


Abstract
The purpose of the present study was to investigate the role of multidrug resistance-associated protein 4 (MRP4) in the tubular secretion of cephalosporin antibiotics. Most of the injectable cephalosporins have an inhibitory effect on the ATP-dependent uptake of [3H]dehydroepiandrosterone sulfate by membrane vesicles expressing hMRP4, whereas cephaloridine, cefsulodin, and cefepime do not. Aminocephalosporins have a weak inhibitory effect. Significant ATP-dependent transport of ceftizoxime (Km, 18 μM), cefazolin (Km, 80 μM), cefotaxime, and cefmetazole has been observed only in the membrane vesicles expressing hMRP4. Ceftizoxime and cefazolin were given by a constant intravenous infusion to wild-type and Mrp4–/– mice. The steady-state plasma concentrations of ceftizoxime and cefazolin were unchanged in Mrp4–/– mice. The urinary recovery of ceftizoxime was significantly reduced in Mrp4–/– mice, whereas it was unchanged for cefazolin. The kidney-to-plasma concentration ratio of ceftizoxime and cefazolin was increased 2.0- and 2.7-fold in Mrp4–/– mice, respectively; thus, the renal clearance with regard to the kidney concentration was reduced in Mrp4–/– mice, to 7.5 and 34% of the corresponding control values, respectively. These results suggest that Mrp4 is involved in the tubular secretion of ceftizoxime and cefazolin, in concert with basolateral uptake transporters.
Cephalosporins are one of the most important groups of antibiotics, and inhibit synthesis of the bacterial peptidoglycan cell wall. They have a cephem nucleus with various side-chains at the 3 and 7 positions of the β-lactam and dihydrothiazine rings, respectively. They are currently classified into four generations based on 1) the general features of their antimicrobial activities and 2) their stability to hydrolysis by β-lactamase. Most cephalosporins are excreted into the urine in unchanged form; only a few cephalosporins, such as cefoperazone, cefpiramide, and cefodizime are excreted predominantly into the bile (Petri, 2006). In the kidney, active secretion by the proximal tubules, together with glomerular filtration, has been suggested to be the renal clearance mechanism of cephalosporins. In particular, inhibition of renal elimination of cephalosporins by probenecid, a well known inhibitor of organic anion transporters, suggests that multispecific organic anion transporters play a major role in their renal elimination (Brown, 1993; Lepsy et al., 2003). Indeed, in vitro transport studies using cDNA-transfected cells or cRNA-injected oocytes have shown that cephalosporins are substrates of renal basolateral multispecific organic anion transporters (OAT1 and OAT3) (Jariyawat et al., 1999; Jung et al., 2002; Uwai et al., 2002; Ueo et al., 2005), and probenecid is a potent inhibitor of these transporters (Tahara et al., 2005). Because cephalosporins are efficiently transported by hOAT3, Ueo et al. (2005) speculated that hOAT3 plays a major role in the renal uptake of cephalosporins.
Because of the hydrophilic nature of cephalosporins, transporters will be involved in the luminal efflux process for efficient directional transport into the urine from the blood. Facilitated diffusion has been suggested as a mechanism for the luminal efflux of cephalosporins. Saturable uptake of cefixime by brush-border membrane (BBM) vesicles exhibits membrane-voltage dependence and is inhibited by probenecid and p-aminohippurate (Tamai et al., 1988). Because benzylpenicillin, cephalothin, and cephradine exhibit trans-stimulation of the efflux of cefixime from the BBM vesicles, the transporter also accepts these compounds as substrates. Renal clearance of aminocephalosporins exhibit nonlinearity that increases as the plasma concentrations increased (Garcia-Carbonell et al., 1993; Granero et al., 1994). This can be explained by saturation of reabsorption in the kidney. The uptake of aminocephalosporins by BBM is pH-dependent, and dipeptide transporters (PEPT1 and PEPT2) have been considered to be responsible for the reabsorption of aminocephalosporins in the kidney (Boll et al., 1996; Terada et al., 1997).
During preliminary studies, we found that some cephalosporins are substrates of an ATP binding cassette transporter, multidrug resistance-associated protein 4 (MRP4/ABCC4). MRP4 is characterized by its broad substrate specificity, and cumulative studies have demonstrated that cAMP, cGMP, p-aminohippurate, urate, dehydroepiandrosterone sulfate, methotrexate, prostaglandins, estradiol-17β-d-glucuronide, and adefovir are MRP4 substrates (Schuetz et al., 1999; van Aubel et al., 2002, 2005; Reid et al., 2003; Zelcer et al., 2003). In addition, glutathione modulates the substrate recognition of MRP4, and MRP4 accepts taurocholate as a substrate only in the presence of reduced glutathione or its S-methyl derivative, which lacks reducing activity (Rius et al., 2003). MRP4 is abundantly expressed in the kidney (Maher et al., 2005; Nishimura and Naito, 2005), where it is localized on the BBM of the proximal tubules (van Aubel et al., 2002; Leggas et al., 2004); thus, it has been considered to play an important role in the tubular secretion of drugs. We have found Mrp4 to be involved in the tubular secretion of diuretics (hydrochlorothiazide and furosemide) and antiviral drugs (adefovir and tenofovir) (Hasegawa et al., 2007; Imaoka et al., 2007).
The purpose of the present study was to elucidate the role of MRP4 in the tubular secretion of cephalosporins in the kidney. The chemical structure of the cephalosporins used in this study are shown in Fig. 1. In vitro transport study using membrane vesicles expressing hMRP4 was carried out to show that these cephalosporins are MRP4 substrate. In addition, to obtain direct evidence, in vivo pharmacokinetic parameters related to the renal elimination of the two cephalosporins, ceftizoxime and cefazolin, were compared in wild-type and Mrp4–/– mice.

Structures of ceftizoxime, cefazolin, cefmetazole, and cefotaxime
Materials and Methods
Materials. Ceftizoxime was a gift from Kowa Co., Ltd. (Tokyo, Japan). Other unlabeled cephalosporins (cefazolin, cefotaxime, and cefmetazole) were purchased from Sigma-Aldrich (St. Louis, MO). [14C]Inulin (8 mCi/mmol) and [3H]dehydroepiandrosterone sulfate (DHEAS) (74.0 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). All other chemicals and reagents were of analytical grade and readily available from commercial sources.
Animals. Female Mrp4–/– and wild-type BL6-129 mice (12–16 weeks old) were used in the present study. Mrp4 knockout mice were established previously (Assem et al., 2004; Leggas et al., 2004). Mrp4–/– mice are fertile and exhibit no physiological abnormalities. The hepatic expression of Sult2a1 is down-regulated (Assem et al., 2004), whereas the mRNA expression of Oat1, Oat3, Mrp2, and Bcrp is unchanged in the kidney of Mrp4–/– mice (Imaoka et al., 2007). All animals were maintained under standard conditions with a dark-light cycle and were treated humanely. Food and water were available ad libitum. The studies reported in this manuscript were carried out in accordance with the guidelines provided by the Institutional Animal Care Committee (Graduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo, Japan).
Cell Culture. HEK293 cells were grown in low-glucose Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Sigma-Aldrich), 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C with 5% CO2 at 95% humidity.
Construction and Infection of Recombinant Adenovirus and Membrane-Vesicle Preparation. Membrane vesicles were prepared from HEK293 cells infected with recombinant adenovirus harboring hMRP4 and GFP gene according to the published method (Hasegawa et al., 2007; Imaoka et al., 2007). For preparation of the isolated membrane vesicles, HEK293 cells cultured in a 15-cm dish were infected by recombinant adenovirus (multiplicity of infection of 5). Forty-eight hours after the infection, the membrane vesicles were isolated from 1 to 2×108 cells. In brief, cells were diluted 40-fold with hypotonic buffer (1 mM Tris-HCl and 0.1 mM EDTA, pH 7.4, at 37°C) containing protease inhibitors (2 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 1 μg/ml pepstatin, and 5 μg/ml aprotinin). After gentle stirring for 1 h on ice, the cell lysates were centrifuged at 100,000g for 30 min at 4°C. The resulting pellets were suspended in 10 ml of isotonic TS buffer (10 mM Tris-HCl and 250 mM sucrose, pH 7.4 at 4°C) and homogenized using a Dounce B homogenizer (glass/glass, tight pestle, 30 strokes). The crude membrane fraction was layered on top of a 38% (w/v) sucrose solution in 5 mM Tris-HEPES, pH 7.4, at 4°C, and centrifuged in a rotor centrifuge (SW41; Beckman Coulter, Fullerton, CA) at 280,000g for 60 min at 4°C. The turbid layer at the interface was collected, diluted to 23 ml with TS buffer, and centrifuged at 100,000g for 30 min at 4°C. The resulting pellet was suspended in 400 μl of TS buffer. Vesicles were formed by passing the suspension 30 times through a 27-gauge needle using a syringe. The membrane vesicles were finally frozen in liquid nitrogen and stored at –80°C until use. Protein concentrations were determined by the Lowry method (Lowry et al., 1951), and bovine serum albumin was used as a standard.
Transport Studies with Membrane Vesicles. The transport studies were performed using a rapid filtration technique. In brief, 15 μl of transport medium (10 mM Tris-HCl, 250 mM sucrose, and 10 mM MgCl2, pH 7.4) containing radiolabeled compound (0.03 μCi), with or without unlabeled substrate, was preincubated at 37°C for 3 min and then rapidly and gently mixed with 5 μl of membrane vesicle suspension (5 μg of protein). The reaction mixture contained 5 mM ATP or AMP along with the ATP-regenerating system (10 mM creatine phosphate and 100 μg/μl creatine phosphokinase). The transport reaction was terminated at designated times by addition of 1 ml of ice-cold buffer containing 10 mM Tris-HCl, 250 mM sucrose, and 0.1 M NaCl, pH 7.4. The stopped reaction mixture was passed through a 0.45-μm hemagglutinin filter (GVWP; Millipore Corporation, Billerica, MA), and then washed twice with 5 ml of stop solution. The radioactivity retained on the filter was determined in a liquid scintillation counter (LS6000SE; Beckman Coulter) after the addition of scintillation cocktail (Clear-sol I; Nacalai Tesque, Tokyo, Japan). For the uptake of cephalosporins, 20 μg of protein was used for each point. The stopped reaction mixture was passed through a 0.45-μm filter (JH-filter; Millipore). Unlike GVWP, JH-filter is not dissolved in organic solvents for extraction of the cephalosporins. Substrates retained on the filter were recovered in 1 ml of methanol containing internal standard by sonication for 15 min. After centrifugation, the supernatants were evaporated using a centrifugal concentrator (CC-105; TOMY, Tokyo, Japan), and dissolved in 45 μl of 0.05% HCOOH. Then, 40-μl aliquots were used for liquid chromatography/mass spectrometry quantification as described below.
In Vivo Infusion Study in Mice. Female BL6-129 and Mrp4–/– mice weighing approximately 20 to 30 g were used. Under pentobarbital anesthesia (30 mg/kg), the femoral vein was cannulated with a polyethylene catheter (PE-50) for the injection. Ceftizoxim (20.8 nmol/min/kg) and cefazolin (12.5 nmol/min/kg) were infused via the jugular vein. Blood samples were collected from the jugular vein at 30, 60, 90, and 120 min after administration and centrifuged. Urine was collected in preweighed test tubes at 30-min intervals throughout the experiment. At the end of the experiment, kidneys were excised. To determine the GFR, [3H]inulin (0.4 mg; 0.9 mCi/min/kg) was infused via the jugular vein. Blood and urine specimens were collected in the same way as for ceftizoxime and cefazolin.
Liquid Chromatography/Mass Spectrometry analysis. The quantification of ceftizoxime, cefazolin, cefmetazole, and cefotaxime was performed by high-performance liquid chromatography (Alliance 2690; Waters, Milford, MA) connected to a mass spectrometer (ZQ; Micromass, Manchester, UK). In this method, 10 μl of plasma, 10 μl of 10-fold-diluted urine, and 10 μl of 30% (w/v) kidney homogenate were deproteinized with 90 μl of acetonitrile containing an internal standard (cephalexin), mixed, and centrifuged. Then, 100-μl samples of the supernatants were concentrated. The concentrated samples were dissolved in 45 μl of 0.05% HCOOH, and 40-μl aliquots were injected into the liquid chromatography/mass spectrometry. High-performance liquid chromatography analysis was performed on a CAPCELL PAK C18 column (MGII, 3 μm, 2.0 mm ID, 50 mm; Shiseido, Tokyo, Japan) at 40°C. Elution was performed with a 95% to 40% linear gradient (for ceftizoxime and cefazolin) or 95% to 5% linear gradient (for cefmetazole) of 0.05% HCOOH over 5 min at 0.4 ml/min. The eluate was introduced into the MS via an electrospray interface. Detection was performed by selected ionization monitoring in positive ion mode (m/z 384, m/z 455, m/z 472, m/z 456, and m/z 348 for ceftizoxime, cefazolin, cefmetazole, cefotaxime, and cephalexin, respectively).
Pharamacokinetic Analysis. Total plasma clearance (CLtotal), renal clearance normalized by the circulating plasma concentration (CLrenal,p), and renal clearance normalized by the kidney concentration (CLrenal,k) were calculated from the equations CLtotal = I/Css,p, CLrenal,p = Vurine/Css,p, and CLrenal,k = (Vurine – Css,p × fb × GFR)/Css,k, where I, Css,p, Vurine, and Css,k represent the infusion rate (nanomoles per minute per kilogram), plasma concentration at steady state (micromolar), urinary excretion rate at steady state (nanomoles per minute per kilogram), and kidney concentration at steady-state (micromolar), respectively. Css,p was determined using the mean value of the plasma concentrations from 30 to 120 min. Vurine was determined as the mean value of the urinary excretion rate from 30 to 60 min, 60 to 90 min, and 90 to 120 min. GFR was determined in a separate experiment and calculated from the mean value of the urinary excretion rate of [3H]inulin from 30 min to 120 min divided by the mean value of the plasma concentration of [3H]inulin from 30 to 120 min.
Statistical Analysis. Statistical differences were determined using one-way analysis of variance followed by Fisher's least significant difference method.
Results
Effects of Various Cephalosporins on hMRP4-Mediated Transport. The uptake of [3H]DHEAS, a typical hMRP4 substrate, in the presence of ATP by membrane vesicles expressing hMRP4 was significantly greater than that in the presence of AMP (204 ± 17 and 4.0 ± 0.7 μl/mg of protein for 2 min), and that obtained in the control vesicles (8.3 ± 1.7 and 4.9 ± 3.5 μl/mg of protein for 2 min). The effect of various cephalosporins on the ATP-dependent uptake of [3H]DHEAS by MRP4-expressing membrane vesicles was examined (Fig. 2). The cephalosporins tested were first- to fourth-generation cephalosporins. Most of the tested cephalosporins had an inhibitory effect on the MRP4-mediated transport of [3H]DHEAS, whereas cephaloridine (a first-generation cephalosporin), cefsulodin (a third-generation cephalosporin), and cefepime (a fourth-generation cephalosporin) had no effect. The inhibitory effect of aminocephalosporins (cefaclor, cephalexin, and cefadroxil) was weak.
ATP-Dependent Transport of Ceftizoxime, Cefazolin, Cefmetazole, and Cefotaxime via Human MRP4. The uptake of cephalosporins by membrane vesicles was determined in the presence of ATP or AMP. Addition of ATP markedly stimulated the uptake of ceftizoxime, cefazolin, cefmetazole, and cefotaxime only in the membrane vesicles expressing hMRP4. AMP lacked this effect, and the uptake by membrane vesicles expressing hMRP4 in the presence of AMP was similar to the that observed in control vesicles determined in the presence of ATP and AMP (Fig. 3). The ATP-dependent uptake of ceftizoxime and cefazolin by membrane vesicles expressing hMRP4 was saturable (Fig. 4). Nonlinear regression analyses showed that the Km values of ceftizoxime and cefazolin, respectively, for hMRP4 were 18.3 ± 2.2 and 80.9 ± 10.9 μM, respectively, and the Vmax values were 0.529 ± 0.026 and 3.24 ± 0.25 nM/mg of protein, respectively.
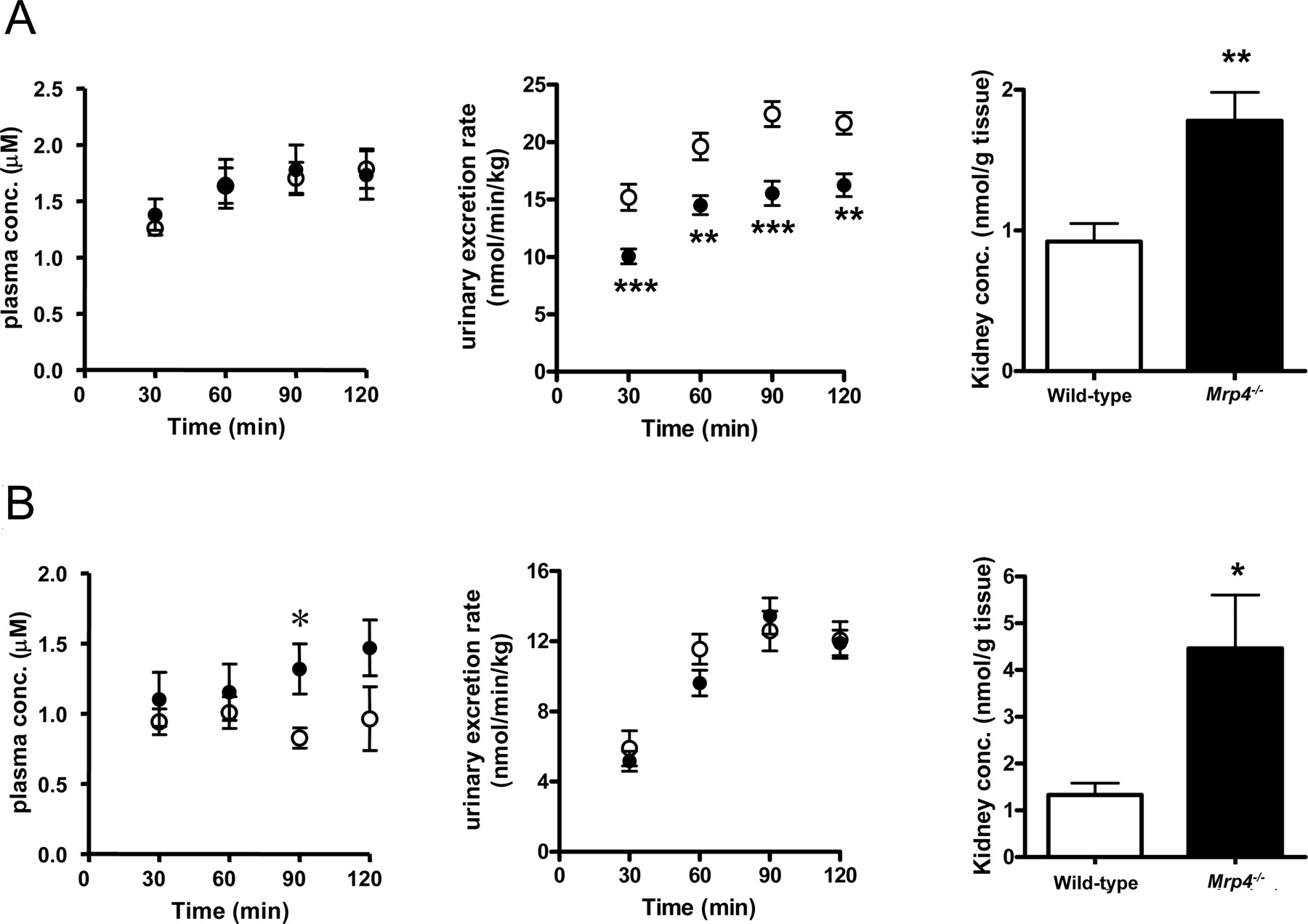
Renal Excretion of Ceftizoxime and Cefazolin in Wild-Type andMrp4–/–Mice. The renal clearance of [3H]inulin in wild-type and Mrp4–/– mice were 8.59 ± 0.80 and 11.9 ± 1.2 ml/min/kg, respectively, and these were used as the GFR. Ceftizoxime and cefazolin were given by intravenous infusion. In the case of ceftizoxime, the plasma concentration after intravenous infusion was similar in wild-type and Mrp4–/– mice (Fig. 4A). The urinary excretion of ceftizoxime was significantly reduced, and the kidney concentration was significantly increased in Mrp4–/– mice (Fig. 5A). The pharmacokinetic parameters are summarized in Table 1. Taking the unbound fraction of ceftizoxime in mouse plasma (0.7) (Tawara et al., 1992) and the GFR into consideration, the renal clearance of ceftizoxime was greater than its glomerular filtration rate (6.0 ml/min/kg). There was no significant difference in the total body and renal clearances between the two strains (Table 1). The kidney-to-plasma concentration ratio (Kp,kidney) was 2.0-fold higher in Mrp4–/– mice than in wild-type mice (Table 1). The tubular secretion clearance with respect to the kidney concentration (CLrenal,k) was significantly reduced in Mrp4–/– mice.
Pharmacokinetic parameters of ceftizoxime and cefazolin during constant infusion into wild-type and Mrp4-/- mice
Data are taken from Figure 5 and represent the mean ± S.E. GFR was determined by the renal clearance of inulin, which was 8.59 ± 0.80 and 11.9 ± 1.2 ml/min/kg in wild-type and Mrp4-/- mice, respectively. The unbound fractions of ceftizoxime and cefazolin have been reported to be 0.7 and 0.2, respectively (Tawara et al., 1992).
The plasma concentration after intravenous infusion of cefazolin was slightly higher in Mrp4–/– mice than in wild-type mice, although there was no significant difference in the urinary excretion rate between Mrp4–/– mice and wild-type mice (Fig. 5B). The kidney concentration of cefazolin was significantly greater in Mrp4–/– mice than in wild-type mice (Fig. 5B). Considering the low unbound fraction of cefazolin in mouse plasma (0.2) (Tawara et al., 1992), the major urinary excretion route of cefazolin is tubular secretion in mice. Similar to ceftizoxime, there was no significant difference in the total body and renal clearances between Mrp4–/– mice and wild-type mice (Table 1). The kidney-to-plasma concentration ratio (Kp,kidney) was 2.9-fold greater in Mrp4–/– mice than in wild-type mice (Table 1). The tubular secretion clearance with respect to the kidney concentration (CLrenal,k) was significantly reduced in Mrp4–/– mice (Table 1).

Effect of various cephalosporins on the ATP-dependent uptake of [3H]DHEAS by hMRP4 expressing membrane vesicles. MRP4-expressing membrane vesicles were incubated with medium containing [3H]DHEAS (0.1 μM) for 2 min in the presence or absence of various cephalosporins at designated concentrations. Each point represents the mean ± S.E. (n = 3). *, p < 0.05; **, p < 0.01, significantly different from control hMRP4-expressing membrane vesicles in the absence of cephalosporins by analysis of variance followed by Dunnett's test.
Discussion
For the first time, in the present study, we have identified cephalosporins (ceftizoxime, cefazolin, cefmetazole, and cefotaxime) as MRP4 substrates. Furthermore, in vivo pharmacokinetic studies using Mrp4–/– mice have shown that Mrp4 is involved in the luminal efflux of ceftizoxime and cefazolin in the kidney.
In the transport studies using membrane vesicles, we found that most of the cephalosporins are inhibitors of MRP4 (Fig. 2). Injectable cephalosporins were more potent inhibitors of hMRP4, except for cefepime, cefsulodin, and cephaloridine, than aminocephalosporins (Fig. 2). The generation of the cephalosporins is probably unrelated to their inhibitory potency. Ceftizoxime, cefazolin, cefmetazole, and cefotaxime could be measured by our LC/MS system with sufficient sensitivity, enabling direct measurement of the accumulation by membrane vesicles. ATP-dependent uptake of these cephalosporins by hMRP4 was observed with similar transport activities only in hMRP4-expressing membrane vesicles (Fig. 3). Thus these four cephalosporins, at least, are substrates of hMRP4.
Among the four cephalosporins, involvement of Mrp4 in the urinary excretion was tested for ceftizoxime and cefazolin. Urinary excretion is the predominant elimination pathway of ceftizoxime and cefazolin in wild-type mice, and this is mediated by both tubular secretion as well as glomerular filtration (Table 1). This is consistent with a previous report in which probenecid and p-aminohippurate inhibited the renal elimination of ceftizoxime and reduced its kidney-to-plasma concentration ratio (Terakawa et al., 1981). The total body clearances of ceftizoxime and cefazolin were almost unchanged, and the renal clearance (CLrenal,p) of ceftizoxime and cefazolin was slightly, but not significantly, reduced in Mrp4–/– mice (Table 1). However, the kidney concentrations were significantly increased in Mrp4–/– mice; consequently, the tubular secretion clearances of ceftizoxime and cefazolin with respect to the kidney concentration (CLrenal,k), representing the intrinsic efflux activity across the BBM, were significantly reduced in Mrp4–/– mice. These results suggest that Mrp4 plays a significant role in the luminal efflux of these cephalosporins in the kidney. It should be noted that the effect of impairment of Mrp4 on the kidney concentrations may exhibit a gender difference because the present study was carried out using female mice, which exhibit a 3-fold higher mRNA expression of Mrp4 in the kidney than male mice (Maher et al., 2005).
As far as cefazolin is concerned, the tubular secretion clearly remained in Mrp4–/– mice (Table 1). Indeed, the tubular secretion clearance of cefazolin with respect to the kidney concentration (CLrenal,k) exhibited moderate reduction (Table 1). Therefore, it is probable that the luminal efflux of cefazolin is also mediated by other transporters. Tamai et al. (1988) found that a membrane voltage-driven transporter is involved in the transport of cefixime in the BBM vesicles from the kidney. It is possible that this transporter is involved in the renal elimination of cefazolin. As membrane voltage-driven transporters for organic anions, OATv1 and RST have been identified in the BBM of the kidney (Jutabha et al., 2003; Imaoka et al., 2004). In addition, MRP2 and BCRP have been also identified on the BBM of the renal tubule cells (Schaub et al., 1999; Jonker et al., 2002; Mizuno et al., 2004). These transporters are candidates involved in the tubular secretion of cephalosporins in conjunction with Mrp4. The minimal change in the renal clearance (CLrenal,p) of cefazolin in Mrp4–/– mice can be explained by speculating that the efflux across the BBM is greater than that across the basolateral membrane, resulting in the uptake being the rate-limiting process of the net secretion. Under these conditions, the net elimination is hardly affected by the reduction in the luminal efflux clearance.

Time-profiles of the uptake of ceftizoxime, cefazolin, cefmetazole, or cefotaxime by human MRP4-expressing vesicles. Membrane vesicles (20 μg) prepared from human embryonic kidney 293 cells infected with hMRP4 (•, ○) or GFP (▪, □) adenovirus were incubated at 37°C in uptake medium containing ceftizoxime (10 μM) (A), cefazolin (10 μM) (B), cefmetazole (5 μM) (C), or cefotaxime (5 μM) (D). •, ▪, uptake from medium containing 5 mM ATP; ○, □, uptake from medium containing 5 mM AMP. Each point represents the mean ± S.E. (n = 3).

Saturation of the ATP-dependent uptake of ceftizoxime and cefazolin by human MRP4-expressing vesicles. ATP-dependent uptake of ceftizoxime (A) or cefazolin (B) by human MRP4 (•)-expressing membrane vesicles was determined for a 2-min incubation at a substrate concentration ranging from 5 to 1000 μM. The results are also given as the Eadie-Hofstee plots. Each point represents the mean ± S.E. (n = 3). The data were fitted to the Michaelis-Menten equation by nonlinear regression analysis, and the solid line represents the fitted curve.
The recovery of ceftizoxime in the urine was significantly reduced in Mrp4–/–, although the plasma concentrations of ceftizoxime were unchanged (Fig. 5). This suggests that impairment of Mrp4 produced a nonrenal elimination pathway for ceftizoxime, which apparently compensated for the reduced renal elimination. The nonrenal elimination pathway remains to be elucidated. Considering that some cephalosporins undergo biliary excretion (Wright and Line, 1980), hepatic elimination would account for the nonrenal elimination pathway of ceftizoxime in Mrp4–/– mice. Indeed, Mrp4 has been identified in the sinusoidal membrane in the liver (Rius et al., 2003), and reduction of sinusoidal efflux could increase the hepatic elimination. It is also possible that this is caused by adaptive regulation of detoxification systems in Mrp4–/–. Unlike ceftizoxime, the urinary recovery of cefazolin was unchanged in Mrp4–/– (Fig. 5), indicating that the nonrenal elimination pathway makes only a limited contribution for cefazolin. Further pharmacokinetic studies are necessary to elucidate the mechanisms underlying such a difference in the pharmacokinetics.
The present study shows that Mrp4 plays an important role in the tubular secretion of ceftizoxime and cefazolin in addition to previously reported Mrp4 substrates, diuretics, and acyclic antivirus drugs (Hasegawa et al., 2007; Imaoka et al., 2007). MRP4 is also expressed in the BBM of the human kidney (van Aubel et al., 2002). It may also make a significant contribution to the luminal efflux of these drugs in the human kidney. Caution must be paid when extrapolating the result of animal studies to humans considering the possibility of species difference in the protein expression, substrate specificity/transport activity of not only MRP4 but also other luminal transporters. It is necessary to obtain clinical data to support the functional role of MRP4 in human kidney. A population pharmacokinetic analysis has suggested a bimodal distribution of the renal clearance of ceftizoxime with regard to the plasma concentration (Facca et al., 1998), although the underlying mechanism remains to be elucidated. Cumulative studies suggest that the allele frequencies non-synonymous with functional change and nonsense mutations of OAT1 and OAT3 are too low to account for the bimodal distribution (Fujita et al., 2005; Erdman et al., 2006). Whether this is associated with interindividual variations in MRP4 activity as a result of genetic polymorphisms and/or other factors is a topic for future investigation.

Time-profiles of the plasma concentration, urinary excretion, and kidney concentration of ceftizoxime and cefazolin in Mrp4–/– and wild-type mice. The plasma concentration, urinary excretion, and kidney concentration of ceftizoxime (A) and cefazolin (B) in Mrp4–/– (•) and wild-type mice (○) were examined. Ceftizoxime and cefazolin were infused at 20.8 and 12.5 nmol/min/kg, respectively. Each symbol represents the mean ± S.E. (n = 4–10). *, p < 0.05; **, p < 0.01, significantly different from wild-type mice.
In conclusion, some cephalosporins are MRP4 substrates, and Mrp4 makes a significant contribution to the luminal efflux of ceftizoxime and cefazolin in the kidney. This is the first report identifying the transporter responsible for the luminal efflux of cephalosporins. In the past, exchanger and/or facilitative transporters have been considered to play major roles in the urinary excretion of drugs, but an ATP binding cassette transporter, MRP4 is involved in the tubular secretion of some diuretics, acyclic nucleoside analogs, and cephalosporins. This finding will contribute to a better understanding of the luminal efflux mechanisms of drugs.
Footnotes
-
This work was supported by the Advanced and Innovational Research Program in Life Sciences from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to Y.S.); a Grant-in-Aid for Scientific Research on Priority Areas (KAKENHI 18059007 to H.K.); National Institutes of Health grants GM60904, ES058571, and CA23099 (to J.D.S.); Cancer Center support grant P30-CA21745 (to J.D.S.); and the American Lebanese-Syrian Associated Charities (to J.D.S.).
-
L.C. and H.K. contributed equally to this work.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.106.031823.
-
ABBREVIATIONS: OAT, organic anion transporter; BBM, brush-border membrane; MRP, multidrug resistance-associated protein; DHEAS, dehydroepiandrosterone sulfate; GFP, green fluorescent protein; TS, Tris-sucrose; GFR, glomerular filtration rate; CL, clearance.
- Received October 16, 2006.
- Accepted March 2, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}