding sites with a matrix similarity >92% were identified (Fig. 5A). To assess the functional significance of these putative C/EBPβ binding sites, we constructed mutants with base substitutions in the core sequence of the C/EBPβ binding sites and performed reporter analysis. The –5950/–5663 bp CYP3A4 wild-type sequence showed a 31.4-fold increase upon LAP transfection. Mutations of the C/EBPβ motifs 1 or 4 caused a significant decrease in the transactivating effect of LAP by 34 and 61%, respectively, whereas mutations of the C/EBPβ motifs 2 or 3 did not cause a statistically significant decrease (Fig. 5B). The more important role of sites 1 and 4 was confirmed by generating deletion mutants lacking several nucleotides of the C/EBPβ consensus sequences. Deletions in sites 1 and 4 confirmed a significant decrease in the transactivating effect of LAP by 32 and 56%, respectively (Del 1 and Del 4; Fig. 5B). To assess whether these sites are working individually or in an integrated pattern, we generated constructs with multiple disruptions of C/EBPβ elements. Sites 2 and 3 are overlapping C/EBPβ binding sites and their role is only evidenced when both sites are simultaneously mutated, suggesting that these two sites may function as redundant alternative sites (Del 2 + 3; Fig. 5B). Therefore, double mutations 2 + 4 and 3 + 4 did not show significant differences with single mutation 4, thus demonstrating that sites 2 and 3 are alternative and need to be mutated simultaneously to abolish their activity. Double mutations 1 + 4 caused a decrease in transactivation by LAP that was more pronounced than the reduction observed with single mutations 1 or 4, although the decrease measured with this double mutation was not proportional to the combined effects of mutation 1 plus mutation 4. Finally, simultaneous disruption of the four C/EBPβ binding elements abolished LAP transactivation to almost background levels (i.e., pGL3-P vector; Fig. 5B), suggesting that these four elements probably account for the major part of the response of the CYP3A4 –5.95-kb enhancer region to LAP.
Endogenous C/EBPβ Binds to the –5950/–5663-bp CYP3A4 5′-Flanking Region in Human Hepatocytes. Gene reporter assays used to examine the role of C/EBPβ in the regulation of CYP3A4 expression are based on recombinant C/EBPβ proteins and chimeric reporter genes. To reinforce the significance of our previous findings by reporter assay, we investigated the occurrence of a direct interaction of endogenous C/EBPβ with the CYP3A4 gene. This was explored by chromatin immunoprecipitation (ChIP) analysis in cultured human hepatocytes, which efficiently express C/EBPβ-LAP. Cross-linked chromatin was immunoprecipitated with an anti-C/EBPβ antibody, and the presence of the CYP3A4 enhancer sequence was analyzed by PCR amplification using primers flanking the –5950/–5663-bp region. As shown in Fig. 6, the antibody-bound DNA fraction contained the –5950/–5663-bp CYP3A4 5′-flanking region. The PCR fragments amplified prove that endogenous C/EBPβ binds in vivo to the far upstream CYP3A4 enhancer (Fig. 6).
As a positive control, we examined the binding of C/EBPβ to the albumin promoter. It has been previously shown that C/EBPβ-LAP strongly activates albumin gene transcription by binding to the promoter D element, a high-affinity binding site for C/EBP proteins (Friedman et al., 1989; Akira et al., 1990; Descombes et al., 1990; Poli et al., 1990). Therefore, the results shown in Fig. 6 confirmed the binding of C/EBPβ to the albumin D element. As a negative control, PCR amplifications were also performed with primers flanking the CYP3A4 exon 2, a region that has no predicted C/EBP binding sites. Immunoprecipitations run with no antibody or with an unrelated β-actin antibody (background DNA fraction) gave no signals or marginal signals, whereas amplification performed from input DNA confirmed that differences were not caused by variations in the amounts of immunoprecipitated chromatin (Fig. 6).
C/BEPβ-LIP Binds to the –5950/–5663-bp CYP3A4 5′-Flanking Region and Down-Regulates CYP3A4 Expression. C/BEPβ-LIP has been suggested to act as a repressor of CYP3A4 transcription. To probe a direct interaction of LIP with CYP3A4 5′-flanking region, we transfected human hepatocytes with a recombinant adenovirus for efficient expression of C/BEPβ-LIP and performed ChIP assay. It is interesting that our results demonstrate increased binding of C/BEPβ to the –5950/–5663-bp CYP3A4 5′-flanking region (Fig. 7B) when recombinant LIP is expressed in the cells (Fig. 7A). In parallel, we analyzed whether recruitment of LIP to the CYP3A4 far upstream enhancer resulted in diminished CYP3A4 expression. Analysis of CYP3A4 mRNA levels by quantitative real-time RT-PCR showed that LIP causes dose-dependent CYP3A4 down-regulation in human hepatocytes (Fig. 7C). Thus, our data suggest that an increased LIP expression results in LIP binding to the –5950/–5663-bp CYP3A4 5′-flanking region, leading consequently to a lower CYP3A4 transcriptional activity.

Identification of 288 bp sequence between –5950 and –5663 bp in the CYP3A4 5′-flanking region responsive to C/EBPβ-LAP. Several deletion fragments from the –3557 to –6954-bp region were obtained and cloned into the pGL3-Promoter vector as described under Materials and Methods. The position relative to the transcription start site and the restriction enzymes used are indicated. The resulting CYP3A4-LUC reporter gene constructs or the enhancerless pGL3-Promoter (0.65 μg) were cotransfected with (▪) or without (□) pAC-C/EBPβ-LAP expression vector (0.1 μg) in HepG2 cells. Normalized luciferase activities were obtained as described in legend to Fig. 1. Data represent the mean ± S.D. from at least three independent experiments performed in triplicate.

Effect of C/EBPβ-LIP on C/EBPβ-LAP mediated-activation at the –5950/–5663 bp CYP3A4 5′-flanking region. Plasmid pGL3-P-5950/-5663 or pGL3-promoter was cotransfected with a constant amount of pAC-C/EBPβ-LAP and increasing doses of C/EBPβ-LIP as indicated in micrograms per plate. Normalized luciferase activities were obtained as described in legend to Fig. 1. Values were expressed as fold-increase over the pGL3-promoter response. Data represent the mean ± S.D. from at least three independent experiments performed in triplicate.

Site-directed mutagenesis of predicted C/EBPβ binding sites in the –5950/–5663-bp CYP3A4 5′-flanking region. A, nucleotide sequence from –5953 to –5667 bp of the CYP3A4 gene. Putative C/EBPβ binding sites with a matrix similarity >92% (as determined with the MatInspector software and TRANSFAC database) are underlined. Sequences deleted to abolish putative C/EBPβ-responsive motifs are boxed and their names indicated. B, transactivation of chimeric CYP3A4-LUC reporter gene constructs by recombinant C/EBPβ-LAP. Plasmids containing base substitution mutations or deletions of C/EBPβ binding sites, or containing the wild-type sequence (wt) (0.65 μg), were cotransfected with pAC-C/EBPβ-LAP expression vector (0.1 μg) in HepG2 cells. Mutants were derived from plasmid pGL3-P-5950/-5663 as described under Materials and Methods. Normalized luciferase activities were obtained as described in legend to Fig. 1. Values were expressed as fold-increase over luciferase activity in the absence of recombinant C/EBPβ-LAP. Data represent the mean ± S.D. from three to seven independent experiments performed in triplicate. ***, p < 0.001; **, p < 0.01, relative to pGL3-P-5950/-5663 wt.
C/BEPβ-LAP and LIP Interact in the –5.95-kb Upstream Enhancer of CYP3A4 and Modulate CYP3A4 Expression Both in Hepatic and Nonhepatic Cells. C/EBPβ-LAP is the predominant C/EBPβ isoform expressed by adult hepatocytes in healthy liver (Descombes et al., 1990), whereas C/EBPβ-LIP is expressed in particular circumstances such as after LPS administration or partial hepatectomy (Rana et al., 1995; An et al., 1996; Hsieh et al., 1998; Welm et al., 2000). The previous data have strongly suggested that both LAP and LIP are able to regulate CYP3A4 expression through binding to the identified –5.95-kb CYP3A4 upstream enhancer. To unequivocally prove this possibility, we transfected nonhepatic HeLa cells with recombinant adenoviruses encoding C/EBPβ-LAP and -LIP and performed ChIP assays. In nontransfected HeLa cells, C/EBPβ binding to the –5950/–5663-bp CYP3A4 5′-flanking region was not detected (Fig. 8B). However, when LAP or LIP was expressed (Fig. 8A), a dose-dependent increase in binding of C/BEPβ proteins was found (Fig. 8B). Recruitment of C/EBPβ-LAP to the CYP3A4 enhancer resulted in an important increase in CYP3A4 mRNA level (Fig. 8C), which is in agreement with the activating role of this transcription factor. LIP binding to the CYP3A4 enhancer, on the contrary, did not change CYP3A4 basal levels (Fig. 8C), which are already extremely low in HeLa cells. We found similar results when recombinant C/EBPβ-LAP and LIP proteins were expressed in human hepatoma HepG2 cells (Fig. 8, D and E), suggesting a key role for C/EBPβ proteins in modulating CYP3A4 expression irrespective of other tissue-specific factors.

ChIP analysis of C/EBPβ binding to the –5950/–5663-bp CYP3A4 5′-flanking region in human hepatocytes. Formaldehyde cross-linked chromatin from 48-h cultured cells was incubated with an antibody raised to the C terminus of human C/EBPβ. Immunoprecipitated DNA (antibody-bound DNA fraction, BN) was analyzed by quantitative real-time PCR with primers specific to the –5950/–5663-bp enhancer region of CYP3A4 (CYP3A4-ER). As positive control, binding of C/EBPβ to the D element in the albumin promoter was analyzed (ALB-PR). As negative control, PCR amplifications with primers flanking the CYP3A4 exon 2 was performed (CYP3A4-Exon2). Parallel PCR reactions were performed with input DNA (input DNA fraction, IN) and with mock-immunoprecipitated DNA (background DNA fraction, BK). Marker, 100-bp DNA ladder.

Expression of C/BEPβ-LIP in human hepatocytes, binding to the CYP3A4 enhancer region at –5.95 kb and effect on CYP3A4 expression. Human hepatocytes were transduced with increasing doses of Ad-LIP (2.5, 10, 25, or 60 m.o.i.) or Ad-pAC (control, insertless adenovirus, 60 m.o.i.), and 48 h later, the levels of expressed LIP protein were analyzed by immunoblotting (A), and the mRNA concentrations of CYP3A4 were measured by quantitative RT-PCR (C). Data represent the mean ± S.D. from three independent cultures. In parallel, ChIP analysis (as described in legend to Fig. 6) was performed in control cells or in hepatocytes transfected with Ad-LIP (25 m.o.i.) to examine the binding of C/EBPβ-LIP in the CYP3A4 enhancer region (B).
Finally, we investigated whether an increase in intracellular LIP levels could negatively affect the binding of LAP to the distal CYP3A4 enhancer element. It has been previously shown that C/EBPβ isoforms LAP and LIP are able to heterodimerize and bind to cognate DNA elements in electromobility shift assays. However, its occurrence in intact living cells and chromatin-condensed genes remained to be demonstrated. We transfected HeLa cells with a constant amount of Ad-LAP and increasing concentrations of Ad-LIP and performed ChIP assays. The results demonstrated that the overall binding of C/EBPβ proteins to the distal CYP3A4 element was not impaired when LIP expression is increased in the cells (Fig. 9A). Parallel analysis of CYP3A4 mRNA confirmed the activating effect of LAP and the repressive effect of coexpressed LIP (Fig. 9B), suggesting that when LIP concentration increases, LAP-LIP heterodimers and LIP-LIP homodimers are probably formed and bound to the distal CYP3A4 enhancer. Binding of LIP, which lacks a transactivation domain, diminishes CYP3A4 transcription and leads to CYP3A4 down-regulation.
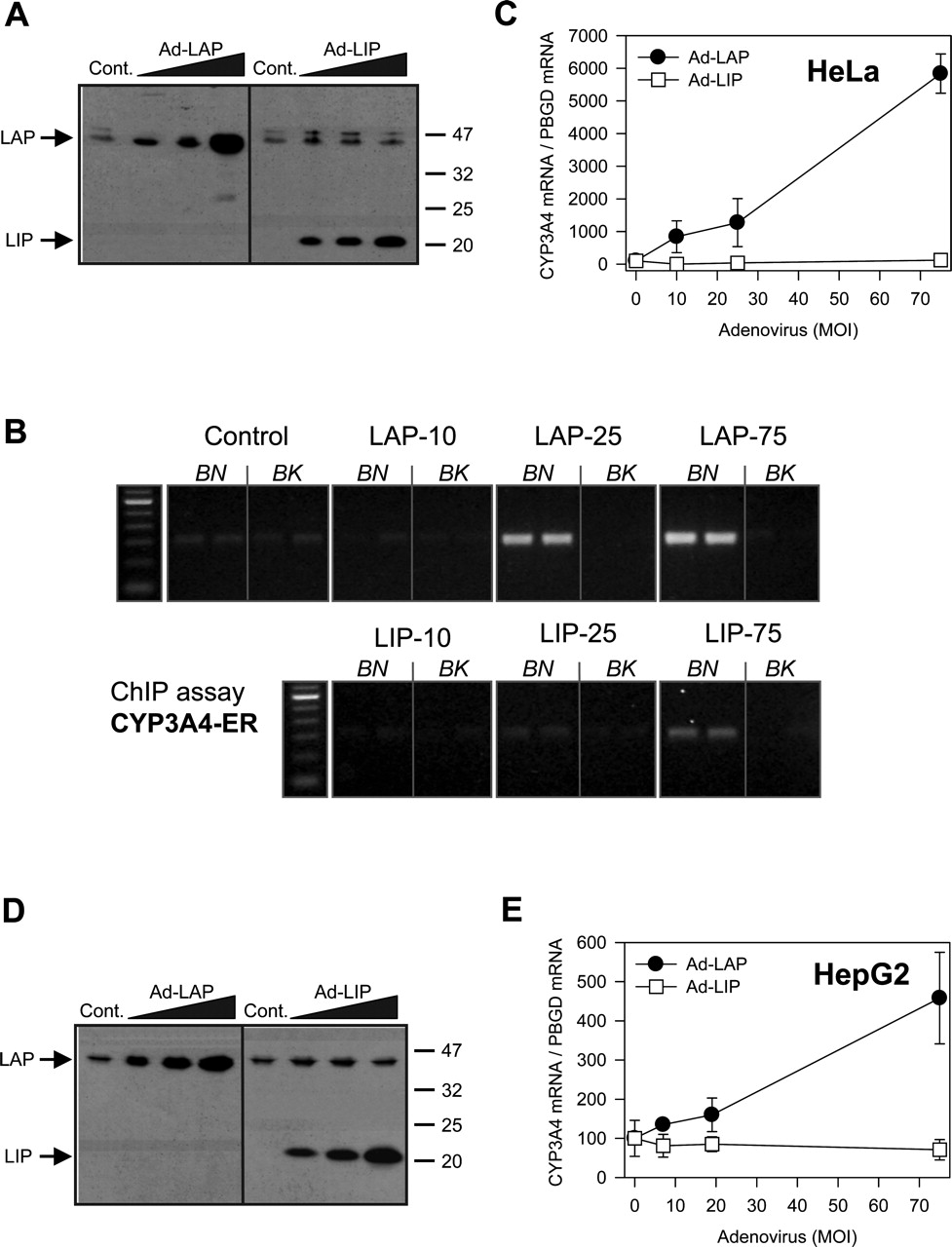
Expression of LAP and LIP in cell lines, binding to the CYP3A4 enhancer region at –5.95 kb and effect on CYP3A4 expression. Hepatic and nonhepatic cell lines were transduced with increasing doses of Ad-LAP or Ad-LIP (10, 25, or 75 m.o.i.) and 48 h latter, the levels of recombinant protein were analyzed by immunoblotting (A, HeLa; D, HepG2), and the mRNA concentrations of CYP3A4 measured by quantitative RT-PCR (C, HeLa; E, HepG2). Data represent the mean ± S.D. from three independent cultures. Binding of expressed LAP and LIP to the –5950/–5663-bp CYP3A4 5′-flanking region was assessed in HeLa cells by ChIP analysis as described in the legend to Fig. 6B).

Coexpression of LAP and LIP in HeLa cells, binding to the CYP3A4 enhancer region at –5.95 kb and effect on CYP3A4 expression. A, 1 × 107 cells were transduced with Ad-LAP and/or Ad-LIP as indicated, and 48 h later, the binding of expressed C/EBPβ to the –5950/–5663-bp CYP3A4 5′-flanking region was assessed by ChIP analysis as described in legend to Fig. 6. B, in parallel plates, the mRNA concentrations of CYP3A4 were measured by quantitative RT-PCR. Data represent the mean ± S.D. from three independent cultures.
Role of LAP and LIP in the Regulation of CYP3A4 in the Context of Induction by Xenobiotics and Repression by Inflammatory Cytokines. Recent reports have demonstrated that the inducibility of CYP3A4 by xenobiotics can be strongly repressed by cytokines released during inflammatory responses (Pascussi et al., 2000), and because inflammatory responses can also induce LIP expression, we investigated whether an increase in LIP levels could be involved in repressing CYP3A4 induction during inflammation. We transfected human hepatocytes with increasing doses of Ad-LIP and incubated with rifampicin for 48 h. Then, we performed real-time RT-PCR to evaluate the dose-dependent effect of LIP on CYP3A4 mRNA induction. Results in Fig. 10A demonstrate that transfection of LIP caused a significant dose-dependent decrease in both basal and rifampicin-induced CYP3A4 levels. The relative induction (fold-increase) of CYP3A4 by rifampicin was not completely blunted by LIP but the extent of induction was significantly reduced. We next investigate whether this effect was also observed in a reporter assay system. HepG2 cells were cotransfected with pGL3-B-9388 (–9388 to +30 bp), and hPXR and LIP expression vectors. Then, rifampicin was added and the effect on reporter activity evaluated. We observed again that LIP was able to dose dependently decrease PXR-mediated transactivation (Fig. 10B), suggesting that binding of LIP to CYP3A4 binding sites can partially inhibit the activation of transcription that PXR elicits through CYP3A4 promoter and enhancer elements.
Inflammatory responses can trigger several mechanisms leading to cytochrome P450 down-regulation. We postulate that one additional mechanism could be the induction of LIP and repression of CYP3A4 through the –5.95-kb enhancer region. To reinforce the relevance of this alternative mechanism in the perspective of inflammation, we treated hepatocytes with the proinflammatory cytokine interleukin-6 and assessed whether transfected LAP can rescue CYP3A4 down-regulation. In human hepatoma cells, interleukin-6 caused a modest, although statistically significant, CYP3A4 down-regulation, and Ad-LAP transfection was able to abolish this negative effect (Fig. 11). In cultured human hepatocytes, forced expression of LAP was able to partially overcome the negative effect of interleukin-6 on CYP3A4 expression. In control hepatocytes, interleukin-6 decreased CYP3A4 mRNA levels from 100 ± 26 to 31 ± 29 (69% decrease), whereas it reduced CYP3A4 mRNA from 160 ± 40 to 117 ± 31 (27% decrease), in LAP-transfected human hepatocytes (n = 2–3 independent cultures). The fact that LAP can partially overcome CYP3A4 down-regulation by interleukin-6 substantiates the hypothesis that LIP could play a relevant role in this event. However, this does not exclude other simultaneous mechanism also contributing to CYP3A4 repression by cytokines.
Discussion
CYP3A4 is both a constitutive and inducible hepatic gene, and its regulation emerges as a complex issue with numerous transcription factors interacting with multiple promoter/enhancer elements spread through more than 11 kb upstream of the transcription start site.
The induction of CYP3A4 by “activated” nuclear receptors, such as PXR and CAR, depends on two specific responsive elements. The first is a proximal everted repeat sequence (ER6), located at –160 bp (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998), and the second is a distal xenobiotic-responsive enhancer module, located between –7.8 and –7.2 kb upstream of the CYP3A4 transcription start site (Goodwin et al., 1999). In the distal enhancer module, at –7.7 kb, the orphan nuclear receptor HNF-4α is capable of binding to a direct repeat sequence to coordinate nuclear receptor-mediated response to xenobiotics (Tirona et al., 2003).

Effect of C/EBPβ LIP in the induction of CYP3A4 by rifampicin. A, cultured human hepatocytes were transfected with increasing doses of Ad-LIP and 24 h latter cells were exposed to 50 μM rifampicin (▪) or DMSO (□) for an additional 48 h. CYP3A4 mRNA levels were analyzed by quantitative RT-PCR. Data represent the mean ± S.D. from three to four independent cultures. ***, p < 0.001; **, p < 0.01; *, p < 0.05, relative to noninduced hepatocytes. B, human hepatoma HepG2 cells were cotransfected with pGL3-B-9388 bp or pGL3-Basic, a constant amount of pcDNA-hPXR and increasing doses of pACC-C/EBPβ-LIP as indicated. The insertless plasmid pAC/CMVpLpA was added to have a constant amount of expression vector. Twenty-four hours after transfection, cells were exposed to 10 μM rifampicin (▪) or DMSO (□) for an additional 48 h. Normalized luciferase activities were obtained as described in legend to Fig. 1. Values were expressed as fold-increase over the pGL3-Basic background level. Data represent the mean ± S.D. from two to three independent experiments performed in triplicate.
The regulation of the constitutive (basal) CYP3A4 expression in the liver is much less known. A region between –10.5 and –11.4 kb that functions as a constitutive liver enhancer module (constitutive liver enhancer module 4) has been identified by Matsumura et al. (2004). This region binds multiple transcription factors, including HNF-1, HNF-4, activator protein-1, and USF1, and is postulated to be an important site for CYP3A4 constitutive transcription. Previous studies from our laboratory have also demonstrated the involvement of C/EBPα in the constitutive transcription of CYP3A4 through three proximal elements at –121, –1393, and –1659 bp (Rodriguez-Antona et al., 2003). Moreover, C/EBPα-mediated transactivation is synergistically activated in hepatic cells by HNF-3γ, which binds at –1718 bp (Rodriguez-Antona et al., 2003). However, not quantitatively important transactivation by C/EBPα was observed when large 5′-flanking regions of the CYP3A4 gene were analyzed (Fig. 1C). This could suggest the existence of other distal regulatory element(s) that would mask the positive effects of C/EBPα on the proximal promoter sites. This suggestion is also supported by other studies that show that activation by C/EBPα decreases when 5′ deletion fragments of the CYP3A4 promoter include more than 160 bp upstream of the transcription start site (Ourlin et al., 1997; Rodriguez-Antona et al., 2003).
Starting from the hypothesis that other members of the C/EBP family could also be involved in the regulation of CYP3A4 gene, we conducted a series of experiments to explore the feasibility of this idea. We have proven the existence of a new distal 288-bp enhancer site at –5.95 kb in the CYP3A4 gene, where C/EBPβ-LAP binds and activates transcription, whereas the truncated form, C/EBPβ-LIP, antagonizes LAP activity and causes gene repression, thus providing a mechanistic explanation for the changes in CYP3A4 expression occurring in various physiopathological states (e.g., inflammation). The functional significance of this regulatory site could be demonstrated by combining chromatin immunoprecipitation, adenoviral expression of C/EBPβ isoforms, and analysis of CYP3A4 expression in human hepatocytes and cell lines.
The fact that LIP acts as a negative regulator of CYP3A4 is likely to have clinical significance. Increased nuclear expression of the transcriptional repressor LIP occurs after the administration of LPS or partial hepatectomy (Diehl and Yang, 1994; Rana et al., 1995; An et al., 1996; Hsieh et al., 1998; Welm et al., 2000). Moreover, it has also been shown that certain proinflammatory cytokines such as IL-6, IL-1, or tumor necrosis factor-α induce LIP expression (Baumann et al., 1992; Iraburu et al., 2000; Jover et al., 2002). Therefore, it is tempting to suggest that CYP3A4 down-regulation in many pathological states, in particular those involving a host inflammatory response (e.g., bacterial and viral infection, trauma, burn injury, tissue necrosis, and autoimmune disease), could be largely caused by LIP up-regulation and binding to the –5.95-kb distal CYP3A4 enhancer.
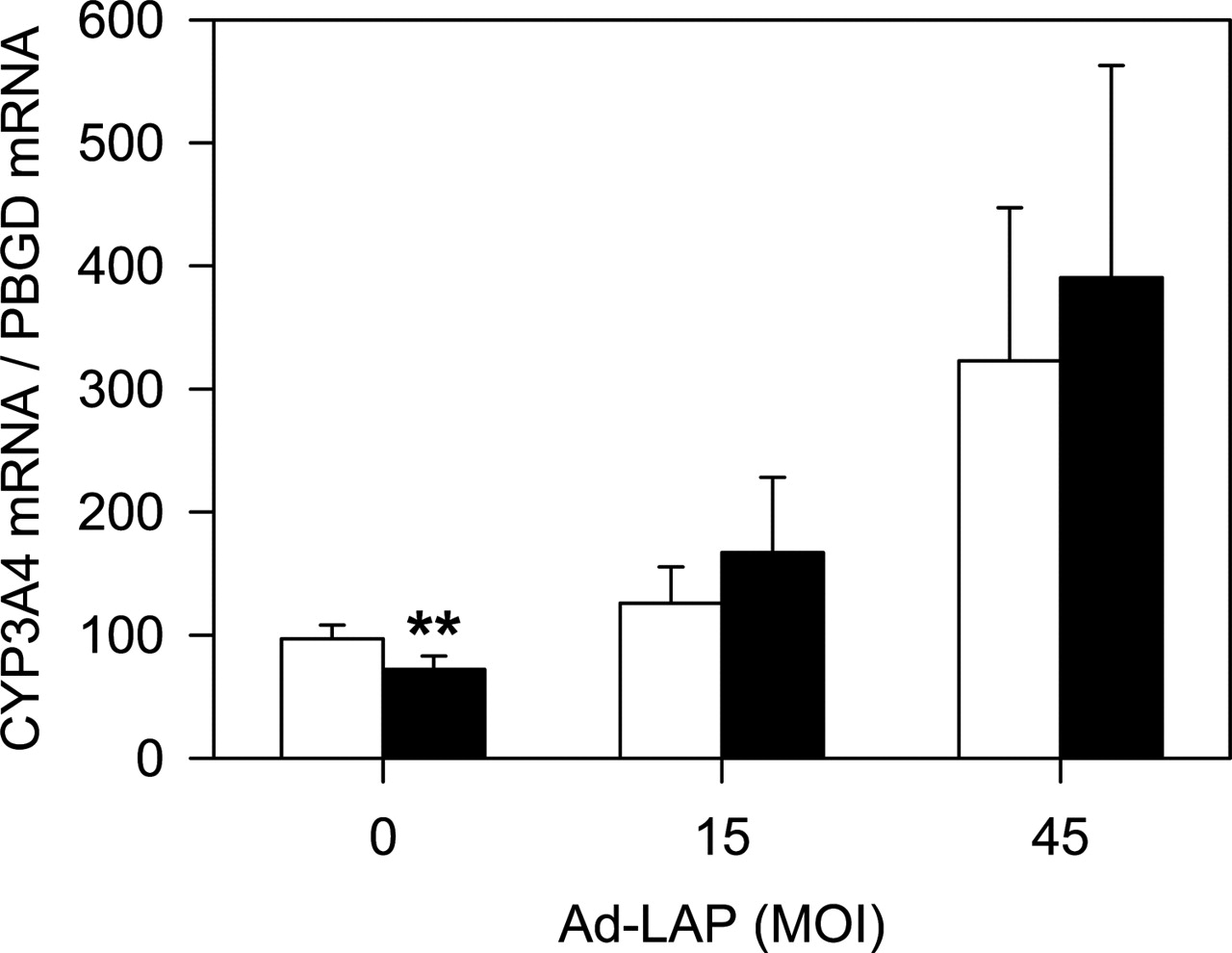
Effect of C/EBPβ LAP in the down-regulation of CYP3A4 by interleukin-6. HepG2 cells were transfected with two different doses of Ad-LAP and exposed to 100 U/ml interleukin-6 (▪) or vehicle (□) for 40 h. CYP3A4 mRNA levels were measured by quantitative RT-PCR. Data represent the mean ± S.D. from three to five independent cultures.
Several alternative mechanisms could also contribute to CYP3A4 down-regulation during cytokine release. It has been shown that tumor necrosis factor-α represses CYP1A1 via redox regulation of nuclear factor-1 (Morel and Barouki, 1998), whereas interleukin 2 down-regulates rat CYP2C11 and CYP3A2 expression via induction of the proto-oncogene c-myc (Tinel et al., 1995), and interleukin 1 inhibits CYP2C11 transcription via binding of nuclear factor-κB to a low-affinity binding site in its promoter (Iber et al., 2000). Nuclear receptors have also been shown to be relevant factors for cytochrome P450 down-regulation, because decreased expression of PXR and CAR was observed after cytokine incubation (Pascussi et al., 2000). In this context, CAR repression is caused via nuclear factor-κB activation followed by interference with the enhancer function of a glucocorticoid response element in the CAR promoter (Assenat et al., 2004).
Our results could also shed some light to explain the high phenotypic variability of CYP3A4 expression. To date no mechanism has been described that could account for such differences. Human variation in CYP3A4 activity is more likely to be caused by regulatory variability rather than structural polymorphism in the CYP3A4 gene (Schuetz, 2004). C/EBPβ protein levels are known to change in the liver not only under pathological situations (e.g., inflammatory response) but also in response to diet and hormones. For example, a high carbohydrate diet reduces C/EBPβ expression in mice and rats, whereas C/EBPβ levels are induced in the livers of diabetic rats (Bosch et al., 1995). Moreover, it is worth noting that insulin increases protein levels of LIP, which concomitantly replaces LAP on target promoters, leading to repression of gene expression (Duong et al., 2002). A similar behavior has been described for CYP3As, which show reduced expression in rats under high sucrose diet (Peters and Teel, 2003), increased expression in rats made diabetics by streptozotocin treatment (Barnett et al., 1990), and down-regulation by insulin administration (Yamazoe et al., 1989; Barnett et al., 1990).
Transcription factors (LAP and LIP) acting on regulatory DNA sequences as the one here described may effectively control CYP3A4 constitutive and inducible expression and ultimately contribute to the different CYP3A4 phenotypes in the human population. Finally, an additional factor for CYP3A4 variability would be the existence of functional single nucleotide polymorphisms in the C/EBPβ binding sites within the distal CYP3A4 enhancer region. This possibility, although not explored yet, has been recently shown to be feasible (Wu et al., 2003).
Acknowledgments
We thank E. Belenchon, C. Corchero, and C. Guzmán for expert technical assistance. Dr. O. Burk kindly provided pcDNA3-hPXR expression vector.
Footnotes
-
This research was supported in part by grants from the Fondo de Investigacion Sanitaria (00/1037 and 00/1038) and Ministry of Science and Technology (SAF 2003-09353). C.P.M.-J. received a predoctoral grant from the Consellería de Educación Ciencia y Cultura, Generalitat Valenciana.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.104.008169.
-
ABBREVIATIONS: PXR, pregnane X receptor; CAR, constitutive androstane receptor; LEFT, liver-enriched transcription factor; HNF, hepatocyte nuclear factor; C/EBP, CCAAT/enhancer-binding protein; LAP, liver activating protein; LIP, liver inhibitory protein; IL, interleukin; LPS, lipopolysaccharide; kb, kilobase(s); bp, base pair(s); PCR, polymerase chain reaction; Ad, recombinant adenovirus; m.o.i., multiplicity of infection; CMV, cytomegalovirus; ChIP, chromatin immunoprecipitation; RT-PCR, reverse transcription-polymerase chain reaction.
- Received October 19, 2004.
- Accepted March 18, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}