


Abstract
The aryl hydrocarbon receptor (AHR) is a ligand-inducible transcription factor that displays interspecies differences with the human and mouse AHR C-terminal region sequences sharing only 58% amino acid sequence identity. Compared with the mouse AHR (mAHR), the human AHR (hAHR) displays ∼10-fold lower relative affinity for prototypical AHR ligands such as 2,3,7,8-tetrachlorodibenzo-p-dioxin, which has been attributed to the amino acid residue valine 381 (alanine 375 in the mAHR) in the ligand binding domain of the hAHR. We investigated whether the 10-fold difference in ligand-binding affinity between the mAHR and hAHR would be observed with a diverse range of AHR ligands. To test this hypothesis, ligand binding assays were performed using the photo-affinity ligand 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin and liver cytosol isolated from hepatocyte-specific transgenic hAHR mice and C57BL/6J mice. It is noteworthy that competitive ligand-binding assays revealed that, compared with the mAHR, the hAHR has a higher relative affinity for certain compounds, including indirubin [(2Z)-2,3-biindole-2,3 (1′H,1′H)-dione and quercetin (2-(3,4dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one]. Electrophoretic mobility shift assays revealed that indirubin was more efficient at transforming the hAHR compared with the mAHR. Indirubin was also a more potent inducer of Cyp1a1 expression in transgenic hAHR mouse hepatocytes compared with C57BL/6J mouse hepatocytes. These observations suggest that indirubin is a potent hAHR ligand that is able to selectively bind to and activate the hAHR. These discoveries imply that there may be a significant degree of structural divergence between mAHR and hAHR ligands and highlights the importance of the hAHR transgenic mouse as a model to study the hAHR in vivo.
The aryl hydrocarbon receptor (AHR) is the only ligand-activated member of the basic-helix-loop-helix Per Arnt Sim domain family of transcription factors. Studies in Ahr-null mice have highlighted the physiological roles of the AHR in liver and cardiac vascularization and development, immune system function, and ovarian follicle maturation (Fernandez-Salguero, 1995; Schmidt et al., 1996; Abbott et al., 1999). The AHR forms a cytoplasmic complex consisting of the heat-shock protein 90, hepatitis B virus-X associated protein 2, and p23. AHR activation leads to concomitant AHR dissociation from the cytoplasmic complex and heterodimerization with the AHR nuclear translocator (ARNT). AHR/ARNT complexes bind to canonical dioxin-responsive elements (DREs) and directly activate the expression of a number genes, including CYP1A1, CYP1A2, CYP1B1, glutathione transferase Ya, UDP-glucuronosyltransferase, epiregulin, and slug (Qiang et al., 1995; Köhle and Bock, 2007).
A myriad of structurally diverse compounds are known to activate the AHR through fitting into a receptor binding pocket with a maximal dimension of 14 × 12 × 5 Å (Waller and McKinney, 1995). AHR ligands are characteristically planar, aromatic, and hydrophobic molecules, including polycyclic aromatic hydrocarbons such as benzo[a]pyrene (B[a]P), halogenated aromatic hydrocarbons such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), the hemoglobin breakdown compounds biliverdin and bilirubin, and a number of naturally occurring flavonols, including quercetin and kaempferal (Denison and Heath-Pagliuso, 1998; Ciolino et al., 1999).
There are substantial differences in dioxin responsiveness among various mice strains, which express structurally divergent mouse AHRs (mAHR). In various inbred mice strains, the Ahrb1-3 and Ahrd alleles are present. AHR expressed by the Ahrb1-3 alleles exhibit higher affinity for dioxin compared with AHR expressed from the Ahrd allele (Poland and Glover, 1990). The human AHR (hAHR) ligand binding domain is most structurally analogous to the mAHRd allele ligand binding domain and therefore has approximately 10-fold lower affinity than the mAHRb allele for TCDD, which has been attributed to the amino acid residue valine 381 (375 in mAHR) in the ligand binding domain of the hAHR (Harper et al., 1988; Ema et al., 1994; Ramadoss and Perdew, 2004).
A number of structural differences exist between the mAHR and hAHR proteins. The human and mouse AHR share only 58% amino acid sequence identity in the C-terminal half, which is the region that contains the transactivation domain of both receptors. It is noteworthy that the hAHR and mAHRb display distinct affinity for LXXLL-coactivator-binding motif, suggesting that each receptor may differentially recruit coactivators and thus may regulate unique subsets of genes (Flaveny et al., 2008). Rodents exposed to the prototypical AHR ligand (TCDD) display a number of symptoms, including thymic atrophy, immunotoxicity, tumor promotion, teratogenicity, reproductive toxicity, hepatotoxicity, and chloracne, whereas human exposure to TCDD primarily causes chloracne (Abbott et al., 1999; Connor and Aylward, 2006). Toxicity studies conducted in the human AHR “knockin” mouse showed that TCDD-mediated toxicity differed between transgenic human AHR and murine Ahrd mice, suggesting that indeed the hAHR may differ in its ability to regulate gene expression (Moriguchi et al., 2003). In addition, previous investigations have also demonstrated that the hAHR can be selectively activated by omeprazole through a yet-to-be-understood mechanism that is independent of ligand binding (Lesca et al., 1995).
In light of the interspecies differences between the hAHR and the mAHR that have thus been elucidated, traditional toxicological and gene regulation studies using C57BL/6J mice may be an inadequate tool for investigating the role of the hAHR in mediating toxicity and regulating gene expression. To address this issue, we developed a transgenic mouse that expresses hAHR under the control of the liver-specific transthyretin (Ttr) promoter. Protein blot analysis has revealed that hAHR protein expression levels in the liver were similar to C57BL/6J AHRb receptor levels. These transgenic AHRTtrAhrb/b mice were bred onto both the Ahr-null mice [Ahr(-/-)] and the albumin promoter-driven Cre recombinase, Ahr-floxed conditional knockout mice (CreAlbAhrfx/fx) backgrounds. Using competitive ligand binding experiments, we found that the hAHR displays a higher relative affinity for certain AHR ligands such as indirubin and quercetin compared with the mAHRb, thus establishing that each receptor has distinct ligand binding characteristics. Indirubin was shown to be more potent at inducing AHR target genes, in primary mouse hepatocytes expressing the hAHR compared with C57BL/6J mouse hepatocytes expressing mAHRb. These discoveries suggest that the hAHR may be distinctly regulated in a species-specific fashion by certain ligands in a manner that cannot be predicted by its relatively lower affinity for prototypic AHR ligands, such as TCDD.
Materials and Methods
Transgenic Mice
The synthetic human AHR cDNA sequence optimized for mammalian codon use and minimal secondary mRNA structure was purchased from GenScript (Piscataway, NJ). The cDNA was amplified by PCR using primers designed with StuI sites, and the resulting product was digested with StuI and inserted into the second exon of the modified pTTR1 vector (obtained from Dr. Terry Van Dyke, University of North Carolina) (Yan et al., 1990), TTRexV3. TTRexV3 was derived by making several point mutations in the first and second exons, which destroy ATGs and introduce unique cloning sites. The TTRexV3-hAHR was digested with HindIII to release the appropriate fragment and purified for microinjection into embryos. TTRexV3-hAHR (AHRTtr) fragment were microinjected into C57BL/6J fertilized eggs at the Penn State University Transgenic Mouse Facility (Department of Dairy and Animal Science, The Pennsylvania State University, University Park, PA). Founder mice and offspring were screened using PCR assays described below. Transgenic mice were mated with Ahr(-/-) and the albumin promoter-driven, Cre recombinase-expressing CreAlbAhrfx/fx mice (obtained from Christopher Bradfield, University of Wisconsin) to produce transgenic AHRTtrAhr(-/-) [strain name, B6.Cg-Ahrtm1Bra Tg (Ttr-AHR)1Ghp] and AHRTtrCreAlbAhrfx/fx [strain name, B6.Cg-Ahrtm3.1Bra Tg (Alb-cre, Ttr-AHR)1Ghp], respectively. We then used the AHRTtrAhr(-/-) and AHRTtrCreAlbAhrfx/fx mice for ligand binding experiments and gene expression analysis, respectively.
Screening of Transgenic Mice. Mouse tail and hepatocyte DNA was isolated using the Wizard SV Genomic DNA isolation system (Promega, Madison, WI) and subjected to PCR using the primers for the AHRTtr, CreAlb, and Ahr(-/-) genes. Analysis of Ahrfx/fx excision was assessed as described previously using the primers OL4062 and OL4064 in combination with the reverse primer OL4088 (Walisser et al., 2005). The Ahrfxfx-excised allele (OL4062/4088) amplified a 180-bp band, whereas amplification from the Ahrfx/fx-unexcised allele (OL4064/4088) resulted in a 140-bp band. The Ahrb allele generated a 106-bp band (OL4064/OL4088). Congenic Ahrd mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were housed on corncob bedding at 70 ± 2°F with a 12-h light/dark cycle. Mice were given access to food and water ad libitum. All primer sequences are listed in Table S1.
Ligand Binding Assays
Photoaffinity Ligand Synthesis. The AHR photoaffinity ligand 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin (PAL) was synthesized as described previously (Poland et al., 1986).
Cytosol Preparation. To generate liver cytosol for ligand binding experiments, mouse livers were homogenized in buffer (25 mM MOPS, 2 mM EDTA, 0.02% NaN3, and 10% glycerol, pH 7.4) containing 20 mM sodium molybdate and protease inhibitors (Sigma, St. Louis, MO) and centrifuged at 100,000g for 1 h.
Ligand Binding Conditions. All binding experiments were conducted in the dark until UV-mediated activation of the PAL. In brief, ligand-treated lysates were incubated at room temperature (except for binding assays involving the mAHRd, which was carried out at 4°C) for 20 min and then photolyzed at 8 cm with 402 nm UV light. Dextran-coated charcoal (1%) was added to the photolyzed samples, which were then centrifuged at 3000g for 10 min to remove free ligand. Labeled samples were resolved using 8% acrylamide-tricine-SDS-PAGE, transferred to PVDF membrane, and visualized using autoradiography. Labeled AHR bands were excised and counted using a γ counter.
Competitive Binding Experiments. A saturating amount of the PAL (0.21 pmol, 8 × 105 cpm per tube) was added to 150 μg of total protein of mouse liver or transiently transfected COS-1 cytosol along with increasing amounts of competing ligands: B[a]P, (2Z)-2,3-biindole-2,3 (1′H,1′H)-dione (indirubin) 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one (quercetin), PCB-126, 5,6-benzoflavone (β-naphthoflavone), 5,7-dimethoxyflavone, and M50354. Samples were then subjected to ligand binding conditions.

The liver-specific transgenic hAHR mouse expresses functional hAHR protein in the liver and hepatocytes at levels comparable with those of mAHRb in C57BL/6J mice. A, hAHR transgenic mice (lines A, B, and C) express hAHR protein at levels comparable with those of mAHRb protein expression in liver. B, whole liver and cultured primary hepatocyte hAHR expression in selected AHRTtr transgenic mouse line (line B) used in ligand binding and gene expression experiments. C, PCR genotyping of DNA isolated from tail clip and hepatocyte DNA from Ahrfx/fx, Ahrfx/fxCreAlb, and AHRTtrAhrfx/fx-CreAlb transgenic mice on a conditional knockout background was conducted using primers for AHRTtr, CreAlb, and Ahrfx. Ahrfx excision was assessed as described previously using the primers OL4062 and OL4064 in combination with the reverse primer OL4088 (Walisser et al., 2005). The Ahrfx-excised allele amplified a 180-bp band, whereas amplification from the Ahrfx-unexcised allele resulted in a 140-bp band. Ahrb C57BL/6J allele generated a 106-bp band. D, saturation ligand binding. Increasing amounts of PAL were added to liver cytosol isolated from the hAHR expressing transgenic line B on an Ahrfx/fxCreAlb background and C57BL/6J, mAHRb-expressing mice. Labeled samples were resolved using 8% acrylamide-tricine-SDS-PAGE, transferred to PVDF membrane, and visualized using autoradiography. Radioactive AHR bands were excised and counted using a γ counter. E, real-time RT-PCR. TCDD-treated cultured primary hepatocytes isolated from AHRTtr-Ahrfx/fxCreAlb, Ahrb mice were treated with increasing amounts of TCDD or vehicle control for 6 h. mRNA expression was quantified using real-time RT-PCR. *, p < 0.05.
Immunoblotting
Whole mouse liver and transiently transfected COS-1 cell cytosol were isolated as described above, and in vitro-translated rabbit reticulocyte lysate proteins (50 μg/well) were resolved using 8% tricine-SDS-polyacrylamide gels. Proteins were transferred to PVDF membrane, and AHR protein was detected using the mouse monoclonal antibody RPT1 (Affinity BioReagents, Golden, CO), goat anti-mouse biotin-conjugated secondary antibody, and [125I]streptavidin and visualized using autoradiography.
Plasmids
pCI-hAHR, pCI-hAHRV381A, pcDNA3-mAHR, and pcDNA3-mAHRA375V, the mAHR-N terminus/hAHR-C terminus (m-hAHR) and hAHR-N terminus/mAHR-C terminus (h-mAHR) chimeric plasmid constructs, were generated previously (Meyer et al., 1998; Ramadoss and Perdew, 2004).
Cell Culture
COS-1 cells were routinely grown in α-minimal essential medium (Sigma) supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin. All cell cultures were maintained under standard conditions in a 37°C incubator at 5% CO2/95% air, and cell culture media were changed every 48 h unless otherwise indicated.
Transient Transfections
COS-1 cells (American Type Culture Collection, Manassas, VA) were seeded in 20-mm tissue culture plates 24 h before transfection and were transfected with 20 μg of either pCI-hAhR, pCI-hAHRV381A, pcDNA3-mAhR, or pcDNA3-mAHRA375V, h-mAHR, and m-hAHR using LipofectAMINE reagent (Invitrogen, Carlsbad, CA), according to manufacturer's instructions.
Primary Hepatocyte Isolation
Liver perfusion and hepatocyte isolation was carried out as described previously (Madden et al., 2000) with some modifications. In brief, mice were anesthetized with 0.1 to 0.3 ml of 2.5% Avertin administered via intraperitoneal injection. Hepatic perfusion was performed with buffer I (5 mM dextrose, 116 mM NaCl, 760 μM NaH2PO4, 5.3 mM KCl, 26 mM NaHCO3, 10 mM HEPES, and 500 μM EGTA, pH 7.2) for 1 min followed by buffer II [0.2 mg/ml type I collagenase (Worthington, Freehold, NJ), 5.3 mM KCl, 116 mM NaCl, 5 mM dextrose, 26 mM NaHCO3, 1.6 mM MgSO4, 900 μM CaCl2, and 48 μg/ml trypsin inhibitor, pH 7.2] for a further 5 to 10 min. Hepatic tissue was excised, transferred, and dissociated in a 100-mm plate containing 9 ml of short-term media (Dulbecco's modified Eagle's medium, 10% fetal bovine serum, 2.5% dimethyl sulfoxide, 10 nM dexamethasone, 100 IU/ml penicillin, and 100 μg/ml streptomycin). Cells were filtered, centrifuged (500g for 1 min), and resuspended in short-term media. Cell viability was assessed via trypan blue staining, and cells were seeded into type-I collagen-coated six-well plates (BD Bioscience, San Jose, CA) at a density of 1 × 106 cells/ml. After 4-h incubation at 37°C, nonadherent cells were aspirated, and fresh short-term media were added. After over-night incubation at 37°C, cells were washed with phosphate-buffered saline, and short-term media were replaced with long-term hepatocyte culture media [Hepatozyme-SFM (Invitrogen), 2.5% dimethyl sulfoxide, 10 nM dexamethasone, 100 IU/ml penicillin, and 100 μg/ml streptomycin].
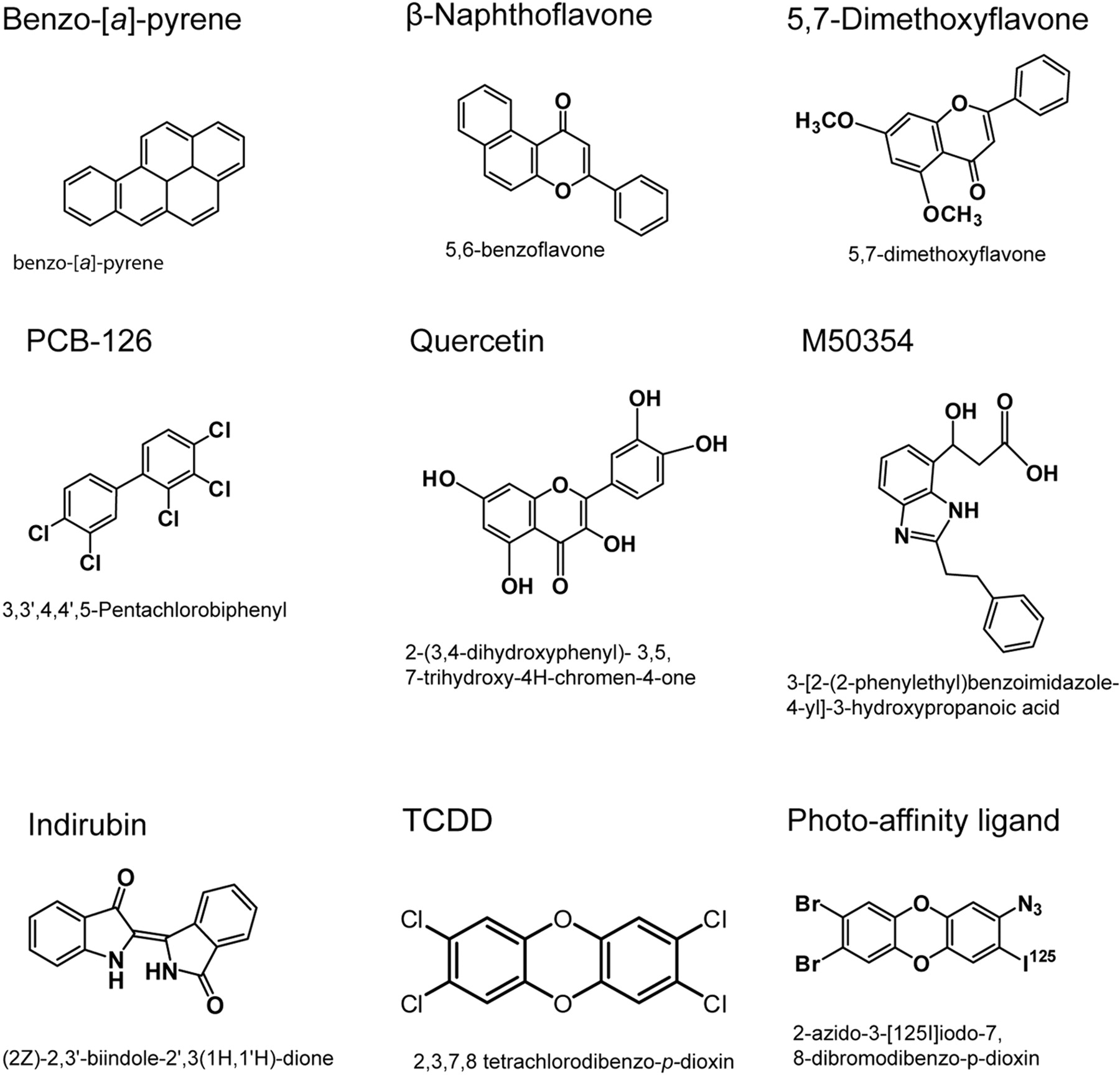
AHR ligands. A structurally diverse set of AHR ligands was chosen for use in competitive ligand binding assays.
Real-Time RT-PCR
Primary hepatocytes isolated from AHRTtrCreAlbAhrfx/fx, Ahrfx/fx, and Ahrb/b mice were treated with AHR ligands (TCDD or indirubin) or vehicle control for 6 h. Total mRNA was isolated from cultured hepatocytes using Tri-reagent (Invitrogen). RNA was then converted to cDNA using ABI cDNA archive synthesis kit (Applied Biosystems, Foster City, CA), and mRNA expression was quantified using real-time RT-PCR with primers listed in Table S1.
Electrophoretic Mobility Shift Assays
pCI-ARNT, pCI-hAHR, pCI-hAHRV381A, pcDNA3-mAHR, and mAHRA375V along with control plasmids were in vitro-translated using the TNT-coupled rabbit reticulocyte lysate system (Promega) in the presence of 1.5 mM sodium molybdate. In vitro-translated AHR proteins (4 μl of lysate) were incubated with ARNT [4 μl of lysate) plus 1.5 μl of HEDG buffer (25 mM HEPES, 1 mM EDTA, 10 mM sodium molybdate, and 10% (v/v) glycerol, pH 7.5] along with either 10 nM B[a]P, 100 nM or 1 μM indirubin, or vehicle control for 15 min at room temperature. 32P-labeled DRE probe was added to each reaction and incubated for 15 min. A total of 16 μl of lysate was then resolved using a 6% DNA-retardation gel (Invitrogen), which was then fixed, vacuum-dried, and visualized using autoradiography. Band intensities were quantified using filmless autoradiographic analysis and OptiQuant software (PerkinElmer Life and Analytical Sciences, Waltham, MA) and presented as digitized light units (DLUs).
Statistical Analysis
Statistical analysis of real-time RT-PCR data were performed using two-way analysis of variance using GraphPad Prism software (GraphPad Software Inc. San Diego, CA). Calculated p values <0.05 were considered statistically significant.
Results
The Liver-Specific Transgenic hAHR Mouse Expresses Functional hAHR Protein in the Liver and Hepatocytes at Levels Comparable with Those of mAHRb. To study the possible unique roles of the hAHR in gene regulation, toxicity, and carcinogenesis in the liver, we generated a liver-specific transgenic hAHR-expressing mouse. hAHR-expressing mice were generated initially on a C57BL/6J background and subsequently crossed with Ahr(-/-) or CreAlbAhrfx/fx mice to transfer the hAHR transgene to an Ahr-null and conditional deletion background. Three AHRTtr transgenic mouse lines were generated that expressed differing amounts of hAHR (Fig. 1A). In transgenic mouse line B, hAHR protein expression levels in whole liver and hepatocytes were comparable (approximately 2-fold higher) with AHRb expression in C57BL/6J mouse liver and hepatocytes (Fig. 1, A and D). Liver cytosol from AHRTtr Ahr(-/-) mice was used in saturation and competitive ligand binding studies (Figs. 1B and 2). An assessment of the binding capacity of the hAHR protein expressed in AHRTtr-Ahr(-/-) transgenic mice showed that the mAHRb had a higher relative ligand binding capacity for the PAL compared with the hAHR (Fig. 1B), a result consistent with previous studies (Ramadoss and Perdew, 2004).

The hAHR and mAHRb displays varied relative ligand binding affinities in competitive ligand binding assays. A to C, a saturating amount of the PAL (0.21 pmol) was added to 150 μg of total protein of mouse liver cytosol along with increasing amounts of competing ligands: B[a]P, indirubin, quercetin, β-naphthoflavone, 5,7-dimethoxyflavone, and M50354. Labeled samples were resolved using 8% acrylamidetricine-SDS-PAGE, transferred to PVDF membrane, and visualized using autoradiography. Labeled AHR bands were excised and counted using a γ counter.
It is noteworthy that the hAHR displayed high levels of constitutive activity and reduced relative inducibility of the AHR responsive genes Cyp1a1, Cyp1a2, and Cyp1b1 when expressed on an Ahr(-/-) background (Supplementary Fig. S1), this result was not observed in AHRTtrCreAlbAhrfx/fx mice, which were on an CreAlbAhrfx/fx background. Therefore, to study hAHR-mediated gene regulation, hepatocytes isolated from AHRTtrCreAlbAhrfx/fx mice were used. Hepatocyte and tail-clip DNA collected from transgenic mice was subjected to PCR genotyping with primers specific for the AHRTtr and CreAlb transgenes and floxed Ahrfx/fx primers to confirm Ahrfx/fx gene excision in CreAlb-positive mice (Fig. 1C). Whole-liver and hepatocyte cytosol isolated from AHRTtrAhr(-/-), AHRTtrCreAlbAhrfx/fx, Ahrfx/fx, and Ahrd/d congenic and C57BL/6J mice were also all subjected to protein blot analysis (Fig. 1, A-D). TCDD was able to induce Cyp1a1 and Cyp1b1 mRNA expression in both mAHRb- and hAHR-expressing hepatocytes in a dose-dependent manner. TCDD at lower doses also demonstrated a notably more potent induction of Cyp1a1 and Cyp1b1 in mAHRb-expressing compared with hAHR-expressing hepatocytes (0.1-1 nM) (Fig. 1E).
The hAHR and mAHRb Display Varied Relative Ligand Binding Affinities in Competitive Ligand Binding Assays. The most commonly used mouse AHR allele in gene expression, toxicity, and carcinogenesis studies is the mAHRb allele. We used this allele to compare ligand binding characteristics of the human and mouse Ah receptors for a structurally diverse subset of AHR ligands (Fig. 2). Liver cytosol isolated from both mAHRb and hAHR transgenic mice were subjected to competitive ligand binding assays using a fixed saturating dose of PAL (0.21 pmol, 8 × 105 cpm per tube) and increasing amounts of known AHR ligands. These competition binding experiments showed that compared with the hAHR, the mAHR had a higher relative affinity for the competing ligands B[a]P, β-naphthoflavone, and PCB-126 (∼10-fold) relative to the PAL (Fig. 3A). Relative to the PAL, the hAHR displayed a reduced but still lower relative ligand binding affinity for 5,7-dimethoxyflavone and M50354 compared with the mAHR (Fig. 3B). We were surprised to find that the hAHR displayed a higher relative affinity for quercetin and the plant tryptophan-derivative indirubin compared with the mAHR (Fig. 3C). Taken together, these data suggest that the human and mouse AHR ligand binding pocket has differential binding properties.

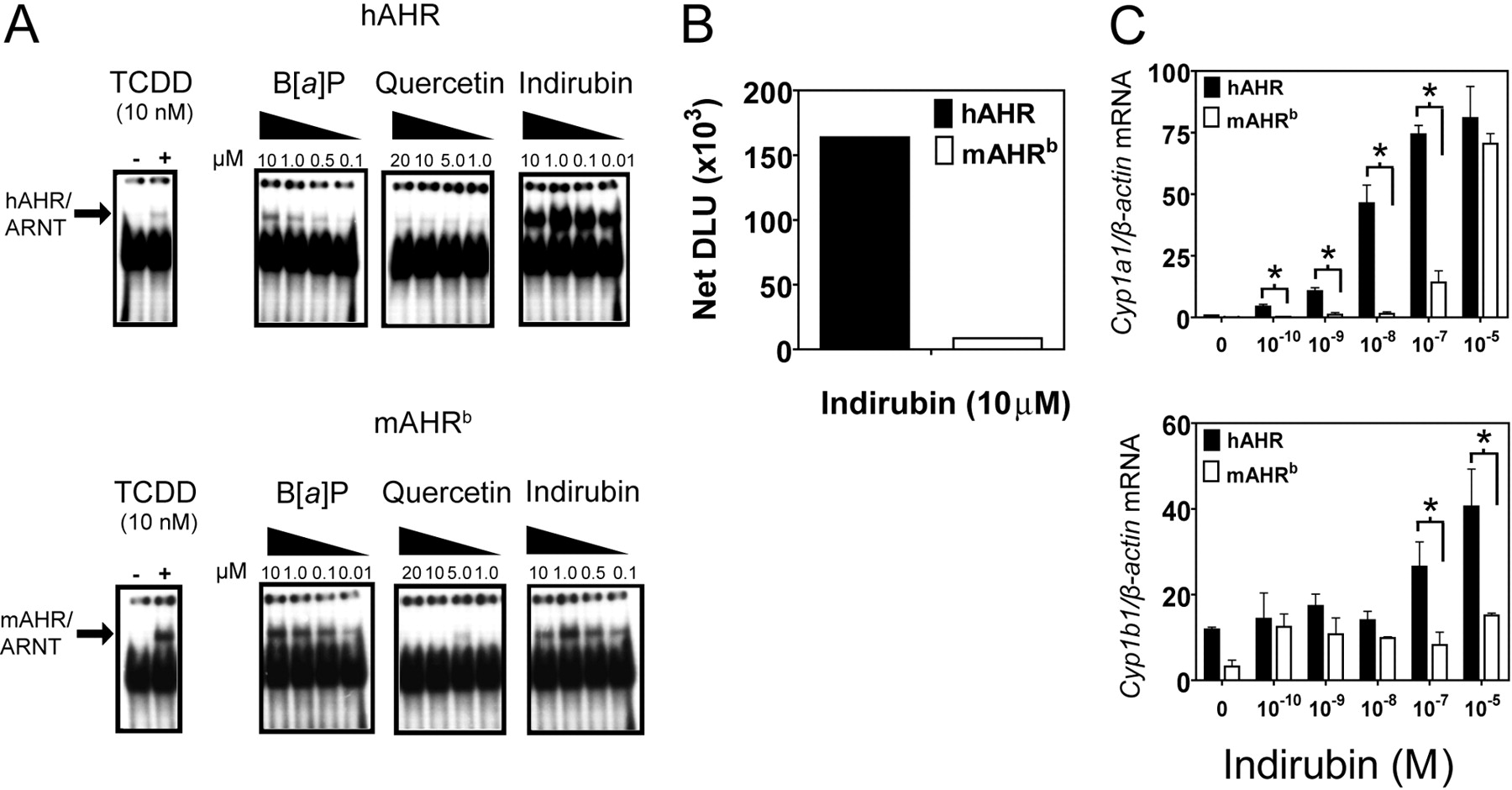
Indirubin more potently induces hAHR transformation and gene activation compared with the mAHRb. A and B, electron mobility shift assays: pCI-ARNT, pCI-hAHR, and pcDNA3-mAHR were in vitro-translated. In vitro-translated AHR proteins were incubated with ARNT plus HEDG buffer along with either 10 nM B[a]P, 100 nM or 1 μM indirubin, or vehicle control and P32-labeled DRE probe. Lysate was then resolved using a DNA retardation gel, fixed, vacuum-dried, and visualized using autoradiography. Band intensities were quantified using filmless autoradiographic analysis and presented as DLUs. C, real-time RT-PCR. Primary hepatocytes isolated from hAHR, and mAHRb-expressing mice were treated with increasing amounts of indirubin or vehicle control for 6 h. Total mRNA was isolated from cultured hepatocytes, and mRNA expression was quantified using real-time RT-PCR.
Indirubin More Potently Induces hAHR Transformation Compared with the mAHR. Ligand binding converts the AHR to a high-affinity DNA binding form that is composed of an AHR/ARNT heterodimer, a process known as transformation. We decided to investigate whether the higher relative affinity the hAHR displayed for indirubin and quercetin in competitive binding experiments would also result in selective transformation of the hAHR by these compounds. EMSAs showed that indirubin was able to more potently induce hAHR heterodimerization and DRE binding compared with the mAHRb (Fig. 4, A and B). It is noteworthy that compared with B[a]P, indirubin was also relatively more effective at inducing hAHR transformation for all of the concentrations at which both ligands were tested (Fig. 4A). It is noteworthy that although quercetin was unable to induce profound mAHR or hAHR transformation, quercetin was able to transform the hAHR at lower ligand concentrations (as low as 1 μM), which failed to stimulate mAHR transformation (Fig. 4A).
Indirubin Is More Potent at Stimulating hAHR-Driven Gene Activation. To test whether indirubin was able to selectively activate the hAHR, primary hepatocytes isolated from mAHRb and hAHR mice were treated with increasing doses of indirubin and assayed for AHR-responsive gene induction using real-time RT-PCR. At lower doses, indirubin selectively induced Cyp1a1 mRNA synthesis only in hAHR-expressing hepatocytes. At higher doses, indirubin also differentially increased Cyp1b1 mRNA levels in hAHR-expressing hepatocytes compared with mAHR hepatocytes (Fig. 4C).
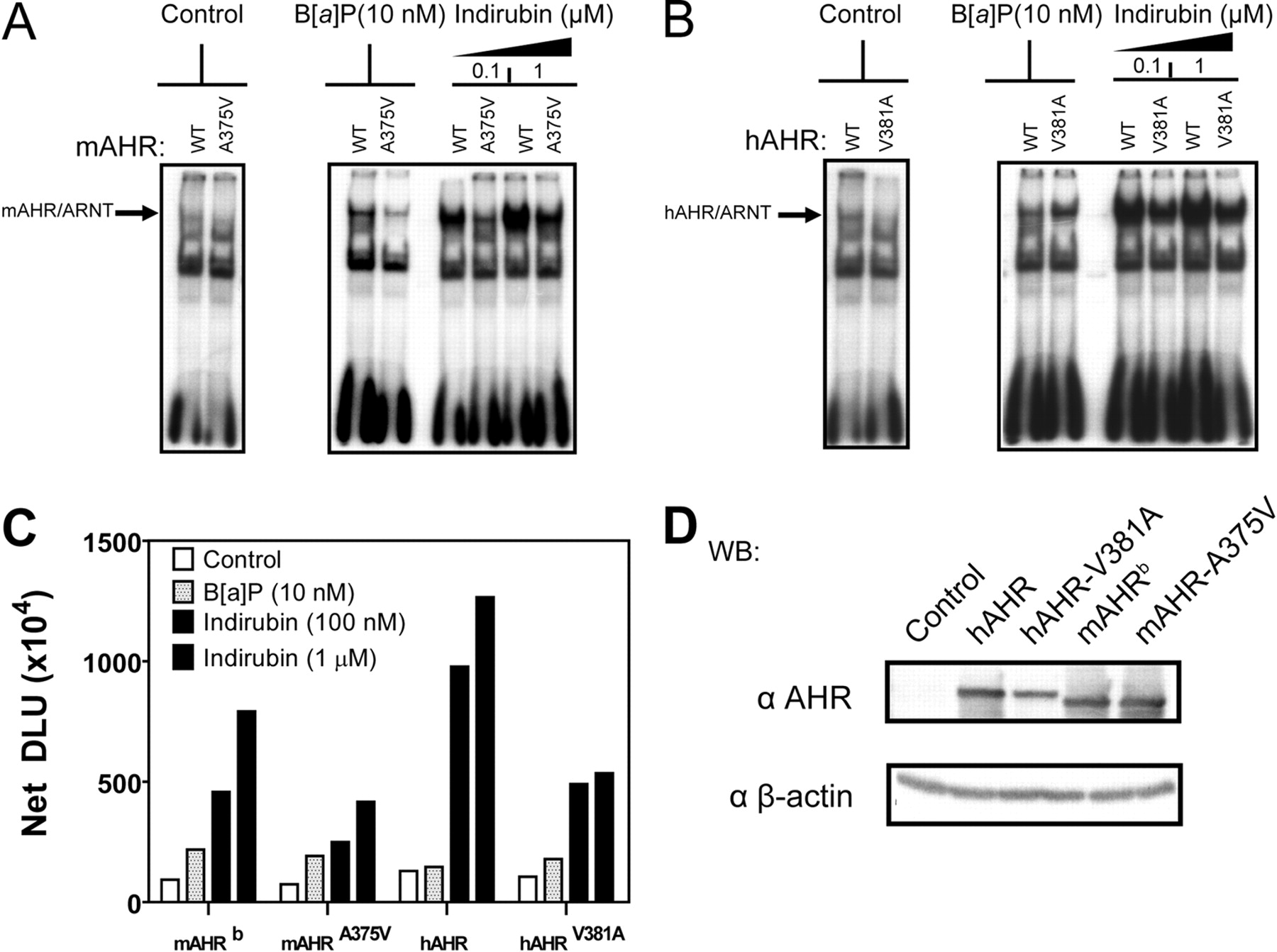
hAHR Relative Affinity for Indirubin Is Not Enhanced by V381A Substitution. The mAHRd has a ligand binding domain that is analogous to that of the hAHR and thus has an ∼10-fold lower affinity than the mAHRb but only a ∼2-fold higher affinity compared with the hAHR for typical AHR ligands like TCDD (Poland and Glover, 1990; Ramadoss and Perdew, 2004). Previous investigations have demonstrated that the low relative ligand binding affinity displayed by the hAHR and mAHRd is due to a valine at residue 381 in the hAHR and residue 375 in mAHRd. Substituting valine 381 with an alanine residue (hAHR-V381A) has therefore been shown to enhance the relative ligand binding affinity of the hAHR to equal that of the mAHRb and enhanced photoaffinity ligand binding capacity (Ema et al., 1994; Ramadoss and Perdew, 2004). Because the mAHRd is thermally unstable at room temperature (Poland and Glover, 1990), the thermally stable yet binding-deficient low-affinity mAHRb point mutant in which alanine 375 is substituted by valine (mAHR-A375V) was instead used in competitive ligand binding experiments. To determine whether valine 381 indeed was responsible for the higher relative affinity the hAHR displayed for indirubin, competitive ligand binding assays were conducted using increasing doses of indirubin and cytosol isolated from Cos-1 cells transiently transfected with either hAHR, hAHR-V381A, mAHR, or mAHR-A375V constructs. The level of expression of each receptor was similar (Fig. 5C). We were surprised to find that, compared with the hAHR, the V381A substitution slightly reduced the relative ligand binding affinity of the hAHR for indirubin (Fig. 5A). In contrast, the mAHR-A375V mutation did not significantly alter the difference in relative binding affinity between the low-affinity mAHR-A375V and high-affinity mAHR for indirubin relative to the PAL.

The high ligand affinity V381A substitution in the hAHR ligand binding domain does not enhance hAHR relative ligand binding affinity for indirubin, and the hAHR C-terminal transactivation domain does not influence hAHR ligand binding affinity for indirubin. A, Val381 substitution does not enhance the relative ligand binding affinity of the hAHR for indirubin. B, the C-terminal transactivation domain of the hAHR or mAHR does not affect the relative affinity of each receptor for indirubin. C, COS-1 cells were transfected with 20 μg of pCI-hAHR, pCI-hAHRV381A, pcDNA3-mAhR, or pcDNA3-mAHRA375V, h-mAHR and m-hAHR, using LipofectAMINE reagent. Cytosol isolation and competitive ligand binding was carried out as described previously.
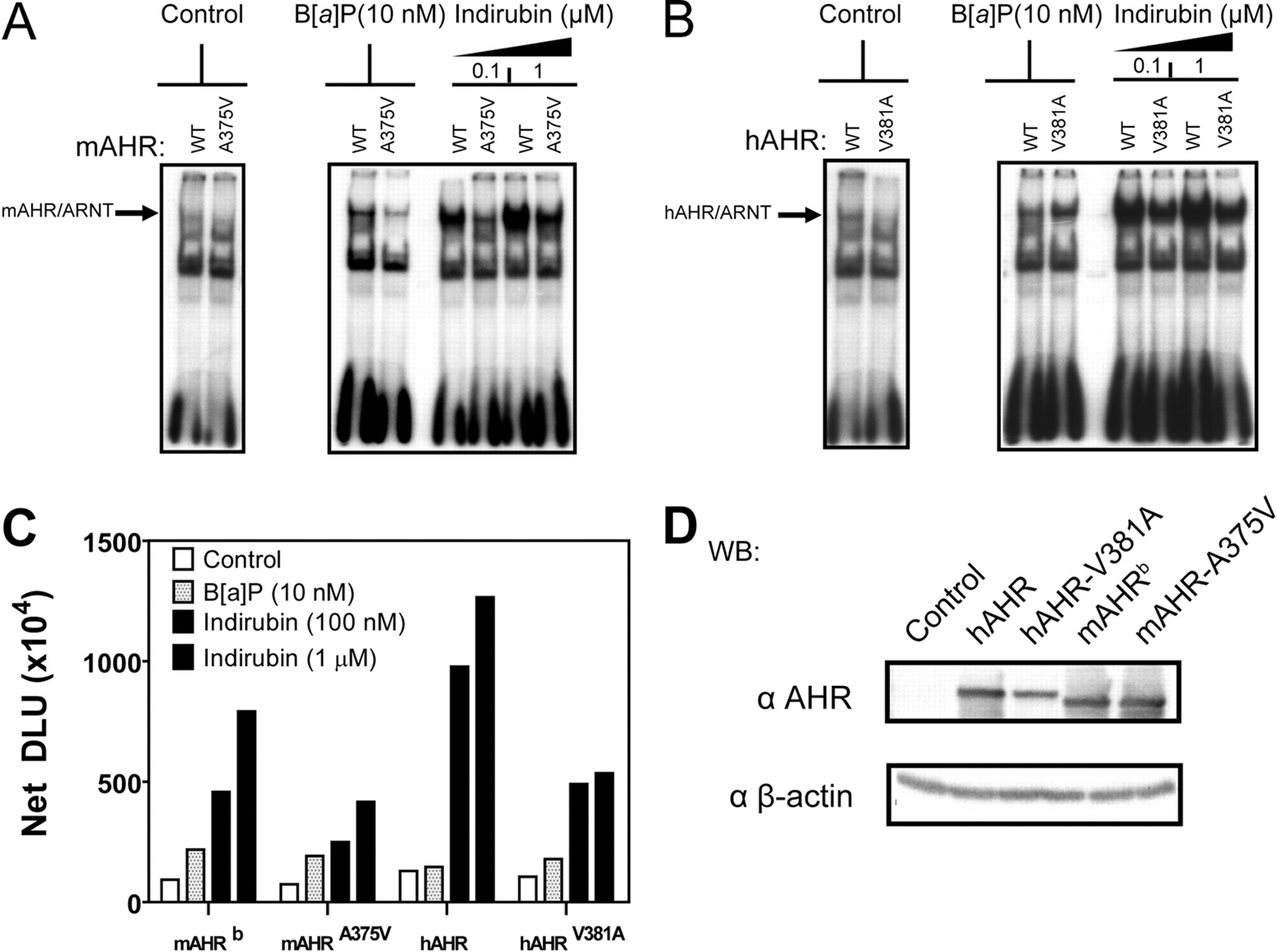
Transformation of hAHR by indirubin is disrupted by V381A substitution. A to C, Electrophoretic mobility shift assays: pCI-ARNT, pCI-hAHR, pCI-hAHRV381A, pcDNA3-mAHRb, and mAHRA375V were in vitro-translated using the TNT-coupled rabbit reticulocyte lysate system. In vitro-translated AHR proteins were subjected to EMSA using 10 nM B[a]P and 0.1 and 1 μM indirubin and then quantified as described previously. D, Western blot of in vitro-translated rabbit reticulocyte lysate. Protein was resolved using 8% tricine-SDS-polyacrylamide gels. Proteins were transferred to PVDF membrane, and AHR protein was detected using the mouse monoclonal antibody RPT1 and visualized using autoradiography. AHR bands were quantified using a multiple purpose filmless autoradiographic analysis and OptiQuant software and presented as DLUs. *, p < 0.05.
The C-Terminal Transactivation Domain of the hAHR and mAHR Does Not Affect the Relative Affinity of Each Receptor for Indirubin. Cos-1 cells were transiently transfected with the chimeric constructs h-mAHR and m-hAHR. These chimeric receptors have the C-terminal domains swapped as described previously (Ramadoss and Perdew, 2005). These receptors were used to investigate whether differences between the human and mouse receptor transactivation domains or three-dimensional folding of the receptors may have contributed to the observed differences in relative ligand binding affinities between the mAHR and hAHR. Competitive ligand binding assays with indirubin involving the hAHR and mAHR protein chimeras h-mAHR and m-hAHR showed no difference in the relative ligand binding affinities compared with that observed with the full-length hAHR and mAHR, respectively (Figs. 3C and 5B). The level of chimeric receptor expression was similar in the cytosol used for ligand binding (Fig. 5C). These results indicate that the C-terminal transactivation domain does not modulate ligand binding affinity.
Transformation of hAHR by Indirubin Is Disrupted by V381A Substitution. To elucidate whether Val381 or Ala375 is critical for efficient indirubin stimulated transformation of hAHR and mAHR, respectively, EMSAs were performed using in vitro-translated hAHR, hAHR-V381A, mAHR, and mAHR-A375V. For the mAHR, the A375V substitution reduced mAHR transformation in response to indirubin (Fig. 6, A and C). We were surprised to find that, for the hAHR, the V381A substitution also reduced (∼2-fold) the efficiency of indirubin-mediated receptor transformation (Fig. 6, B and C). This suggests that the presence of the valine residue at position 381 does not reduce hAHR receptor affinity for indirubin but does decrease B[a]P binding and subsequent transformation.
Discussion
In the past, studies aimed at examining the physiological function of the AHR and its role in mediating polycyclic aromatic hydrocarbon/halogenated aromatic hydrocarbon-driven toxicity and carcinogenesis have used rodent models, with the expectation that the data can be extrapolated to humans. The hAHR has a 10-fold lower affinity for typical AHR ligands like TCDD compared with the most studied mAHRb allele. However, a number of recent investigations have demonstrated that the hAHR may possess a significant number of contrasting properties compared with the mAHR, which may limit the reliability of using rodent model systems to predict hAHR function. High homology of the hAHR to the guinea pig AHR, the rodent most sensitive to TCDD, suggested that humans might also be highly responsive to TCDD (Korkalainen et al., 2001). In addition, examination of hAHR ligand specificity compared with those of zebrafish and rainbow trout AHRs revealed that mono-ortho polychlorinated biphenyls activated hAHR but were not very effective in activating either zebrafish or rainbow trout AHRs (Abnet et al., 1999), suggesting that the hAHR may specifically bind a structurally unique subset of ligands. It is noteworthy that CYP1A1 induction in response to TCDD treatment was also shown to be most potent in human lymphocytes compared with mouse and rat lymphocytes (Nohara et al., 2006). Conversely, using a “knock-in” hAHR mouse, Moriguchi et al. (2003) demonstrated that the hAHR was resistant to TCDD-mediated toxicity compared with the mAHRd, suggesting that indeed, humans might also be resistant to TCDD-mediated toxicity. However it should be noted that this humanized AHR mouse failed to show hAHR protein expression data, thus casting doubt on the validity of this model. Likewise, studies that examined various toxicological endpoints in a myriad of animal models provided evidence supporting the traditional conclusion that impaired hAHR ligand binding correlates with TCDD resistance in vivo (Connor and Aylward, 2006). The hAHR has also been shown to differentially recruit coactivator-LXXLL motifs, which suggests that the hAHR and mAHR may actually regulate gene expression through recruitment of distinct coactivators (Flaveny et al., 2008). To comprehend the physiological and toxicological role of the hAHR, we developed a transgenic hAHR mouse that expresses hAHR only in the liver. Real-time RT-PCR analysis showed that the hAHR is less responsive to TCDD-induced activation at lower doses compared with the mAHRb in primary hepatocytes, which is consistent with numerous published studies in human cell lines. This transgenic mouse model is unique in that it allows a direct comparison of hAHR and mAHR function within the same cellular background (e.g., coactivators, response elements, etc.) in an in vivo system. Furthermore, the use of the Ahr fx/fx/CreAlb conditional knockout system as a background on which to express the hAHR in hepatocytes permits the study of the hAHR, whereas circumventing the physiological problems encountered with transgenic mice in which hAHR is expressed on an Ahr(-/-) background. For example, the isolation of hepatocytes from Ahr(-/-) mice is difficult, possibly because of aberrant hepatic vascularization (data not shown). In contrast, isolation of hAHR-expressing hepatocytes in transgenic mice on the Ahr fx/fx/CreAlb background is similar to that of C57BL/6J mice. In addition, the hAHR displayed high levels of constitutive activity and limited inducibility when expressed on an Ahr(-/-) background (Supplementary Fig. S1), which was not observed when hAHR was expressed on an Ahr fx/fx/CreAlb background.
Competitive ligand binding experiments involving a number of AHR ligands highlighted that, in contrast with the mAHR, the hAHR may display high affinity to a distinct subset of ligands that are structurally divergent from typical exogenous AHR ligands like TCDD. As expected, the mAHRb showed a higher relative ligand binding affinity for the AHR ligands B[a]P, PCB-126, and β-naphthoflavone. Yet compared with the mAHRb, the hAHR demonstrated a higher relative ligand binding affinity for quercetin and indirubin specifically. Indirubin has been shown previously to be 35- to 140-fold more potent at inducing hAHR than mAHR activity in a yeast reporter system (Kawanishi et al., 2003). In these competitive ligand binding studies, gene expression analysis and EMSA analysis all suggest that indirubin is a high-potency, high-affinity hAHR-specific ligand. The “high ligand affinity” hAHR-V381A substitution in the hAHR ligand binding domain instead partially inhibited the high binding affinity interaction and potent transformation of the hAHR by indirubin. This was in contrast with expected observations based on previous mutagenesis analysis of the ligand binding domain of the hAHR. Illuminating the specific amino acid residues that are critical to high-affinity hAHR/indirubin interaction requires further investigation. Another possible explanation is that despite the hAHR Val381 ligand binding pocket mutation, binding of the endogenous hAHR ligand(s) is still conserved. Indeed, the hAHR ligand binding pocket may actually use distinct residues to stably bind to traditionally defined low-affinity and high-affinity ligands (Backlund and Ingelman-Sundberg, 2004). These results suggest that the hAHR ligand binding pocket may not actually be functionally impaired but instead structurally adapted to binding ligands, which are in structural contrast with known high-affinity exogenous mAHR ligands. This fact has implications for future investigations aimed at discovering high-affinity endogenous hAHR ligands. Future studies therefore should take into account that potential hAHR ligands may be structurally distinct from high-affinity mAHR ligands. Furthermore, the hAHR may indeed have unique molecular properties that contrast with the mAHR, which highlight the limitations of using rodent AHRs in model systems geared toward understanding the role of the hAHR in gene regulation, toxicity, and carcinogenesis. The hAHR-expressing transgenic mouse lines described here may therefore be valuable for testing several hypotheses relevant to hAHR activity that may not be discerned in typical rodent models.
Acknowledgments
We thank Dr. Christopher Bradfield for kindly providing the Ahrfx/fxCreAlb mice and helpful discussions.
Footnotes
-
This work was supported by National Institutes of Health National Institute of Environmental Health Sciences [Grant ES04869]; the Natioanl Institutes of Health National Center for Research Resources [Grant 1C06-RR14520]; and the Dow Chemical Company.
-
ABBREVIATIONS: AHR, aryl hydrocarbon receptor; PAL, 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin; EMSA, electrophoretic mobility shift assay; ARNT, aryl hydrocarbon receptor nuclear translocator; DRE, dioxin-responsive element; B[a]P, benzo[a]pyrene; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; Ttr, transthyretin; PCR, polymerase chain reaction; RT-PCR, reverse transcriptase polymerase chain reaction; bp, base pair; MOPS, 3-(N-morpholino)propanesulfonic acid; PVDF, polyvinylidene difluoride; HEDG, HEPES/EDTA/sodium molybdate/glycerol; DLU, digitized light unit; PAGE, polyacrylamide gel electrophoresis.
-
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material. - Received January 15, 2009.
- Accepted March 19, 2009.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}