


Abstract
During the last several years, evidence that various enzymes hydrolyze NAD into bioactive products prompted scientists to revisit or design strategies able to increase intracellular availability of the dinucleotide. However, plasma membrane permeability to NAD and the mitochondrial origin of the dinucleotide still wait to be clearly defined. Here, we report that intracellular NAD contents increased upon exposure of cell lines or primary cultures to exogenous NAD (eNAD). NAD precursors could not reproduce the effects of eNAD, and they were not found in the incubating medium containing eNAD, thereby suggesting direct cellular eNAD uptake. We found that in mitochondria of cells exposed to eNAD, NAD and NADH as well as oxygen consumption and ATP production were increased. Conversely, DNA repair, a well known NAD-dependent process, was unaltered upon eNAD exposure. We also report that eNAD conferred significant cytoprotection from apoptosis triggered by staurosporine, C2-ceramide, or N-methyl-N′-nitro-N-nitrosoguanidine. In particular, eNAD reduced staurosporine-induced loss of mitochondrial membrane potential and ensuing caspase activation. Of importance, pharmacological inhibition or silencing of the NAD-dependent enzyme SIRT1 abrogated the ability of eNAD to provide protection from staurosporine, having no effect on eNAD-dependent protection from C2-ceramide or N-methyl-N′-nitro-N-nitrosoguanidine. Taken together, our findings, on the one hand, strengthen the hypothesis that eNAD crosses the plasma membrane intact and, on the other hand, provide evidence that increased NAD contents significantly affects mitochondrial bioenergetics and sensitivity to apoptosis.
Introduction
During the last several years we witnessed renewed interest in the biochemistry and pharmacological potential of pyridine nucleotides NAD(H) and NADP(H). Rescued from the oblivion in which they had fallen because of the widespread view of their exclusive participation in redox metabolism, pyridine nucleotides are now known to be involved in pleiotypic cellular processes of key relevance to cell homeostasis and death (Imai, 2009; Koch-Nolte et al., 2009). Recent evidence that expression of NAD-synthesizing and -consuming enzymes and the ensuing cellular NAD contents follow circadian rhythms (Nakahata et al., 2009) further emphasizes the relevance of the dinucleotide to cell physiology.
NAD is now known to be a bona fide substrate of various enzymes involved in survival, death, signaling, inflammation, and neoplastic transformation. Therefore, the pyridine nucleotide pool is not stable as previously envisaged, but is highly dynamic, being continuously resynthesized upon its hydrolysis operated by various NAD-consuming enzymes. This knowledge prompted strong interest in the biological properties of products originating from NAD hydrolysis and also encouraged scientists in different fields to revisit the biochemistry of NAD neosynthesis from tryptophan, as well as NAD rescue from nicotinamide (Bogan and Brenner, 2008).
Poly(ADP-ribose) polymerases (PARPs) are proteins responsible for the majority of NAD consumption both under constitutive conditions and when hyperactivated by genotoxic stress (Rouleau et al., 2010). It is noteworthy that basal activity of PARP-1 has been reported at complex DNA architectures such as hairpins or cruciform structures in the absence of DNA damage (Potaman et al., 2005). PARP-1-dependent poly(ADP-ribosyl)ation has been the focus of intense investigation because of its active role in DNA repair, inflammation, and cell death. Potent PARP-1 inhibitors recently reached the clinical arena and are used in clinical trials for treatment of various types of solid neoplasms (Rouleau et al., 2010). At variance with poly(ADP-ribosyl)ation, mono(ADP-ribose) transfer is less understood (Di Girolamo et al., 2005), and evidence is accumulating that putative PARPs are indeed enzymes able to exclusively catalyze mono(ADP-ribose) transfer reactions (Hottiger et al., 2010). Additional NAD-consuming enzymes are sirtuins, a group of seven proteins (SIRT1–7) with various intracellular distribution able to catalyze the transfer of an acetyl group from an acetylated protein to the ADP-ribose moiety of NAD with the formation of O-acetyl-ADP-ribose (Finkel et al., 2009). The NAD consumption potential of sirtuins is thought to be significantly lower than that of PARPs, even though sirtuins are emerging regulators of numerous cellular processes spanning from cell death to life span elongation (Finkel et al., 2009). The ADP-ribosyl cyclases/NAD glycohydrolases CD38 and CD157 are supplementary NAD-consuming enzymes able to form ADP-ribose, cyclic ADP-ribose, and nicotinic acid adenine dinucleotide phosphate (Malavasi et al., 2008). Although the first two compounds appear structurally similar, they are endowed with different functional properties, with ADP-ribose being able to bind the macro domain of proteins and also activate Ca2+-permeable TRPM2 membrane receptors (Perraud et al., 2001; Till and Ladurner, 2009), and cyclic-ADP-ribose acting as an agonist of the endoplasmic reticulum Ca2+-channel ryanodine receptor (Guse et al., 1999). Nicotinic acid adenine dinucleotide phosphate also regulates intracellular calcium release (Koch-Nolte et al., 2009).
Ongoing intracellular NAD consumption allows us to hypothesize that under basal or stressful conditions the dinucleotide content may become limiting in specific cell compartments. Addressing this issue is of remarkable relevance to cell biology because we would identify an unprecedented regulatory step of different metabolic pathways. In addition, limiting NAD concentrations would also be a pharmacological target of potential therapeutic significance. Compounds able to boost or reduce NAD contents, indeed, might be original tools to pharmacologically modulate specific cellular functions (Belenky et al., 2007; Sauve, 2008). Metabolic NAD precursors have traditionally been used to increase its intracellular contents and, recently, also to boost the activities of the NAD-dependent enzymes. However, it is still debated whether NAD per se can cross the plasma membrane. Although the canonical view considers NAD unable to permeates lipid bilayers (Di Lisa and Ziegler, 2001), several studies report evidence for exogenous NAD (eNAD) uptake by different cells (Bruzzone et al., 2001; Ying et al., 2003; Alano et al., 2004, 2010; Araki et al., 2004; Pillai et al., 2005; Qin et al., 2006; Billington et al., 2008; Wang et al., 2008). In the present study, we further investigated cellular uptake of eNAD and also focused on its possible functional effects as indirect evidence of eNAD entrance.
Materials and Methods
Cell Culture Conditions, Transfection, Death Assay, and ROS Production.
HeLa, RAW, and HepG2 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 2 mM glutamine, 10% fetal bovine serum, and antibiotics. Cultures were brought to 50 to 70% confluence and used for the experiments. Pure neuronal cultures were prepared by seeding cortical cells obtained from 16-day-old rat embryos as described previously (Chiarugi, 2002). Neurons were cultured in Neurobasal medium with B-27 supplement (Invitrogen, Carlsbad, CA) and 0.5 mM glutamine onto poly-d-lysine-coated multiwell plates. Cells were used after 7 and 15 days in vitro (DIV) from preparation. Rat glia cultures were prepared as described previously (Chiarugi and Moskowitz, 2003) and used at 15 DIV. Cell cultures were exposed to NAD, ADP-ribose, nicotinamide (Nam), nicotinamide mononucleotide (NMN), nicotinamide riboside (NR), AMP, N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), C2-ceramide, staurosporine (STP), H2O2, 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide (EX-257), or other compounds directly dissolved in the culture media. NR was synthesized as described by Yang et al. (2007b). Cell transfection was performed as described previously (Cipriani et al., 2005) by using siRNA for SIRT1 from QIAGEN (Milan, Italy). Cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay and phase-contrast microscopy (Cipriani et al., 2005). Caspase 3 activity was evaluated by means of a fluorimetric kit (Invitrogen). An inverted Nikon TE-2000U microscope equipped with a change-coupled device camera was used for cell visualization. ROS have been quantified by means of the dichlorofluorescein assay.
NAD, NADH, and ATP Measurement.
NAD(H) contents were quantified by means of an enzymatic cycling procedure according to Cipriani et al. (2005). In brief, cells grown in a 48-well plate were killed with 50 μl of 1 N HClO4 and then neutralized with an equal volume of 1 N KOH for NAD measurement, whereas the opposite was done for NADH extraction with an additional heating (60°C, 30 min) between KOH and HClO4. After the addition of 100 μl of 100 mM Bicine (pH 8), 50 μl of the cell extract was mixed with an equal volume of Bicine buffer containing 23 μl/ml ethanol, 0.17 mg/ml MTT, 0.57 mg/ml phenazine ethosulfate, and 10 μg of alcohol dehydrogenase. The mixture was kept at room temperature for 20 min and then absorbance at 550 nm was measured. The amount of NAD consumed upon exposure to H2O2 was calculated by subtracting the amount of NAD measured in cells at various time points after H2O2 challenge to that present in control cells. A standard curve allowed quantification of NAD. Intracellular NADH content were evaluated by analyzing cell autofluorescence as reported previously (Cipriani et al., 2005). In brief, autofluorescence was visualized or measured with a microscopic apparatus (Nikon 2000-TE; Nikon, Tokyo, Japan) equipped with a UV filter (excitation 365 nm) and a charge-coupled device camera or spectrometer. The cellular ATP content and mitochondrial ATP production were measured by means of an ATPlite kit from PerkinElmer Life and Analytical Sciences (Zaventem, Belgium) as described previously (Cipriani et al., 2005).
Mitochondrial Isolation and Membrane Potential Analysis.
Mitochondria were isolated from cells using a glass/glass homogenizer in 500 μl of extraction buffer and centrifuged at 600g as described previously (Formentini et al., 2009). In brief, supernatants were centrifuged at 7000g to obtain the mitochondrial pellet, which was denatured with perchloric acid for NAD content determination. The mitochondrial membrane potential (ΔΨm) was evaluated by means of a spectrofluorometer (Victor3; PerkinElmer Life and Analytical Sciences, Milan, Italy). In brief, tetramethylrhodamine ethyl ester perchlorate fluorescence was quantitated in cells grown in a 48-well plate, exposed or not to staurosporine 300 nM in the presence or absence of NAD for 2 and 6 h. Tetramethylrhodamine ethyl ester perchlorate was added to the culture media at 2.5 nM 30 min before fluorescence determination.
Western Blotting.
For Western blotting, cells were scraped, collected in Eppendorf tubes, centrifuged (1500g for 5 min at 4°C) and resuspended in lysis buffer (50 mM Tris, pH 7.4, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 4 μg/ml aprotinin and leupeptin, and 1% SDS). Twenty to 40 μg of protein/lane were loaded. After 4 to 20% SDS-polyacrylamide gel electrophoresis and blotting, membranes (Immobilon-P; Millipore Corporation, Bedford, MA) were blocked with phosphate-buffered saline (PBS) containing 0.1% Tween 20 and 5% skim milk (PBST-5% milk) and then probed overnight with primary antibodies (1:1000 in PBST-5% milk). The anti-PAR monoclonal antibody (10H) was from Alexis (Vinci, Italy). Membranes were then washed with PBST and incubated for 1 h in PBST-5% milk containing an anti-mouse peroxidase-conjugated secondary antibody (1:2000). After washing in PBST, ECL reagent (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) was used to visualize the peroxidase-coated bands.
PCR Assays.
One microgram of total RNA isolated from treated or untreated cells was retro-transcribed using an iScript kit (Bio-Rad Laboratories, Hercules, CA) and amplified with the following specific primers: SIRT1, forward 5′-GAACAGGTTGCGGGAATCC-3′ and reverse 5′-CAAGCGGTTCATCAGCTGG-3′; and 18S ribosomal RNA forward 5′-CGGCTACCACATCCAAGGAA-3′ and reverse 5′-GCTGGAATTACCGCGGCT-3′. Real-time PCR assays were performed with a Rotor-Gene SYBR Green PCR Kit (QIAGEN, Milan, Germany) and analyzed using the Rotor-Gene 3000 cycler system (Corbett Research, Mortlake, VIC, Australia). Semiquantitative PCR was performed as described previously (Formentini et al., 2009). For semiquantitative PCR, the protocol described by Formentini et al. (2009) was adopted using the primers described above.
Mitochondrial and Cytosolic ATP Measurement.
ATP was indirectly quantified by means of cytosol- or mitochondria-targeted luciferase. Transfection with cytosolic or mitochondrial luciferase cDNA (4 μg/ml) was performed by the standard calcium-phosphate procedure. Cell luminescence was evaluated 36 h after transfection by means of a luminometer. As previously reported, recordings did not exceed 15 min because longer measurements may be biased by factors other than intracellular ATP such as CoA or luciferin availability, pH as well as oxyluciferin, and oxygen radical formation.
Comet Assay.
DNA strand breaks were evaluated by the comet assay. Cells were treated or not with 100 μM H2O2 in PBS on ice for 5 min. Later, cells were trypsinized, whereas those used for evaluation of DNA repair were incubated for a further 2 h at 37°C after substitution of the H2O2 solution with fresh culture medium and then run through the comet assay. In brief, after trypsinization cells were centrifuged and then resuspended in low melting point agarose and layered on microscopy slides to be run through the comet assay. Microscopic analysis was performed using a Labophot-2 microscope (Nikon) provided with epifluorescence. Each experimental point was run in duplicate, and the images of 50 randomly chosen nuclei per slide were captured and analyzed using the comet assay IV image analysis system (Perceptive Instruments, Haverhill, Suffolk, UK). The program calculated the total fluorescence of each nucleus and the fluorescence distribution of the head and tail of the comet. Data are expressed as a percentage of total fluorescence migrated in the tail for each nucleus (percent DNA in tail), a parameter linearly related to the number of DNA breaks. The mean percent DNA in tail of 50 nuclei/slide was calculated, and the duplicate values were further averaged. The value of % DNA in tail obtained estimated the basal and induced DNA damage in terms of strand breaks.
Oxygen Consumption Analysis.
Quantitation of oxygen consumption by cells exposed or not to NAD for 6 h was conducted by means of the Oxygraph system (Hansatech Instruments, Norfolk, UK). Cells (250,000) were loaded in the chamber in 400 μl of respiration buffer (70 mM sucrose, 220 mM mannitol, 2 mM HEPES, pH 7.4, 5 mM MgCl2, 5 mM K2HPO4, 1 mM EDTA, and 0.1% bovine serum albumin), and oxygen consumption was monitored for 10 min at 37°C.
NAD Precursor Quantitation by LC-Tandem Mass Spectrometry.
Growth media were replaced with PBS, and cells were exposed to 1 mM NAD for 1 h, directly dissolved in the solution. After 1 h, the culture medium was extracted with methanol for protein precipitation, centrifuged at 14,000 rpm for 15 min, lyophilized, and then resuspended in 100 μl of water-3% CH3CN. Five microliters of the solution were injected into a high-performance liquid chromatography series 200 micro pump apparatus (PerkinElmer Life and Analytical Sciences) and chromatographed using a ZIC-HILIC column (50 × 2.1 mm i.d. 3.5-μm particle size 200 Å; Merck SeQuant; Umeå, Sweden). Mobile phase A consisted of water containing 3% CH3CN and 10 mM ammonium acetate, and mobile phase B was CH3CN containing 3% water and 10 mM ammonium acetate. An elution gradient was performed going from 100 to 40% B in 20 min; B was held at 40% for 7 min, and then brought back to the starting value in 1 min. The equilibration time before the next injection was 10 min. The eluate, at a flow rate of 200 μl/min, was directly introduced into the LC-mass spectrometry system interface. The LC-tandem mass spectrometry system consisted of a Perkin Elmer Sciex (Thornhill, ON, Canada) API 365 triple quadrupole mass spectrometer equipped with a TurboIonSpray interface. The compounds were ionized by positive ion electrospray and detected using multiple reaction monitoring. The needle and orifice voltages were adjusted to 6.0 kV and 61 V, respectively. The drying gas flow rate and temperature were 7 l/min and 250°C, respectively. The first and third quadrupole mass analyzers were operated at unit mass resolution. Fragmentation was accomplished with a collision energy of 40 eV; nitrogen was used as the collision gas at a collision gas thickness of 2.6 × 1015 molecules · cm−2. The ion transitions monitored were m/z 123.0 → 79.9 for nicotinamide, 335.1 → 123.1 for NMN, 348.1 → 135.9 for AMP, 560.0 → 349 for ADP-ribose, and 664.0 → 428.2 for NAD. These transitions were selected on the basis of the triple quadrupole collision-induced dissociation product ion spectrum of the analyte.
Results
eNAD Is Transported Intact across the Plasma Membrane.
We first attempted to investigate whether eNAD can cross the plasma membrane and increase intracellular NAD (iNAD) content. Cells were incubated in the growth medium, and the nucleotide dissolved in saline was directly added to the medium itself. As shown in Fig. 1A, exposure of HeLa cells to different concentrations of eNAD led to significant increases of iNAD. In particular, 1 μM eNAD did not increase the cellular pool even after prolonged incubation times. When eNAD was raised up to 10 μM, a tendency for an increase in iNAD contents was noted after 6 h. Exposure to 100 μM or 1 mM concentrations caused significant increases in iNAD (221 ± 18 and 342 ± 28% after 6 h, respectively). It is noteworthy that iNAD contents were not increased when cell exposure to 1 mM eNAD was conducted at 4°C (Fig. 1A). When cultures of mouse macrophages (RAW242 cells), human hepatoma cells (HepG2), or rat primary astrocytes were exposed to 1 mM eNAD for 6 h, iNAD increments were also obtained (Fig. 1B). Of interest, iNAD increased in cortical neurons exposed to 1 mM eNAD only in cultures of 15 DIV, whereas it was ineffective in cultures of 7 DIV (Fig. 1B).

Effects of eNAD and metabolic precursors on iNAD contents in various cell lines. A, iNAD increase upon exposure of HeLa cells to eNAD is time-, concentration-, and temperature-dependent. B, increase of iNAD upon exposure of different cell lines (RAW mouse macrophages or C6 rat glioma cells) or primary astrocyte or neuronal cultures to 1 mM NAD for 6 h. Neuronal cultures of 7 or 15 DIV were used. C, effect of different NAD precursors added to the culture medium (1 mM for 6 h) on iNAD content in HeLa cells. D, effect of 1 mM eNAD on iNAD depletion induced by FK866 (100 μM for 6 h). In A, each point represents the mean ± S.E.M. of four experiments conducted in duplicate. In B to D, each column represents the mean of three experiments conducted in duplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001, versus control (i.e., without eNAD addition); analysis of variance + Tukey post hoc test. ADPR, ADP-ribose.
Although previous findings demonstrated that iNAD contents increased upon exposure to eNAD, they do not prove that eNAD crosses the plasma membrane intact. In principle, eNAD might be hydrolyzed into cell-permeable metabolites that, once they have crossed the plasma membrane, reform NAD through the rescue pathway. To address this issue, we exposed HeLa cells to different NAD precursors alone or in combination, at concentrations identical to that of eNAD (1 mM) and measured their effects on iNAD contents. We reasoned that eNAD could be hydrolyzed by phosphodiesterases/pyrophosphatases into AMP and NMN or by NADases/NAD glycohydrolases into ADP-ribose and nicotinamide. The possibility that NMN could undergo dephosphorylation by membrane nucleotidases forming nicotinamide riboside was also taken into account. We found that, as expected, nicotinamide, nicotinamide riboside, and NMN exposure augmented iNAD, although to an extent several fold lower than that prompted by NAD. Of interest, ADP-ribose reduced both basal NAD contents and those increased by nicotinamide, whereas AMP had no effect (Fig. 1C). These findings are at odds with the hypothesis that eNAD increases iNAD contents because of extracellularly formed NAD precursors. To corroborate this assumption, by means of LC-mass spectrometry we also checked whether these NAD derivatives were present in the medium of cells exposed to eNAD. We optimized the analytical method to reach a sensitivity threshold of 5.3, 1.16, 1.15, 3.6, and 2.18 ng for ADP-ribose, AMP, nicotinamide, NMN, and nicotinamide riboside, respectively. Under our experimental settings, despite cells being incubated for 2 or 6 h in 1 ml of incubating solution (PBS added with 5 mM glucose and 10% fetal bovine serum) containing 1 mM NAD (corresponding to 666 μg of NAD), none of the above-mentioned NAD metabolites was found in the incubating medium (not shown). Taken together, these findings suggested that eNAD permeates the plasma membrane intact. Finally, we analyzed the effect of eNAD on iNAD depletion induced by N-[4-(1-benzoyl-4-piperidinyl)butyl]-3-(3-pyridinyl)-2E-propenamide (FK866), a nicotinamide phosphoribosyltransferase inhibitor. We found that the effect of FK866 [used at 100 μM to prompt rapid depletion of iNAD (Pittelli et al., 2010)] was completely abolished in the presence of eNAD (Fig. 1D).
Mitochondria Sense eNAD.
Mitochondria contain conspicuous amounts of intracellular NAD, although the contents vary according to the different cell types (Alano et al., 2007). It is also accepted that the internal membrane of the organelle is impermeable to pyridine nucleotides, thereby leaving unanswered the key question of the origin of mitochondrial NAD. Another question of central relevance to cellular homeostasis that still waits an answer is whether mitochondrial NAD(H) content is limiting the enzymatic reactions that use the nucleotide within the matrix, including the Krebs' cycle. To our knowledge, whether eNAD alters mitochondrial bioenergetics has not been investigated. It is remarkable that we found that eNAD was able to increase the mitochondrial content from 3.1 ± 0.2 to 7.3 ± 0.7 nmol/mg protein, whereas nucleocytosolic increments were more contained, increasing from 1.3 ± 0.3 to 2.3 ± 0.2 nmol/mg protein (Fig. 2A). Considering that the Krebs' cycle rapidly converts NAD into NADH, we then evaluated the cellular content of the latter. Figure 2B shows that NADH levels were also increased in eNAD-exposed cells. Therefore, cell autofluorescence, a classic index of reduced pyridine dinucleotide contents (Cipriani et al., 2005), increased in cells exposed to eNAD. As shown in Fig. 2C, autofluorescence was present in round- and rod-shaped bodies scattered throughout the cytoplasm showing the prototypical mitochondrial morphology. It is noteworthy that mitochondrial autofluorescence substantially increased in HeLa cells exposed to 1 mM NAD for 6 h (Fig. 2C). Acquisition of the autofluorescence emission spectrum obtained by means of a 356-nm excitation filter gave a peak of 450 nm, which is typical of NAD(P)H, further suggesting that the increase in fluorescence intensity in cells exposed to eNAD is due to reduced pyridine dinucleotide accumulation (Fig. 2D). A concomitant mitochondrial increase of NAD and NAD(P)H in eNAD-exposed cells rules out the possibility that increased NADH content in the organelles is due to cytosolic transfer of reducing equivalents through the mitochondrial malate-aspartate shuttle. Given that cytosolic NAD is essential for glycolysis and that NADH plays a key role in mitochondrial bioenergetics by donating electrons to respiratory complex I, we next investigated whether eNAD affects cellular energy production. ATP contents readily increased in cells exposed to 1 mM NAD for 1 to 6 h (Fig. 2E). To ascertain whether the increase of ATP was due to mitochondrial production, we starved cells overnight in the absence of glucose and in the presence of the mitochondrial energy substrates glutamine and pyruvate. Under these experimental conditions, eNAD was still able to increase cellular ATP contents (Fig. 2F), suggesting that mitochondria contribute to the eNAD-dependent ATP increase. Furthermore, given that ATP increase at 1 h did not differ in the absence or presence of glucose (4.9 ± 1.1 and 4.8 ± 0.7 nmol of ATP/mg protein, respectively) (Fig. 2F), data suggested that mitochondria were mainly responsible for eNAD-dependent increased energy production. To substantiate this assumption, we next transfected HeLa cells with cytosolic or mitochondrial luciferase, a strategy allowing discrimination of ATP production in the two cell compartments. As shown in Fig. 2G, the eNAD-dependent increase of ATP production occurred not only in the cytosol but also in the mitochondria. As further confirmation of the increased metabolic activity of mitochondria of eNAD-exposed cells, we found that oxygen consumption was higher in cells exposed for 6 h to 1 mM eNAD (Fig. 3, A and B). However, despite the increased respiratory activity, eNAD did not increase reactive oxygen species formation (Fig. 3C). As a whole, these findings suggest that eNAD reaches the mitochondrial matrix and boosts oxidative phosphorylation and ATP production upon conversion into NADH. In this regard, it is worth noting that the increased availability of ATP might promote NMNAT activity that, in turn, contributes to the increased iNAD contents.

eNAD affects mitochondrial bioenergetics of HeLa cells. Effects of eNAD (1 mM for 6 h) on nucleocytosolic and mitochondrial NAD content (A) or intracellular NADH levels (B). Rotenone (ROT, 10 μM for 1 h) is used as a positive control for the NADH increase. C, effects of eNAD (1 mM for 6 h) on cell autofluorescence. D, autofluorescence emission spectra of control or eNAD-exposed (1 mM for 6 h) cells. E, effects of 1 mM eNAD on whole-cell ATP contents. F, ATP contents in cells exposed to 1 mM eNAD for 30 or 60 min under control or glucose starvation (overnight) conditions. G, effects of eNAD (1 mM for 5 h) on photon emission from cytosolic (Cyt-Lucif) or mitochondrial (Mit-Lucif) luciferase-transfected cells. Each column represents the mean ± S.E.M. of at least three experiments. In images (C) or spectra (D), representative results of three experiments are shown. *, p < 0.05; **, p < 0.01; ***, p < 0.001, versus control (A, B, E, and F) or luciferase-transfected cells (G). Analysis of variance + Tukey post hoc test. Scale bar, 10 μm.

Effects of eNAD on oxygen consumption by HeLa cells. A, representative experiment of the effects of eNAD (6-h preincubation at 1 mM) on oxygen consumption. One experiment representative of six is shown. The arrow indicates the time when cells were added to the respiration buffer. B, oxygen consumption rate in control or eNAD-exposed (6-h preincubation at 1 mM) cells is shown. C, ROS production in cells exposed to eNAD (1 mM for 6 h) or to the complex III inhibitor antimycin (100 μM for 2 h, positive control). Each column represents the mean ± S.E.M. of six (B) or three (C) experiments. **, p < 0.01, versus control. Analysis of variance + Tukey post hoc test.
eNAD Does Not Improve DNA Repair.
Because of the key role of PARP-1 in single and double DNA strand break repair and the large consumption of NAD attributed to the enzyme during genotoxic stress (Rouleau et al., 2010), we wondered whether eNAD exposure could promote PARP-1-dependent DNA repair. HeLa cells were exposed to H2O2, a prototypical PARP-1 activator leading to activation of the base excision repair apparatus. A large body of evidence demonstrates that PARP-1 activity is instrumental for proper assembly and function of the base excision repair machinery (Fisher et al., 2007). We first wondered whether the kinetics of PARP-1-dependent NAD depletion differed between control and eNAD-exposed cells. As evidenced by the increased PAR content, the challenge of HeLa cells with H2O2 led to a significant increase of PARP-1 activity (Fig. 4A). Of interest, we found that, upon H2O2 exposure, the amount of NAD consumed by PARP-1 was higher in eNAD-challenged cells, suggesting that nuclear NAD is limiting PARP-1 activity, at least under these experimental conditions (Fig. 4B).

Effects of eNAD on H2O2-induced poly(ADP-ribosyl)ation and DNA repair. A, Western blotting of PAR content in control (C) or H2O2-exposed cells (100 μM for 5 min). PAR appears as a smear because of the increased molecular weight of poly(ADP-ribosyl)ated proteins. B, iNAD consumption in cells exposed to 100 μM H2O2 for different times and preincubated or not with eNAD (1 mM for 6 h). The amount of NAD consumed upon exposure to H2O2 was calculated by subtracting the amount of NAD measured in cells at various time points after H2O2 challenge to that present in control cells. C, representative images of cell nuclei visualized with the comet assay of control cells (CRL), exposed for 5 min to 100 μM H2O2 or exposed to 100 μM H2O2 and then allowed to repair for an additional 2 h without or with a 6-h preincubation to 1 mM eNAD. D, measurement of DNA damage (percent DNA damage in tail) in cells treated as in C. A, one blot representative of four. B, each point represents the mean ± S.E.M. of three experiments conducted in duplicate. D, each column represents the mean S.E.M. of three experiments conducted in duplicate. *, p < 0.05; **, p < 0.01 versus control; #, p < 0.05, versus H2O2. Analysis of variance + Tukey post hoc test. Scale bar, 5 μm.
Next, to understand whether the increased synthesis of PAR had a functional correlate, we analyzed DNA repair in control and eNAD-exposed cells. By means of the comet assay, we first determined the lower concentrations of H2O2 able to induce an extent of DNA damage that could be repaired within a reasonably short period of time. As shown in Fig. 4C, 100 μM H2O2 caused damage that could be significantly repaired within 2 h. It is noteworthy that DNA repair did not differ in control or eNAD-exposed cells (1 mM for 6 h) (Fig. 4, C and D).
Effects of eNAD on Apoptotic Cell Death.
Because of the key role of NAD-consuming enzymes such as PARP-1 or SIRT1 in apoptotic cell death, as well as the bioenergetic status of mitochondria in activation of the apoptotic machinery, we next wondered whether eNAD exposure could affect this type of cell demise. To this end, we exposed cells to three different apoptotic triggers such as staurosporine, C2-ceramide, and MNNG. Staurosporine is a potent, nonspecific protein kinase inhibitor prompting type II apoptosis; C2-ceramide also activates mitochondria-dependent apoptosis through unknown mechanisms, whereas MNNG activates a caspase-independent apoptotic program, mainly because of mitochondrial apoptosis-inducing factor release. MNNG is an alkylating agent typically used to prompt PARP-1-dependent apoptosis (Cipriani et al., 2005). We found that eNAD exposure reduced cell death triggered by the three cytotoxic chemicals (Fig. 5, A–C). In particular, the effects of eNAD on staurosporine-dependent cell death was significant at concentrations of 100 μM and further increased at 1 mM (Fig. 5C). It is noteworthy that the cell death assay we used (MTT reduction) is based on dye reduction by respiratory complex IV, therefore, measuring electron transfer from succinate to cytochrome oxidase (Berridge and Tan, 1993) that could be biased by the increased iNAD concentrations. However, as shown in Fig. 5C, we found that MTT reduction did not differ in cells exposed 24 h to eNAD compared with control cells. One possible explanation is that the increased mitochondrial NAD availability boosts the Krebs' cycle without reaching the succinate dehydrogenase step or that increased mitochondrial NAD selectively promotes the activity of pyruvate dehydrogenase or additional NAD-dependent enzymes of the Krebs' cycle, which have a different Km for NAD. Regardless, we also evaluated eNAD cytoprotection by phase-contrast microscopy. Figure 5D shows that at 100 μM and 1 mM eNAD reduced the apoptotic appearance of adherent cells, which appeared healthy, with diminished nuclear condensation. Given the ability of eNAD to increase iNAD content, we wondered whether cytoprotection could be, at least in part, mediated by prevention of NAD depletion induced by the cell stressor. We found that NAD did not decrease up to 6 h in cells exposed to C2-ceramide or staurosporine (not shown). In contrast, MNNG induced robust NAD depletion in control cells. It is noteworthy that cells exposed to MNNG and preincubated with eNAD had NAD levels that were almost twice those of control cells (Fig. 5E), an event that could underlay eNAD-dependent protection from MNNG.
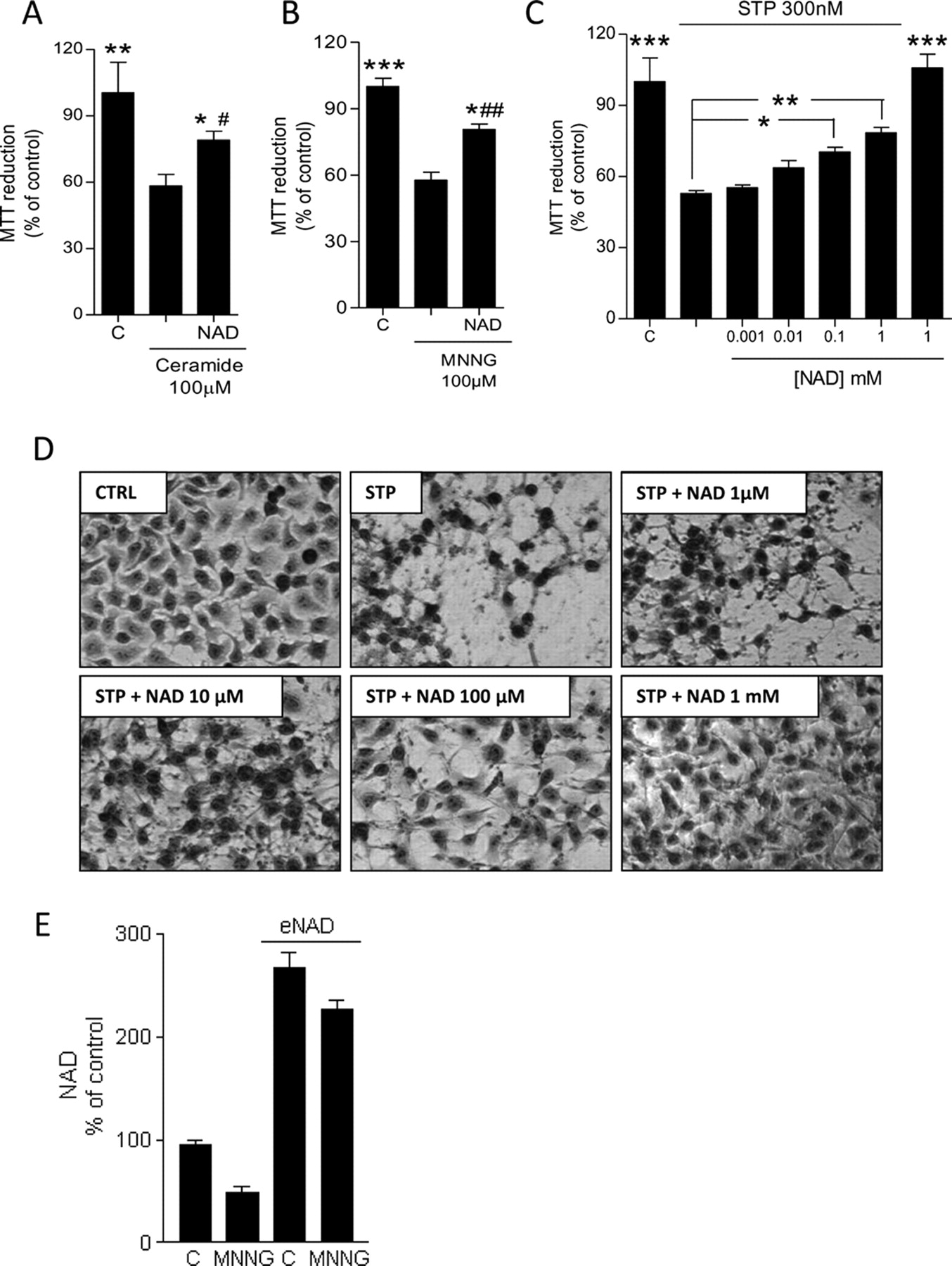
Effects of eNAD on apoptotic cell death. Effects of exposure to the indicated concentrations of eNAD (6 h) to cell death induced by C2-ceramide (A), MNNG (B), or staurosporine (C). D, phase-contrast visualization of the effects of eNAD at the indicated concentrations on staurosporine (300 nM)-induced cell death evaluated after an overnight incubation. E, effect of MNNG exposure (100 μM for 1 h) on NAD content in control or eNAD (1 mM for 6 h)-exposed cells. In A to C and E, each column represents the mean ± S.E.M. of at least three experiments conducted in duplicate. In D, representative images of three experiments are shown. *, p < 0.05; **, p < 0.01, versus the apoptotic inducer; #, p < 0.05; ##, p < 0.01 versus control. Analysis of variance + Tukey post hoc test. Scale bar, 10 μm. C, CTRL, control.
Next we attempted to investigate the mechanisms responsible for eNAD-dependent protection from C2-ceramide or staurosporine. We first focused on mitochondria because of their active role in apoptosis and the ability of eNAD to improve their bioenergetics. A key event in activation of the intrinsic apoptotic route is mitochondrial permeability transition (MPT) as a result of opening of the permeability transition pore (PTP), an event regulated by pyridine nucleotide content and their redox status. In particular, mitochondrial NADH reduces PTP opening, thereby protecting cells from apoptosis. Hence, to test whether MPT was affected by eNAD, we measured mitochondrial membrane potential in cells exposed for different times to staurosporine in the presence or absence of eNAD. Figure 6A shows that mitochondrial membrane potential steeply decreased upon 2 and 6 h of exposure to staurosporine to an extent similar to that induced by the protonophore carbonyl cyanide p-trifluoromethoxyphenylhydrazone. It is noteworthy that staurosporine-dependent depletion of mitochondrial membrane potential was in part prevented in cells exposed to eNAD (1 mM for 6 h). To gather further information that eNAD impaired activation of the intrinsic apoptotic pathway, we evaluated the dinucleotide effect on activity of caspase 3, a protein typically activated upon mitochondrial permeability transition pore opening. Consistent with data on the effect of eNAD on staurosporine-dependent loss of mitochondrial membrane potential, we found that caspase activity was drastically reduced in cells exposed to staurosporine in the presence of eNAD (Fig. 6B). It is noteworthy that eNAD-dependent reduction of caspase 3 activation was more pronounced than the ability of the nucleotide to prevent loss of mitochondrial membrane potential. These findings suggested that additional molecular mechanisms underpinned eNAD-dependent impairment of the apoptotic machinery.

Mechanisms underlying the antiapoptotic effects of eNAD in HeLa cells. A, effects of 1 mM eNAD on reduction of mitochondrial membrane potential in cells exposed for 2 or 6 h to 300 nM staurosporine. The protonophore carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (1 μM) was used as a positive control of mitochondrial membrane depolarization. B, effects of eNAD 1 mM on STP (300 nM for 6 h)-induced caspase activation. C, effects of the SIRT1 inhibitor EX-257 on eNAD-dependent STP (300 nM for overnight)-induced cell death. eNAD was incubated at the concentration of 1 mM. D, real time (a) and semiquantitative (b) PCR evaluation of SIRT1 transcript levels in control and silenced cells. 18S ribosomal RNA is shown as a loading control. D, lost of protection from staurosporine-induced apoptosis by eNAD in SIRT1-silenced cells. Cells were exposed to 1 mM eNAD and challenged with 300 nM staurosporine overnight. Each column represents the mean ± S.E.M. of at least three experiments conducted in duplicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001, versus control (B and E) or staurosporine (A and C); #, p < 0.05 versus STP. Analysis of variance + Tukey post hoc test. C, control.
We therefore reasoned that the antiapoptotic effect of eNAD could also be mediated by the increased availability of the dinucleotide to NAD-consuming enzymes. We ruled out PARP-1 involvement because inhibition, rather than activation, of the enzyme protects from apoptosis in general (Hong et al., 2004) and, in particular, from that induced by staurosporine (Simbulan-Rosenthal et al., 1999). In contrast, a key antiapoptotic role of SIRT1 emerged from numerous studies (Finkel et al., 2009). We therefore investigated whether inhibition of SIRT1 affected eNAD-dependent protection from apoptosis. To this end, we exposed cells to eNAD and staurosporine in the absence or presence of the potent and selective SIRT1 inhibitor EX-257. Figure 6C shows that the compound prevented the ability of eNAD to protect from staurosporine-induced apoptosis. It is noteworthy that EX-257 per se did not display any cytotoxic effect on resting cells. To rule out possible nonspecific effects underlying EX-257 cytoprotection and to further underscore the relevance of SIRT1 to the antiapoptotic effects of eNAD, we silenced SIRT1 by means of siRNA. Upon 48 h of exposure to siRNA, both quantitative and semiquantitative PCR demonstrated that SIRT1 transcript levels decreased to approximately 8% of control (Fig. 6D). We found that eNAD could not rescue SIRT1-silenced cells from staurosporine-induced apoptosis (Fig. 6E). However, neither inhibition nor silencing of SIRT1 affected eNAD-dependent protection form cell death induced by C2-ceramide or MNNG (not shown).
Discussion
In the present study we report that exposure to eNAD substantially increases the dinucleotide cellular pool, suggesting plasma membrane permeability. It is noteworthy that these increments take place in different cellular compartments and alter specific NAD-dependent reactions. As a whole, our findings indicate that some cellular functions are limited by pyridine nucleotide availability and can be pharmacologically enhanced by exposing cells to eNAD. In keeping with this hypothesis, a recent study demonstrated that genetic or pharmacological suppression of PARP-1 leads to increased availability of iNAD that is responsible for improved mitochondrial bioenergetics (Bai et al., 2011).
Plasma membrane permeability to pyridine nucleotides is controversial. It has been suggested that evidence for intracellular entrance of eNAD may be due to 1) binding to the plastic of the plates, 2) binding to plasma membrane without transport, and 3) extracellular hydrolysis followed by intracellular reconstitution. As for the latter possibility, we have been unable to detect NAD catabolites in the incubating media of cells exposed to eNAD, suggesting lack of extracellular NAD metabolism. Therefore, none of the catabolites added alone or in combination to the culture media is able to reproduce the effect of eNAD. It might be possible, however, that degradation of eNAD below the detection limit still produces metabolites able to enter the cell, such as nicotinamide riboside. Additional evidence that eNAD alters specific cellular functions indicates that it is bioactive and not inertly bound to plasma membrane or other surfaces. Last, data showing that eNAD entrance is abrogated in cells incubated at 4°C rules out passive diffusion and suggests the involvement of specific transport mechanisms. This is in keeping with the finding that NAD enters neurons of 15 but not of 7 DIV, suggesting that expression of specific membrane proteins during in vitro neuronal differentiation is needed to take up eNAD. In this regard, plasma membrane transporters and/or channels involved in NAD uptake have been proposed (Billington et al., 2008; Alano et al., 2010).
The ability of eNAD to increase the mitochondrial NAD content and energy production is of particular relevance. At present, indeed, we still wait for a clear picture of the origin of mitochondrial NAD. Although fluxes of NAD through membranes of isolated mitochondria have been reported (Rustin et al., 1996, 1997), it has been questioned whether membrane integrity is conserved during organelle isolation. Thus, the view that mitochondria are not permeable to pyridine nucleotides received large consensus. It is also not clear whether mitochondria have a functional NAD rescue pathway originating from nicotinamide or other precursors such as NMN or nicotinamide riboside (Berger et al., 2005; Yang et al., 2007a; Pittelli et al., 2010). Regardless, the identification of membrane NAD transporters in yeast mitochondria (Todisco et al., 2006) and intracellular vesicles of eukaryotic cells (Davis et al., 2008), together with the key biological relevance of the dinucleotide, undermines the view that mammalian mitochondria lost this carrier and became impermeable to NAD. Our findings allow us to speculate that either mitochondria take up intact NAD directly from cytoplasm or they take up NAD precursors, which are then transformed into NAD in the matrix. Although we cannot discriminate between these two possibilities, it is worth noting that the present study indicates that mitochondria sense cytoplasmic concentrations of NAD and/or its precursors, resetting their pyridine nucleotide pool accordingly. Also important is the notion that mitochondrial fluctuation of NAD contents according to those present in the cytoplasm does not seem bidirectional. Indeed, work from our group reports that reduction of cytosolic NAD is not followed by concomitant reduction of NAD in mitochondria (Yang et al., 2007a; Pittelli et al., 2010). It seems, therefore, that these organelles are able to maintain their NAD content when that of cytosol decreases but readily increase the pyridine nucleotide pool when the cytoplasmic availability of NAD and/or its precursors increases. Taken together, these findings suggest that mitochondria allow NAD resetting monodirectionally toward an increase and not vice versa. Raising of mitochondrial NADH coupled with augmented ATP production in cells exposed to eNAD is a clear indication that increased concentrations of NAD in mitochondria are translated into improved bioenergetics. Data indicating that mitochondrial NAD concentrations are limiting both the Krebs' cycle and oxidative phosphorylation. In principle, this knowledge might have important implications in cell biology and treatment of mitochondrial dysfunction, such as those occurring in mitochondrial disorders, neurodegeneration, diabetes, and aging. It is known, however, that almost complete (98% at 8 min) single-pass transformation of NAD infused into the portal vein or hepatic artery takes place in the perfused rat liver (Broetto-Biazon et al., 2008), indicating rapid degradation by hepatocytes. In addition, intracellular Ca2+ waves triggered by extracellular NAD degradation by CD38 can severely compromise cellular homeostasis.
Data showing that eNAD increases H2O2-induced PARP-1-dependent iNAD consumption but does not improve DNA repair suggest that not all NAD-dependent processes are limited by the intracellular dinucleotide concentrations. We interpret these findings, considering that free nuclear NAD contents are limiting PARP-1 activity and PAR formation but not genome repair. This is in keeping with evidence that 1) the intracellular pyridine nucleotide pool is mostly bound to proteins (Zhang et al., 2002), 2) proximity of the NAD producing enzyme NMNAT-1 to PARP-1 enhances poly(ADP-ribosyl)ation (Berger et al., 2007), and 3) eNAD increases DNA-damage-dependent PAR formation (Alano et al., 2010). On the other hand, our data hint that the amount of PAR produced with the physiological iNAD concentrations suffices to signal DNA repair. In apparent contrast with this hypothesis, eNAD improves efficiency of DNA repair in postischemic neurons in vitro (Wang et al., 2008). Although we cannot explain the reasons for this inconsistency, it is worth noting that the molecular mechanisms involved in oxygen glucose deprivation-induced DNA damage and repair differ qualitatively and quantitatively from those activated by H2O2. It is possible, therefore, that the ability of eNAD to assist DNA repair is stress- and cell type-dependent.
A key finding of our study is that apoptosis is reduced by increasing the extracellular concentrations of NAD. On the one hand, data underscore the functional effects of increased iNAD concentrations and on the other provide important information on molecular mechanisms regulating the apoptotic machinery. Although several mechanisms could underpin eNAD cytoprotection, we report here that the dinucleotide reduces staurosporine-induced loss of mitochondrial membrane potential, a prototypical trigger of the intrinsic apoptotic pathway. Data are in keeping with prior work indicating a key role of pyridine nucleotides in regulating PTP opening (Halestrap et al., 2002). We also show that inhibition or silencing of the NAD-consuming, antiapoptotic deacetylase SIRT1 abrogates eNAD-dependent cytoprotection from staurosporine but not from C2-ceramide or MNNG. Albeit at present the reason for such a specific role of SIRT1 is unclear, data are in keeping with the notion that staurosporine, C2-ceramide, and MNNG prompt apoptosis through different mechanisms that, in turn, activate specific cell death pathways. In addition, as shown in Fig. 5E, eNAD may protect from MNNG-induced cell death by reducing iNAD depletion. Our data are consistent with previous reports showing that eNAD affords protection from various stresses such as β-amyloid (Qin et al., 2006), ischemia (Wang et al., 2008), NMNAT inactivation (Wang et al., 2005), or PARP-1-dependent cell death (Pillai et al., 2005; Alano et al., 2010). The study by Pillai et al. (2005) showing that SIRT1 activity mediates the cytoprotective effects of eNAD addition in myocyte cell culture undergoing PARP-1 activation further strengthens the link between eNAD availability and SIRT1-dependent protection from apoptosis. Likewise, recent work shows that increased iNAD availability as a result of PARP-1 inhibition promotes mitochondrial bioenergetics in a SIRT1-dependent manner (Bai et al., 2011). Finally, evidence that eNAD post-treatment provides neuroprotection when PARP-1 inhibitors no longer have effects (Alano et al., 2010) underscores the therapeutic potential of pharmacological strategies increasing the dinucleotide extracellular contents.
In conclusion, the present study furthers our understanding of the biochemistry of NAD, underscoring its pharmacotherapeutic potential. Development of strategies able to increase the intracellular pool of NAD in specific cellular compartments may represent a pharmacological challenge for the next future.
Authorship Contributions
Participated in research design: Pittelli, Giovannelli, Moroni, and Chiarugi.
Conducted experiments: Pittelli, Felici, Pitozzi, Bigagli, Cialdai, and Romano.
Performed data analysis: Pittelli.
Wrote or contributed to the writing of the manuscript: Pittelli and Chiarugi.
Footnotes
This study was supported by the Federazione Italiana Sclerosi Multipla, Regione Toscana (Progetto Salute 2009) and Ente Cassa di Risparmio di Firenze.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.111.073916.
-
ABBREVIATIONS:
- PARP-1
- poly(ADP-ribose) polymerase-1
- eNAD
- exogenous NAD
- ROS
- reactive oxygen species
- DIV
- days in vitro
- Nam
- nicotinamide
- NMN
- nicotinamide mononucleotide
- NR
- nicotinamide riboside
- MNNG
- N-methyl-N′-nitro-N-nitrosoguanidine
- STP
- staurosporine
- EX-25
- 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide
- siRNA
- small interfering RNA
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- PBS
- phosphate-buffered saline
- PBST
- phosphate-buffered saline containing 0.1% Tween 20
- PAR
- poly(ADP-ribose)
- PCR
- polymerase chain reaction
- LC
- liquid chromatography
- iNAD
- intracellular NAD
- FK866
- N-[4-(1-benzoyl-4-piperidinyl)butyl]-3-(3-pyridinyl)-2E-propenamide
- NMNAT
- nicotinamide mononucleotide adenylyltransferase
- MPT
- mitochondrial permeability transition
- PTP
- permeability transition pore.
- Received June 1, 2011.
- Accepted September 14, 2011.
- Copyright © 2011 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}