


Abstract
Angiotensin II type 1 receptor antagonists (ARBs) have become an important drug class in the treatment of hypertension and heart failure and the protection from diabetic nephropathy. Eight ARBs are clinically available [azilsartan, candesartan, eprosartan, irbesartan, losartan, olmesartan, telmisartan, valsartan]. Azilsartan (in some countries), candesartan, and olmesartan are orally administered as prodrugs, whereas the blocking action of some is mediated through active metabolites. On the basis of their chemical structures, ARBs use different binding pockets in the receptor, which are associated with differences in dissociation times and, in most cases, apparently insurmountable antagonism. The physicochemical differences between ARBs also manifest in different tissue penetration, including passage through the blood-brain barrier. Differences in binding mode and tissue penetration are also associated with differences in pharmacokinetic profile, particularly duration of action. Although generally highly specific for angiotensin II type 1 receptors, some ARBs, particularly telmisartan, are partial agonists at peroxisome proliferator-activated receptor-γ. All of these properties are comprehensively reviewed in this article. Although there is general consensus that a continuous receptor blockade over a 24-hour period is desirable, the clinical relevance of other pharmacological differences between individual ARBs remains to be assessed.
I. Introduction
Angiotensin II [angiotensin(1–8), ANG] is an important regulator of homeostasis, particularly with regard to electrolyte balance, by affecting, e.g., thirst, blood pressure, sympathetic nervous activity, and renal function. Some of these effects occur directly, i.e., via ANG receptors in target tissues such as vascular smooth muscle or renal tubular cells, whereas others occur indirectly. Indirect effects include those mediated via adrenal release of aldosterone, acting on specific aldosterone receptors (Horisberger and Rossier, 1992), via prejunctional release of noradrenaline, acting on adrenoceptors (Nap et al., 2003), and via elevation of blood pressure, acting on renal sodium excretion by pressure natriuresis (Kline and Liu, 1994). Moreover, ANG has both acute, e.g., vascular smooth muscle contraction, and chronic effects, e.g., vascular smooth muscle hypertrophy, which often funnel into the same responses at the organism level, e.g., blood pressure elevation.
Originally, it had been assumed that ANG is mainly acting systemically as a hormone. In this view the enzyme renin released from the kidneys cleaves angiotensin I [angiotensin(1–10)] from angiotensinogen, which is produced in the liver. Angiotensin I is then metabolized by angiotensin converting enzyme (ACE), a membrane-bound enzyme largely expressed in the lungs, to form ANG. Additionally, 20–30% of the systemic ANG production is believed to come from alternative pathways involving cathepsin G, chymase, and other serine proteases (Tsukamoto and Kitakaze, 2013). However, meanwhile it has become clear that renin, angiotensinogen, and ACE are not only formed in kidney, liver, and lung, respectively, but can also be expressed in many other tissues including heart (Tamura et al., 1997b), vessel wall, kidney (Siragy and Carey, 2010), adipose tissue (Cassis et al., 2008), gastrointestinal tract (Wong et al., 2007), or urogenital tract (Comiter, 2012), yielding a tissue renin-angiotensin system (RAS). Of note, there is also a tissue RAS in the brain, which mediates important physiologic functions, e.g., in the regulation of thirst or cognition (Culman et al., 2002; Pelisch et al., 2011). The relative roles of the classic systemic, the brain, and the other tissue RAS in the control of renal function, blood pressure, and other physiologic effects remain to be fully elucidated.
The direct molecular target for all of the above effects are the ANG receptors, of which the subtypes 1 (AT1R) and 2 (AT2R) exist. In rats and mice, but not in most other species including humans, two subtypes of AT1R exist that are encoded by distinct genes but apparently have the same ligand recognition profile for all ARBs that have been tested (de Gasparo et al., 2000); accordingly, ARBs such as candesartan and losartan blocked ANG-induced blood pressure elevations in AT1AR knockout mice (Oliverio et al., 1997). The ANG receptor family also includes an AT4 receptor, but the natural ligand for this subtype is not ANG but rather its breakdown product angiotensin IV, i.e., angiotensin(3–8) (de Gasparo et al., 2000).
On the basis of the important role of ANG and specifically AT1R in the regulation of blood pressure and renal function, AT1R antagonists, also named angiotensin receptor blockers (ARBs) or “sartans,” have become a cornerstone of blood pressure-lowering and renoprotective therapy. The aim of this article is a comprehensive comparison of the physicochemical, pharmacological, and pharmacokinetic properties of the clinically available ARBs followed by a critical discussion of the clinical implications of existing differences. Specifically, we will cover the ARBs azilsartan (also known as TAK-536 for the prodrug or TAK 491 for the active compound), candesartan (also known as TCV-116 for the prodrug or CV-11974 for the active metabolite), eprosartan (also known as SK&F 108566), irbesartan (also known as SR 47436 or BMS 186295), losartan (also known as DUP-753 or MK-954 or EXP3174 for the active metabolite), olmesartan (also known as CS-866 for the prodrug and RNH-6270 for the active metabolite), telmisartan (also known as BIBR 277), and valsartan (also known as CGP 48,933). Clinical rather than pharmacokinetic studies will only be discussed where they directly link to the experimental or pharmacokinetic studies and have not been reviewed systematically for this article.
Some clinically used ARBs for oral administration such as candesartan cilexitil, losartan, and olmesartan medoxomil are prodrugs of candesartan, EXP3174, and olmesartan, respectively, and/or have additional active metabolites (Schmidt and Schieffer, 2003). Azilsartan medoxomil (also known as azilsartan kamedoxomil) is a prodrug for azilsartan, but in some countries azilsartan rather than its prodrug is used for oral treatment. The prodrugs and their respective conversion to the active metabolites will be discussed in more detail in the pharmacokinetics section of this article. Unless the prodrug is specifically mentioned, all subsequent text relates to the active metabolites of the ARBs.
Our article is based on a systematic Medline search using the key words azilsartan, candesartan, eprosartan, irbesartan, losartan, olmesartan, telmisartan, and valsartan, which was completed in July 2012; abstract references were not considered. Additionally pharmacokinetic data were obtained from the prescribing information as approved by the U.S. Food and Drug Administration. Not surprisingly, the number of available studies was greatest for losartan, as this has been the first clinically available ARB; very large amounts of data were also available for candesartan, telmisartan, and valsartan, somewhat less for eprosartan and irbesartan, and the least for the very recently developed azilsartan. Although the number of studies on a given compound increases the level of confidence, it does not necessarily make one compound superior to another.
II. Physicochemical Properties
Most clinically used ARBs have common molecular structures resembling the first marketed ARB losartan (Fig. 1). These components are known to be critical for binding to AT1R and may be the basis for differences in oral bioavailability (Kohara et al., 1996), binding affinity, dissociation rates, insurmountability (Fujino et al., 2010), inverse agonism (Miura et al., 2006), and even other effects that are not mediated by binding to AT1R (Fujino et al., 2010). These properties and their possible relationship to clinical effects will be discussed in later chapters.

Structures of clinically used ARBs and their active metabolites.
Candesartan, irbesartan, valsartan, and olmesartan also contain the biphenyl moiety with the attached, acidic tetrazole seen in losartan. Other marketed ARBs have substitutions for the tetrazole that maintain the acidic property. Telmisartan replaces tetrazole with a carboxyl group, whereas the most recently introduced ARB, azilsartan, features another modification, biphenyl-5-oxo-1,2,4-oxadiazole, that may increase lipophilicity and bioavailability compared with candesartan (Kohara et al., 1996). Eprosartan, the ARB with the most differentiated structure, replaces biphenyl-tetrazole with benzoic acid. Thus, ARBs are similar in this region (Cappelli et al., 2004), with each of the ARBs sharing a hydrophobic interaction between the phenyl rings and the receptor along with an ionic interaction of the acidic moiety with basic residues (Mire et al., 2005). At the other end of the molecule, where losartan has imidazole with Cl and COOH substituents, ARBs have a greater variety of structures that may explain differences. Valsartan is unique in not having a nitrogen containing heterocycle. Eprosartan has a large substituent on the imidazole ring, whereas olmesartan is more closely related to losartan, and irbesartan has a cyclopentyl ring incorporated in place of the Cl. Candesartan and azilsartan substitute benzimidazole, whereas telmisartan is unusual in that it contains benzimidazole with a second benzimidazole attached. Although all of the ARBs bind to the same or a very similar site on AT1R, as a consequence of these differences in structure, each of the ARBs can bind in slightly different ways (Ohno et al., 2011). This will be further discussed in Section IV.F.
III. Tissue Distribution
A. Lipophilicity
A physical property related to the chemical structure of the various ARBs is lipophilicity, typically expressed as logP (partition coefficient) or logD (distribution coefficient) values. Of note this nomenclature is handled quite inconsistently in the literature. Lipophilicity is an important factor in the ability of drugs to cross cell membranes and accordingly is relevant for diverse processes such as absorption upon oral administration or tissue penetration including access to special body compartments such as the brain (Liu et al., 2008). Although all the ARBs are lipophilic to some extent, their lipophilicity varies markedly, with telmisartan being the most lipophilic (Table 1). Olmesartan and losartan/EXP3174 are least lipophilic depending on the measurement or calculation selected. Since acidic groups at both ends of the molecule are required for high affinity binding to the receptor (see Section IV.F), some ARBs are too polar and thus have poor oral bioavailability. To increase bioavailability, several are orally dosed as ester prodrugs that break down rapidly in the body to produce the active moiety. Only the lipophilicity of the active circulating drug is relevant for tissue penetration after the drug has been absorbed. Thus, prodrug lipophilicity, where applicable, is important for absorption, whereas active molecular lipophilicity is important for distribution within the body.
Lipophilicity, interaction with P-gp, and CNS penetration of clinically used ARBs (active metabolite/parent compound, where applicable)
Note that lipophilicity has been reported in a range of different formats; the table uses that which was reported by the cited investigators.
Comparing the ARBs on the basis of lipophilicity is complicated by the fact that the complete group has never been tested head to head using a single method. Values can be determined experimentally or by using one of a variety of available algorithms to calculate lipophilicity based on structure (Lin et al., 1992; Erbe et al., 2006; Gardiner and Paine, 2011). Values determined by different methods are often significantly different from each other. Experimental logD values determined at physiologic pH are the most predictive of behavior in vivo, and where available, they have been included in Table 1. A column of calculated values using a consistent algorithm (ALOGPS) is included in Table 1 as an attempt to rank order the ARBs, but these values are in all cases different from the experimental data. Both by measured logD at physiologic pH and by the ALOGPS calculation, telmisartan is more lipophilic than other ARBs, and it should be better able to penetrate membranes (Wienen et al., 2000). This may be reflected in its clinical effects.
Components of the RAS exist in tissues, such as the kidney, heart, and brain, as well as in the circulation, and inhibition of “tissue RAS” may contribute to the effects of ARBs (see Section I). Effects outside the blood vessel or renal tubular lumen require that the drug can penetrate into the tissue. On the basis of the above data on lipophilicity (Table 1) it can be expected that all ARBs penetrate into tissues to various degrees. One measure that may reflect tissue penetration is volume of distribution (Vd; see also Section VI.B). Of note, Vd can be assessed and expressed in different ways. It can be calculated after oral administration as an apparent value after correcting for bioavailability and then typically is expressed as Vz/f. However, calculation after i.v. administration is more informative as it is independent of bioavailability and then often is expressed as Vss. For drugs with low bioavailability Vz/f typically is considerably greater than Vss. Unless otherwise specified, Vd in the following text refers to Vss estimates. Vd varies from about 10 liters (0.13 l/kg) for candesartan to 500 liters for telmisartan, with most ARBs in the range of 10–93 liters. The distinction between telmisartan and irbesartan, for example, with a Vd value of 93 liters, is unlikely to be of clinical relevance as both compounds display significant tissue distribution. Vd values for azilsartan, candesartan, EXP3174, olmesartan, and valsartan are no higher than 17 liters (∼0.2 l/kg). This suggests that the protein binding of these compounds is restrictive. Low Vd values indicate that an agent is confined to albumin (0.1 l/kg) or total body water space (0.6 l/kg) and probably does not distribute readily into the tissue compartment, which acts as an additional drug reservoir. The distinction between telmisartan and other ARBs can possibly be explained by the highly lipophilic nature of telmisartan in relation to the other compounds (Wienen et al., 2000).
High Vd values suggest that these drugs efficiently enter tissue compartments, but this needs to be confirmed by direct testing by injecting a drug, e.g., in its radiolabeled form, and determining its presence in various tissues or by demonstrating tissue specific activity in animal models. One question of particular interest is whether a drug crosses the blood-brain barrier, because the brain has the components of the RAS, including AT1R (Culman et al., 2002; Pelisch et al., 2011), and several effects of ANG such as body water balance, blood pressure maintenance, and cognition, are at least partly centrally mediated (Wright and Harding, 2011). This will be discussed further in Section III.C.
B. Transporter Molecules Involved in Tissue Distribution
This section will summarize the data on ARBs as substrates and inhibitors of transporter molecules that play a role in absorption, tissue distribution, and excretion of ARBs. These transporter interactions can affect the pharmacokinetics of the respective drugs (see Section VI) and also drug-drug-interactions (see Section VI.G), and polymorphisms of these transporters apparently can play a role in ARB pharmacokinetics (see Section VI.H). ARB interactions with urate transporters in the kidney proximal tubule have also been postulated to explain differential effects on serum uric acid levels, which will be discussed below.
Tissue access by xenobiotics not only depends on penetration into the tissue, which largely is under the control of lipophilicity, but also on active extrusion from tissues. This typically involves transporters such as P-glycoprotein (P-gp; see Table 1), also known from oncology studies as multidrug resistance protein 1. P-gp is an ATP-dependent transport protein with a broad range of substrates that pumps drugs out of cells (Fromm, 2002). It is expressed in intestinal enterocytes and can limit the oral bioavailability of substrate molecules. In addition, drug extrusion by P-gp is an important functional part of the blood-brain barrier (Liu et al., 2008). Of note, P-gp is a genetically polymorphic protein (Gerloff, 2004). Losartan, but not the active metabolite EXP3174, has been reported to be a substrate for P-gp (Soldner et al., 2000). Candesartan efflux from a Caco-2 monolayer is concentration dependent and saturable and inhibited by cyclosporine A, a potent inhibitor of P-gp, suggesting that candesartan also is a substrate (Zhou et al., 2009). Similarly, valsartan transport from the serosal to the mucosal side of everted rat ileum is reduced in the presence of P-gp inhibitors quercetin and verapamil (Challa et al., 2013). Olmesartan medoxomil is a substrate, whereas olmesartan is not (Yamada et al., 2007). Moreover, some ARBs are inhibitors of P-gp that may lead to drug-drug interactions (see Section VI.G). Thus, telmisartan was shown to block digoxin transport in vitro with an IC50 of 2.19 μM (Kamiyama et al., 2010), whereas candesartan cilexetil and irbesartan were less potent (IC50 14.7 and 34 μM, respectively), and losartan, eprosartan, and candesartan did not inhibit P-gp (Kamiyama et al., 2010; Weiss et al., 2010). Given the intestinal concentration of telmisartan, telmisartan is potent enough to be expected to affect oral bioavailability of P-gp substrates in the intestine. Telmisartan is known to increase digoxin maximal concentrations in clinical studies (Stangier et al., 2000b), which could be attributed to P-gp inhibition. Although telmisartan marginally increases ATPase activity in an in vitro study of P-gp activity suggesting it could be a P-gp substrate (Chang et al., 2006), it is not clear whether telmisartan itself is transported by P-gp (Deppe et al., 2010). Telmisartan acylglucuronide, a main metabolite of telmisartan, is transported into the bile by P-gp and other transporters (Ishiguro et al., 2008). According to an EMA assessment report (EMEA/H/C/002293) azilsartan medoxomil is not a substrate for P-gp, but the transport of azilsartan itself was difficult to evaluate due to low transport in Caco-2 cells.
Several ARBs are eliminated from the body through bile excretion. Organic acid transporting polypeptide (OATP) family members OATP1B1 and OATP1B3, the primary isoforms in the liver (Klaassen and Aleksunes, 2010), are responsible for uptake of ARBs studied to date. Olmesartan is 60% excreted unchanged via bile, and valsartan is also secreted in the bile, mainly unchanged. In vitro studies show that for both these ARBs, OATP1B1 and OATP1B3 are responsible for uptake, and canalicular multispecific organic anion transporter (cMOAT/MRP2) plays a role in biliary excretion (Nakagomi-Hagihara et al., 2006; Yamashiro et al., 2006; Yamada et al., 2007). Telmisartan and telmisartan acylglucuronide are primarily taken up by the liver by OATP1B3 and subsequently secreted into the bile as telmisartan acylglucuronide by P-gp, MRP2, and breast cancer resistance protein (Ishiguro et al., 2008).
Elevated serum uric acid has been linked to hypertension, cardiovascular disease, and renal disease (Feig et al., 2008). Losartan, but not the active metabolite EXP3174, has been shown to lower serum uric acid (Nakashima et al., 1992), whereas valsartan has no effect (Gonzalez-Ortiz et al., 2000), candesartan increases serum uric acid (Manolis et al., 2000), irbesartan increases serum uric acid nonsignificantly (Dang et al., 2006), and telmisartan has no effect (Aranda et al., 2005). These effects may be mediated by differential interactions of ARBs with kidney uric acid transporters. Uric acid is filtered in the glomerulus and reabsorbed into proximal tubule endothelial cells by the transporter URAT1 located in the luminal membrane (Sato et al., 2008). Tubular secretion of uric acid is mediated by several organic acid transporters, OAT1, OAT3, OAT4, and MRP4, in the basolateral membrane (Sato et al., 2008). In addition, the glucose transporter, GLUT9 (also known as URATv1), also in the basolateral membrane, plays a role in transporting urate into the blood (Anzai et al., 2008). In rat proximal tubule membranes vesicles, losartan inhibits urate uptake competitively with an IC50 of 9.5 µM, whereas EXP3174 and eprosartan were 6-fold less potent (65 and 60 µM), respectively (Edwards et al., 1996). This suggested the URAT1 inhibition as the explanation for the decrease in serum uric acid and pointed to an AT1R-independent mechanism because EXP3174 is more potent than losartan at the receptor (see Section IV.A). Losartan also inhibits URATv1, whereas valsartan does not (Anzai et al., 2008). Irbesartan and telmisartan also inhibit both URAT1 and URATv1 in vitro (Nakamura et al., 2010), whereas candesartan does not (Nakamura et al., 2010). However, these experiments were performed at concentrations that are high relative to actual plasma concentrations or estimated kidney concentrations of the drugs (Sato et al., 2008). Iwanaga et al. (2007) tested a series of ARBs at more relevant nanomolar concentrations using Xenopus laevis oocytes expressing URAT1. Losartan and telmisartan inhibited urate uptake in this model, whereas EXP3174, olmesartan, and valsartan did not. URAT1 and URATv1 are the major transporters involved in uric acid transport (Lipkowitz, 2012), but ARBs circulate as organic anions that also interact with OATs and can be secreted by OATs in the kidney (Edwards et al., 1996). Olmesartan is primarily taken up into human kidney slices by OAT3 (Yamada et al., 2007; Watanabe et al., 2011), and candesartan, losartan, and valsartan all inhibit estrone-3-sulfate uptake by OAT4 in transfected HEK cells (Yamashita et al., 2006), although the Ki values are significantly higher than expected plasma concentrations. In vitro assays using membrane systems expressing OAT1, OAT3, OAT4, or MRP4 demonstrate that valsartan, olmesartan, and losartan each inhibit one or more of these transporters at concentrations that are achieved in blood or kidney tissue after oral dosing, whereas telmisartan and candesartan inhibit only at higher concentrations (Sato et al., 2008). Therefore, interactions between some ARBs and uric acid at these renal transporters could also contribute to effects on serum uric acid (Sato et al., 2008).
C. Penetration into Brain
Label information, when available, indicates that most ARBs, including azilsartan, candesartan, losartan, olmesartan, telmisartan, and valsartan, cross the blood-brain barrier poorly or not at all (see Table 1 and Section VI.B). These claims are typically based on studies using radiolabeled compound, which upon i.v. administration yielded little radioactivity in the rat central nervous system as determined by whole body autoradiography (Wienen et al., 2000). Nevertheless, apparent central nervous system effects have been observed in preclinical studies with several ARBs, suggesting that ARBs can cross the blood-brain barrier under certain conditions. Examples of these preclinical studies are discussed below. Penetration into the brain may also have beneficial effects on the development of Alzheimer’s disease beyond those of blood pressure lowering (Duron and Hanon, 2010).
Candesartan, administered systemically by osmotic minipump (Nishimura et al., 2000) or orally (Song et al., 1999), reduced [125I]-(Sar1-Ile8) ANG binding to brain slices examined ex vivo by quantitative autoradiography, indicating some brain penetration. Several i.v. doses of candesartan inhibited pressure responses to ANG administered intracerebroventricularly in conscious rats (Gohlke et al., 2002). After chronic subcutaneous (but not acute i.v.) administration, candesartan reduced infarct size after middle cerebral artery occlusion and reperfusion in rats although both administration routes caused similar blood pressure reduction (Groth et al., 2003).
Eprosartan, chronically administered peripherally by osmotic minipump in rats, also reduced [125I]-(Sar1-Ile8) ANG binding to brain slices from inside and outside the blood-brain barrier as examined ex vivo by quantitative autoradiography, indicating some brain penetration (Muders et al., 2001). However, this study had used eprosartan doses of 30 and 60 mg/kg, whereas a significant blood pressure effect is observed at 10 mg/kg (Brooks et al., 1992), questioning the presence of central effects at therapeutic doses.
Although centrally administered irbesartan did not attenuate acute blood pressure increases by i.v. ANG, chronic peripherally given irbesartan attenuated those to centrally administered ANG (Leenen and Yuan, 2001). Possible brain penetration by orally administered irbesartan and losartan was also explored by a different approach, i.e., by testing inhibition of the dipsogenic response to ANG injected intracerebroventricularly in normotensive and spontaneously hypertensive rats (Polidori et al., 1998). Upon intragastric dosing both ARBs inhibited the response, but higher doses of irbesartan compared with losartan were required. In addition, higher doses of both drugs were needed in normotensive rats compared with hypertensive rats, suggesting that hypertension may change the blood-brain barrier in some way, allowing easier access to drugs. Irbesartan and losartan were also compared in a mouse middle cerebral artery occlusion model. Addition of a dose of irbesartan that was not protective on its own to an effective dose of a CCR2 antagonist (propagermanium) produced an improved reduction in infarct area, while adding losartan did not (Tsukuda et al., 2011). In fact, studies with losartan have given a variety of results, sometimes suggesting that losartan or its metabolite cannot cross the blood-brain barrier, sometimes demonstrating central effects. The difference may depend on dosing or on the models involved (Wong et al., 1990a; Bui et al., 1992; Li et al., 1993; Culman et al., 1999). However, the [125I]-(Sar1, Ile8) ANG brain autoradiography technique described above for candesartan and eprosartan indicates that losartan does penetrate the blood-brain barrier in rats (Song et al., 1991).
Olmesartan administered chronically by mini-pump also showed beneficial effects in the brain to reduce damage from transient cerebral ischemia and reperfusion in rats, independent of a blood pressure lowering (Hosomi et al., 2005), and also had positive effects in an occlusive stroke model in mouse (Iwai et al., 2006) and gerbil (Faure et al., 2006; Scott and McCormack, 2008).
In early studies, when [14C]telmisartan was dosed in rats for 7 days, radioactivity was low to nondetectable in brain tissue (Shimasaki et al., 1999), consistent with the result using whole body autoradiography (Wienen et al., 2000); however, autoradiography is not a very sensitive technique in this regard and may not detect low ligand concentrations. More recent evidence comes from PET studies in rats and humans with [11C]telmisartan, demonstrating penetration into the brain, although to a lesser extent than into many peripheral tissues (Shimizu et al., 2012). Similar PET studies in monkeys showed that telmisartan reaches therapeutically relevant concentrations in the brain; it undergoes a slow clearance but does not show signs of accumulation in the brain (Noda et al., 2012). Accordingly, telmisartan was detected in rat cerebrospinal fluid after 8 days of oral treatment (Gohlke et al., 2001), a direct indication that the drug penetrates the blood-brain barrier. As a functional correlate of such penetration, telmisartan, given orally or intravenously, blocked the drinking and pressor responses to intracerebroventricular ANG in rats (Gohlke et al., 2001). Telmisartan was more effective in inhibiting this central response at lower doses than either losartan or irbesartan (Culman et al., 1999). Orally administered telmisartan also reduced oxidative stress and sympathetic nervous system activation in the rat rostral ventrolateral medulla, whereas candesartan was ineffective in this model (Kishi et al., 2012).
No published nonclinical studies addressing blood-brain barrier penetration were found for azilsartan or valsartan.
Thus, the overall data on blood-brain barrier penetration are not fully conclusive. Most ARBs exhibit little penetration into the brain, but may do so to some extent upon chronic dosing and/or when the blood-brain barrier has become more permissive under pathologic conditions. In this regard the strongest evidence for penetration into the brain has been presented for telmisartan, which is consistent with its greater lipophilicity as compared with other ARBs (see Sections III.A and VI.B). At least some ARBs in some studies have shown beneficial effects in animal models of Alzheimer’s disease, implying penetration through the blood-brain barrier (Mogi et al., 2008; Tsukuda et al., 2009), but the overall literature in this regard is controversial (Ferrington et al., 2011).
Of interest, not only does passage through the blood-brain barrier potentially contribute to clinical effects of ARBs, but the RAS can actually modulate blood-brain barrier permeability. Thus, ANG can make it leakier, whereas ARBs such as olmesartan and telmisartan have been shown to improve blood-brain barrier function in vitro and in vivo, and this may result in improved cognition (Fleegal-DeMotta et al., 2009; Pelisch et al., 2011). Telmisartan’s ability to improve blood-brain barrier function may also depend in part on peroxisome proliferator-activated receptor (PPAR)-γ activation (Min et al., 2012).
IV. Direct AT1R Effects
The affinity of a drug for its molecular target is most often derived from radioligand binding experiments under equilibrium conditions, either performed as competition experiment against a radioligand labeling the target or as saturation experiment using a radiolabeled form of the drug of interest. Alternatively, the affinity of a drug for its molecular target can be derived from kinetic radioligand binding experiments, i.e., association and dissociation studies typically involving a radiolabeled form of the drug. This approach is less common because it is technically more cumbersome but has the added advantage of directly providing dissociation rates, which may contribute to the duration of action of the drug. If the dissociation rate of a drug from a receptor is very slow, equilibrium may not be achieved during standard incubation periods; in such cases the potency estimate of a drug may differ dependent on the specific experimental conditions being chosen, particularly incubation time before a response is assessed. Therefore, true affinity estimates of a drug, typically expressed as Ki or Kd values, often are difficult to determine for drugs with slow dissociation rates. In these cases the concentration producing 50% inhibition under the chosen experimental conditions (IC50) often is reported, although this is only an approximation and most often an underestimation of the true affinity.
Of note, radioligand binding studies do not provide information about antagonist properties of a drug unless very specific assay conditions are chosen. Antagonism is primarily determined by functional experiments, e.g., for formation of inositol trisphosphate or elevation of intracellular Ca2+ concentrations at the cellular level or vasoconstriction in an organ bath at the tissue level. Such data can show whether antagonism is competitive or not and can also be used to determine drug affinities functionally. For the latter purpose the most robust antagonist potency estimates come from experiments testing various antagonist concentrations on the concentration-response curve of an agonist for a chosen physiologic response. If a sufficient number of antagonist concentrations is tested, apparent antagonist affinity (pKB value) can be determined by Schild-plots (Arunlakshana and Schild, 1959); these have the advantage of additionally providing insight into the mode of antagonism, i.e., competitive versus noncompetitive. However, in some cases only a single antagonist concentration is tested against the concentration-response curve of the agonist, which can also result in an affinity estimate, an apparent pA2 value (Neubig et al., 2003). In cases of insurmountable antagonism, which is displayed by some ARBs (see below), analysis techniques such as the van Rossum procedure (van Rossum, 1963) or double-reciprocal regression (Kenakin, 1987) can be used to obtain antagonist potency estimates. All of these approaches have been applied to ARBs, but not each approach has been used for every ARB. Of note, affinity estimates obtained by any of these methods can differ to some degree depending on the type of assay or specific assay conditions, a phenomenon also known from other drug classes (Michel et al., 1995). Therefore, minor affinity differences within a drug class typically can only be detected in direct comparative studies.
A. Radioligand Binding Studies
Numerous studies have determined the affinity of various ARBs in competition radioligand binding assays, and many of them have compared multiple ARBs within the same study. Most competition radioligand binding assays were based on the agonist radioligand [125I]ANG or peptidic antagonist radioligands such as [125I]-(Sar1,Ile8)-ANG, but in some cases also nonpeptidic antagonist radioligands such as [125I]EXP985 have been used (Chiu et al., 1992); the results obtained with both approaches were similar.
With the exception of azilsartan, many competition binding studies have been reported for each ARB using a range of model systems (transfected cells, natively AT1R expressing cells and tissue homogenates from humans and various other mammalian species). Widely used model systems include rat (Aguilera, 1992; Chiu et al., 1992) and bovine adrenals (Boulay et al., 1992; Ouali et al., 1992). Studies in which multiple tissues and/or multiple species with expression of bona fide AT1R have been compared directly show that variability of reported ARB affinities largely represents interstudy differences rather than those between cell/tissue types or species (de Gasparo and Whitebread, 1995).
A naturally occurring human AT1R mutant (A163T, rs12721226) exhibits rather similar affinity for most ARBs compared with the human wild-type receptor but an approximately sevenfold lower affinity for losartan and its active metabolite (Arsenault et al., 2010). Engineered AT1R mutations can also affect the affinity of ARBs (Feng et al., 1995; Noda et al., 1996; 2006, 2008), but a given amino acid exchange may differentially affect the affinity for various ARBs (Bhuiyan et al., 2010a), indicating that they use overlapping but distinct binding pockets (see Section IV.F). ARB affinities are largely the same across mammalian species but dogs may be an exception, as both canine adrenal and liver had a significantly lower affinity for both losartan and valsartan compared with the same tissues in rats, marmosets, and humans (de Gasparo and Whitebread, 1995).
On the basis of these considerations, Fig. 2 shows a summary of reported affinity estimates for various ARBs across all reported competition binding studies, regardless which species and tissue/cell type have been used and equally accepting Ki and IC50 values. Only data from experiments in dogs were excluded as they may exhibit systematically different ARB affinity compared with other mammals (de Gasparo and Whitebread, 1995). On the basis of these studies, the various ARBs appear to have the following affinity estimates (pKi or pIC50; median with 95% confidence intervals) at mammalian AT1R: azilsartan (8.51; 4.71–10.94; n = 3), candesartan (8.43; 8.01–8.80; n = 26), eprosartan (8.26; 8.02–8.58; n = 12), irbesartan (8.72; 8.42–8.87; n = 14), losartan (7.71; 7.50–7.73; n = 63), EXP3174 (8.17; 7.93–8.87; n = 21), olmesartan (8.17; 7.99–8.60; n = 5), telmisartan (8.33; 8.12–8.53; n = 14), and valsartan (8.46; 7.95–8.48; n = 17). ARB affinity estimates based on quantitative autoradiography have yielded similar results (Jin et al., 1997). Affinity estimates based on association and dissociation rate constants yielded similar values as in the competition binding studies, e.g., for candesartan and telmisartan (Fierens et al., 1999; Maillard et al., 2002a).
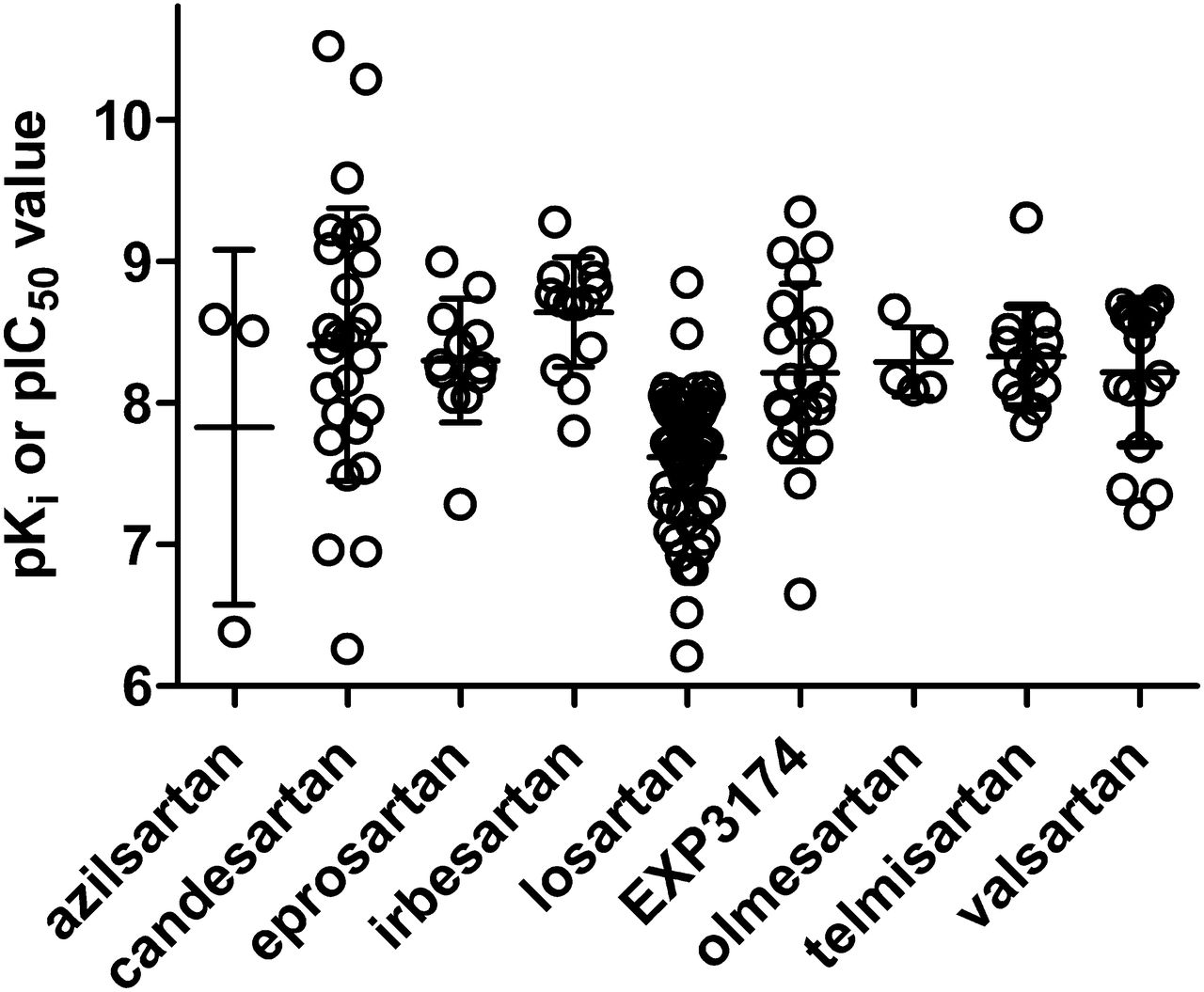
Affinity estimates for various ARBs at AT1R as determined in competition radioligand binding studies. Each data point represents a single study, but most of these studies have assessed multiple ARBs in comparison. Data are from a wide range of cell types, tissues, and species as well as from heterologously expressed human AT1R, but were pooled for this figure because no major differences in affinity estimates based on these model systems were observed. However, canine data were excluded from this figure, because in multiple dog tissues AT1R affinities for multiple ARBs are lower than in the same tissues from other mammals (de Gasparo and Whitebread, 1995). Data are pKi or, in approximation thereof, pIC50 values as reported in (1990a,b,c, 1992; Speth and Kim, 1990; Wong et al., 1990b,d; Buhlmayer et al., 1991; Obermüller et al., 1991; Weinstock et al., 1991; Aguilera, 1992; Boulay et al., 1992; Crawford et al., 1992; Edwards et al., 1992a,b; Ernsberger et al., 1992; Leung et al., 1992; Lin et al., 1992; Lyall et al., 1992; Noda et al., 1993,1996; Ouali et al., 1992; 1992,1993; Bernhart et al., 1993; Cazaubon et al., 1993; Criscione et al., 1993; Feolde et al., 1993; Ries et al., 1993; Shibouta et al., 1993; Tanabe et al., 1993; Dickinson et al., 1994; Herbert et al., 1994; Nishikawa et al., 1994; Schambye et al., 1994,1995; Nakamura et al., 1994; Wienen and Entzeroth, 1994; de Gasparo and Whitebread, 1995; Feng et al., 1995; Flesch et al., 1995; Itazaki et al., 1995; Keiser et al., 1995; Mizuno et al., 1995; Kohara et al., 1996; Yanagisawa et al., 1996; Almansa et al., 1997; Garcia-Sainz et al., 1997; Hashimoto et al., 1997; Jin et al., 1997; Ojima et al., 1997; Tamura et al., 1997a; Virone-Oddos et al., 1997; Häuser et al., 1998; Inada et al., 1999; Vanderheyden et al., 1999; Fierens et al., 1999; Hines et al., 1999; Fabiani et al., 2000; Le Bourdonnec et al., 2000; Vanderheyden et al., 2000; Burnier, 2001; Koike et al., 2001; Maillard et al., 2002a; Sudoh et al., 2003; Nussberger and Koike, 2004; Mire et al., 2005; Miura et al., 2006, 2011, 2013; Arsenault et al., 2010; Bhuiyan et al., 2009b, 2010a; Casimiro-Garcia et al., 2011).
Some ARBs are clinically administered as prodrugs that are converted into an active metabolite in vivo. In the case of losartan, both the parent compound and the active metabolite EXP3174 are detectable in plasma upon oral losartan administration (see Section VI.A); in studies that have directly compared both, the active metabolite was on average approximately four times as potent as the parent compound (median pKi/pIC50 8.17 versus 7.59; n = 19). For the prodrugs azilsartan medoxomil, candesartan cilexetil, and olmesartan medoxomil, only the active metabolite is detectable in plasma upon oral administration of the prodrug (see Section VI.A). Olmesartan is somewhat more potent than its parent drug (8.0 versus 33 nM) (Koike et al., 2001) and candesartan much more so than its parent drug [0.6 versus 167 nM in one study (Noda et al., 1993), 0.03 versus 1 nM in a second study (Flesch et al., 1995)]. No direct comparison of binding affinity between azilsartan and azilsartan medoxomil has been reported.
Although most of the above data are based on competition binding experiments, other studies have attempted to use tritiated versions of an ARB as the radioligand and then determine its apparent affinity from saturation binding studies. This approach has been applied mainly to losartan (Chiu et al., 1990a; Chansel et al., 1993; Feng et al., 1995) and candesartan (Ojima et al., 1997; Inada et al., 1999; Maillard et al., 2002a) but in some studies also to eprosartan (Aiyar et al., 1993), irbesartan (Delisee et al., 1993; Fierens et al., 1999), olmesartan (Le et al., 2007), telmisartan (Maillard et al., 2002a; Le et al., 2007), valsartan (de Gasparo and Whitebread, 1995; Verheijen et al., 2000), or the experimental ARB EXP985 (Chiu et al., 1992). The affinity estimates from the saturation approach have generally been in good agreement with those of the competition studies.
An ANG binding site on rat hepatocyte nuclear membranes has also been reported that, based on losartan and other compounds, exhibits an AT1R like ligand recognition profile (Booz et al., 1992), but the physiologic relevance of this site remains unclear.
In conclusion, the active forms of all clinically used ARBs have rather similar affinities at AT1Rs, with losartan and its active metabolite, EXP3174, having the lowest and irbesartan the highest affinity (median of reported values 19 and 2 nM, respectively) in radioligand binding studies; where applicable, the corresponding prodrugs exhibit a somewhat lower affinity. With the possible exception of dogs, these values are remarkably consistent across mammalian species, tissues, and experimental approaches.
B. Antagonism at Cellular Level
AT1Rs couple to a variety of prototypical signaling responses, mostly via pertussis toxin-insensitive G-proteins of the Gq/11 family, but in some cases also via pertussis toxin-sensitive Gi/o proteins, and both pathways can coexist within the same cell (Crawford et al., 1992; Poggioli et al., 1992; Huwiler et al., 1998). Cellular signaling responses mediated by such G-proteins include an activation of phospholipase C with subsequent formation of inositol phosphates (Crawford et al., 1992; Ouali et al., 1992; Poggioli et al., 1992; Garcia-Sainz et al., 1997; Vanderheyden et al., 1999; Ojima et al., 2011; Miura et al., 2013) and elevation of free intracellular Ca2+ concentrations (Leung et al., 1992; Poggioli et al., 1992; Ransom et al., 1992; Delisee et al., 1993; Pepperell et al., 1993; Herbert et al., 1994; Itazaki et al., 1995; Ko et al., 1997; Zhang and Mayeux, 2012) or inhibition of adenylyl cyclase (Crawford et al., 1992). Arrestin recruitment has been used as a proximal step to test cellular signal transduction antagonism by ARBs (Sanni et al., 2010).
These signaling responses result in acute cellular responses such as phosphorylase stimulation in hepatocytes (Garcia-Sainz et al., 1997) or hormone production and release from adrenal cells (Aguilera, 1992; Criscione et al., 1993; Wada et al., 1994). Prolonged AT1R stimulation results in protein and DNA synthesis and proliferation (Herbert et al., 1994; Sung et al., 1994; Flesch et al., 1995; Itazaki et al., 1995; Virone-Oddos et al., 1997; Hines et al., 1999). Inhibition of such responses was demonstrated in cells from rat, rabbit, guinea pig, pig, cow, and human sources, including vascular smooth muscle cells (Leung et al., 1992; Herbert et al., 1994; Sung et al., 1994; Flesch et al., 1995; Itazaki et al., 1995; Virone-Oddos et al., 1997; Fortuno et al., 1999), cardiomyocytes (Delisee et al., 1993), hepatocytes (Garcia-Sainz et al., 1997) and hepatoma cell lines (Le Bourdonnec et al., 2000), renal tubule cells (Poggioli et al., 1992; Zhang and Mayeux, 2012), adrenal cells (Aguilera, 1992; Ouali et al., 1992; Criscione et al., 1993; Wada et al., 1994), ovarial cells (Pepperell et al., 1993), and various cell lines natively expressing AT1R (Crawford et al., 1992; Ransom et al., 1992) or having been transfected with them (Vanderheyden et al., 1999). The ARB affinity estimates obtained in functional assays at the cellular level, largely in signal transduction assays, are generally in good agreement with those determined by competition radioligand binding assays, at least for ARBs where more than two studies have been reported (candesartan 9.36, 8.23–10.00, n = 6; irbesartan 8.52, 7.90–8.90, n = 11; losartan 7.45, 6.87–7.61, n = 26; EXP3174 8.08, 6.82–8.91, n = 6). This holds not only true for an overall comparison of published studies but also when a given ARB was tested in binding and functional assays within one study (Aguilera, 1992; Crawford et al., 1992; Ouali et al., 1992). An exception are late cellular responses such as protein and DNA synthesis, where ARBs were less potent than in binding or acute signaling assays (Ca2+ elevation) (Herbert et al., 1994; Virone-Oddos et al., 1997), possibly reflecting some degradation of the ARBs being studied (irbesartan, losartan, EXP3174) during long incubation times.
Interestingly, it has been reported that telmisartan can cause downregulation of AT1R mRNA and protein expression in vascular smooth muscle cells (Imayama et al., 2006). As this effect apparently is not mediated by telmisartan binding to the AT1R, it is not expected to be mimicked by other ARBs; whether and to which extent this may contribute to the overall inhibition of AT1R-mediated responses upon chronic treatment in vivo remains to be established.
C. Antagonism at Tissue Level
AT1R antagonism by ARBs has been shown and quantified in a wide variety of isolated tissues. Most of this work has been carried out in isolated blood vessels. The vast majority of in vitro studies on ARB effects on ANG-induced vasoconstriction has been performed with isolated rabbit aorta, but some findings have also been reported, e.g., for rat aorta (Inada et al., 1994), rat portal vein (Zhang et al., 1993; Morsing et al., 1999), guinea pig aorta (Mizuno et al., 1995; Hashimoto et al., 1997), rabbit mesenteric artery (Balt et al., 2002), dog pulmonary artery (Guimarães et al., 2011), pig and human coronary artery (Maassen vandenBrink et al., 1999), or human gastroepiploic artery (Jin et al., 1997) or human subcutaneous microvessels (Garcha et al., 1999); data with compounds that have been tested in multiple preparations indicate that the antagonist potency of a given ARB is comparable across species and vascular beds. On the basis of these studies the various ARBs appear to have the following affinity estimates (pA2 for surmountable or pD2 for insurmountable ARBs; median with 95% confidence intervals) at mammalian vascular AT1R (Fig. 3): azilsartan (9.90, n = 1), candesartan (10.08; 9.87–10.71; n = 9), eprosartan (8.80; 7.25–10.53; n = 3), irbesartan (8.52; 7.00–10.04; n = 2), losartan (8.15; 7.83–8.23; n = 21), EXP3174 (9.62; 9.12–9.92; n = 10), olmesartan (9.90; n = 2), telmisartan (9.48; n = 2), and valsartan (9.26; n = 1). Notably, these functional data have generally yielded somewhat higher affinity estimates than the radioligand binding studies (see Section IV.A). However, at least for the ARBs exhibiting insurmountable antagonism, this may at least partly relate to the observation that the apparent antagonist potency increases with incubation time (see Section IV.E). In a similar vein it was found that the in vitro responses of isolated aorta to ANG obtained from rats having undergone in vivo treatment with candesartan for up to 24 hours prior to tissue removal also exhibited reduced responses (Inada et al., 1994). Of note, ARBs not only can inhibit vasoconstriction but also hypertrophy and/or proliferation responses of vascular smooth muscle growth (Herbert et al., 1994) or endothelium (Varty et al., 1994). They also can prevent agonist-induced desensitization of vascular responses (Robertson et al., 1994b).

Affinity estimates for various ARBs based on antagonism of ANG-induced vasoconstriction, largely in isolated rabbit aorta. Each data point represents a single study, but many of these studies have assessed multiple ARBs in comparison. Data are shown as pA2 values for competitive and as pD2 values for insurmountable antagonists as reported in (Wong et al., 1990b,d; Buhlmayer et al., 1991; Edwards et al., 1992a; Lin et al., 1992; Liu et al., 1992b; Bernhart et al., 1993; Cazaubon et al., 1993; Criscione et al., 1993; Leung et al., 1993; Noda et al., 1993; Shibouta et al., 1993; Dickinson et al., 1994; Schambye et al., 1994; Keiser et al., 1995; Mizuno et al., 1995; Hashimoto et al., 1997; Jin et al., 1997; Tamura et al., 1997a; Garcha et al., 1999; Inada et al., 1999; Maassen vandenBrink et al., 1999; Morsing et al., 1999; Balt et al., 2002; Guimarães et al., 2011; Wienen et al., 1992, 1993; Zhang et al., 1993, 1994; Ojima et al., 1997, 2011).
Some ARBs, specifically losartan, have also been tested for their antagonism of nonvascular muscle contraction induced by ANG, e.g., in guinea pig heart (Feolde et al., 1993), guinea pig esophagus, stomach, gall bladder, ileum and colon (Leung et al., 1993), rat urinary bladder (Tanabe et al., 1993), or in rat and guinea pig ileum and guinea pig stomach and trachea (Liu, 1993). The affinity estimates obtained in those studies were generally in good agreement with those from the radioligand binding, cellular, and vascular antagonism studies. Interestingly, in the latter study losartan behaved as a competitive and surmountable antagonist in some but not other smooth muscle preparations.
Finally, the potency of ARBs has also been quantified for antagonism of neuronal, specifically prejunctional neurotransmitter release enhancing effects, e.g., in rabbit thoracic aorta (Nap et al., 2002) and mesenteric artery (Balt et al., 2002), rat mesenteric artery (Balt et al., 2001a), atrium (Shetty and DelGrande, 2000), and ventricle (Guimarães et al., 2011) or canine pulmonary artery (Guimarães et al., 2011). In such studies, eprosartan, irbesartan, losartan, and valsartan typically exhibited potencies in line with other model systems. In electrophysiological studies in rat superior cervical ganglion in vitro losartan also had a potency in line with other model systems (Hawcock et al., 1992). The relative contribution of prejunctional AT1R to the overall ARB effects remains to be explored.
D. Antagonism In Vivo
Although in vivo studies ultimately have a greater therapeutic relevance than in vitro studies, their mechanistic interpretation is more difficult because an observed effect, e.g., on blood pressure, cannot necessarily be attributed to a specific cell type mediating it. For example, ARBs can acutely modify transmitter release from cardiac and vascular sympathetic nerve endings (Nap et al., 2003) and that may contribute to blood pressure effects. By far the most frequently used way to determine AT1R antagonism in vivo is measurement of blood pressure responses to i.v. injections of ANG in the absence and presence of one or more doses of an ARB. Such studies have mostly been performed in rats (see below), but some studies have also been performed in hamsters (Trippodo et al., 1995; Jin et al., 1997), dogs (Cazaubon et al., 1993; Kubo et al., 1993; Christ et al., 1994; Ito et al., 1995; Hashimoto et al., 1997; Hayashi et al., 1997), cats (Champion and Kadowitz, 1997), or monkeys (Cazaubon et al., 1993; Criscione et al., 1993; Roccon et al., 1994) (for human studies see below).
The ability of ARBs to inhibit ANG-induced blood pressure elevations in rats has been tested under a variety of experimental conditions, i.e., in conscious, anesthetized, or pithed animals and with oral or intravenous ARB administration. Such inhibition has been reported for azilsartan (Kohara et al., 1996), candesartan (Kubo et al., 1993; Wada et al., 1994, 1996; Kohara et al., 1996; Nakano et al., 1997; Champion et al., 1998; Koike et al., 2001; Maillard et al., 2002a), eprosartan (Wang and Brooks, 1992), irbesartan (Cazaubon et al., 1993; Lacour et al., 1994; Christophe et al., 1995; Culman et al., 1999; Maillard et al., 2002a), losartan (Wong et al., 1990a,c; Abdelrahman and Pang, 1992; Gorbea-Oppliger et al., 1994; Christophe et al., 1995; Mizuno et al., 1995; Kohara et al., 1996; Almansa et al., 1997; Culman et al., 1999; Koike et al., 2001), olmesartan (Mizuno et al., 1995; Koike et al., 2001), telmisartan (Wienen et al., 1993; Maillard et al., 2002a), and valsartan (Criscione et al., 1993). Although most of these studies have quantified the degree of antagonism by one or more ARB doses over time, their findings are difficult to compare across studies. A better comparison is possible by another set of studies in rats, in which an ED50 for ANG-induced blood pressure elevation was determined. As summarized in Table 2, such studies show remarkable consistency for ED50 of a given ARB administered by a given route of administration. This consistency stretches across studies, even when performed by different investigators, and across the use of, e.g., conscious versus pithed rats. However, in some cases such as eprosartan and losartan, the i.v. administration was much more potent than oral dosing, possibly reflecting differences in bioavailability and/or active metabolites. However, such differences in antagonistic potency between oral and intravenous dosing were not observed with candesartan. Although all ARBs except the parent compound losartan and perhaps azilsartan have a rather similar molecular weight and affinity for the AT1R (Fig. 2), their antagonist potency upon i.v. administration differs considerably with irbesartan, EXP3174, and olmesartan being least and telmisartan most potent (Table 2), possibly reflecting pharmacokinetic differences between them (see Section VI).
In vivo antagonism of ANG-induced blood pressure elevations in rats
Data refer to the parent compound for oral administration and to the active metabolite for parenteral administration.
Such quantitative differences in in vivo antagonism were also observed in direct comparative studies of multiple ARBs. For example, candesartan caused more potent and longer lasting inhibition than losartan with both oral and intravenous administration (Shibouta et al., 1993), whereas irbesartan and losartan were equipotent in another study (Culman et al., 1999). At doses yielding similar peak inhibition of ANG-induced blood pressure elevation, candesartan and telmisartan yielded much longer lasting inhibition than irbesartan (Maillard et al., 2002a). On the other hand, losartan and olmesartan had a similar duration of action upon intravenous and oral administration to conscious rats or intravenous administration to anesthetized rats (Mizuno et al., 1995). The antagonism of ANG-induced blood pressure elevation by oral azilsartan medoxomil was less potent but longer lasting than that by olmesartan medoxomil (Kusumoto et al., 2011), whereas the active metabolite azilsartan had longer lasting effects than those of candesartan (Kohara et al., 1996). With regard to in vivo antagonist potency, these findings from direct comparative studies are largely consistent with those from the indirect comparisons shown in Table 2.
In conscious monkeys the degree of antagonism of the ANG blood pressure response by irbesartan was correlated to its plasma concentration (Roccon et al., 1994). Accordingly, in some studies the ED50 for inhibiting the ANG-induced BP elevation was not expressed based on dose but rather based on the corresponding total and free plasma concentration, e.g., for EXP3174 in conscious rats (Wong et al., 1996).
In vivo antagonism studies were performed not only in healthy normotensive rats, but, e.g., also with chronic oral eprosartan treatment in conscious 5/6 nephrectomy rats (Gandhi et al., 1999) or in spontaneously hypertensive rats with losartan (Wong et al., 1990c). In some cases they also were not limited to the inhibition of the systemic blood pressure response to ANG but, e.g., with valsartan have also explored antagonism for specific vasculature in the microcirculation, such as ciliary arteries and isolated porcine eye (Meyer et al., 1995).
Although most in vivo antagonism studies have focused on blood pressure, several studies have also looked into inhibition of other ANG responses. For example, candesartan was shown to inhibit the ANG-induced increase in plasma aldosterone (Wada et al., 1994), and losartan and telmisartan inhibited the ANG-induced enhancement of renal expression of pro-inflammatory molecules (Kumar et al., 2010).
However, most in vivo studies on nonvascular AT1R antagonism have been performed with the neuronal (prejunctional) receptors modulating catecholamine release. Thus, locally administered irbesartan was shown to inhibit ANG-stimulated catecholamine secretion from canine adrenals (Martineau et al., 1995). Studies on prejunctional AT1R were largely performed in pithed rats, where ANG enhances electrical stimulation-induced noradrenaline release. One such study reported an antagonist order of potency of telmisartan > losartan > irbesartan (Balt et al., 2001b), whereas another study from the same investigators reported a potency of candesartan > valsartan = eprosartan (Balt et al., 2001c). Other investigators reported a potency order of candesartan > eprosartan > EXP3174 > irbesartan and noted that this potency profile was similar to that observed for inhibition of ANG-induced vasoconstriction in the same study (Dendorfer et al., 2002). In contrast, yet other investigators found that enhancement of sympathetic outflow as enhanced by low-dose ANG was inhibited by eprosartan, but not irbesartan, losartan, or valsartan (Ohlstein et al., 1997); however, these findings are difficult to interpret as no such differences have been reported for any other in vivo study quantifying AT1R antagonism by ARBs.
1. Antagonism In Vivo in Humans.
Antagonism of AT1R by various ARBs has also been tested in vivo in humans, mostly healthy volunteers, and, similar to the above animal studies, this was largely done for ANG-induced blood pressure elevations. Following the original study with losartan (Christen et al., 1991), such studies have been performed with candesartan (Delacretaz et al., 1995; Ogihara et al., 1995; Belz et al., 1997, 2000; Malerczyk et al., 1998; Fuchs et al., 2000; Gleiter et al., 2004), irbesartan (Belz et al., 1999, 2000; Maillard et al., 1999; Mazzolai et al., 1999), losartan (Munafo et al., 1992; Belz et al., 1997, 1999, 2000; Maillard et al., 1999; Mazzolai et al., 1999; Fuchs et al., 2000; Gleiter et al., 2004), telmisartan (Belz et al., 2000; Stangier et al., 2001), and valsartan (Müller et al., 1994; Morgan et al., 1997; Belz et al., 1999, 2000; Maillard et al., 1999; Mazzolai et al., 1999). Most of these studies were based on single or repeated oral ARB administration and have assessed the degree of antagonism at multiple time points.
Some of these studies have used a combined in vivo and ex vivo approach to further the understanding of AT1R antagonism. In this approach, plasma samples were obtained at multiple time points in line with those of the ANG infusions. These plasma samples were added to ex vivo AT1R competition radioligand binding assays. After correction for effects of plasma in the absence of ARB, these assays allow measuring AT1R occupancy by the ARB with autocorrection for plasma protein binding and also taking into account any pharmacologically active compound, i.e., parent compound and active metabolites, to the degree to which it contributes to receptor occupancy. This approach even allows for the construction of Schild plots, which otherwise can only be done for in vitro assays, to determine drug affinity in vivo. Moreover, such radioreceptor assays can be seen as a more relevant way to study pharmacokinetics, and this has been validated against classic pharmacokinetic measurements not only for ARBs (Malerczyk et al., 1998) but also for antagonists at other types of receptors such as adrenoceptors (Wellstein et al., 1988; de Mey et al., 1993; Taguchi et al., 1998).
By using such approaches it was found that candesartan yields a stronger inhibition of ANG-induced blood pressure elevation than losartan for a given level of receptor occupancy, indicating greater affinity and/or slower off-rate from receptor (Belz et al., 1997; Fuchs et al., 2000). Direct comparative studies from these investigators also showed an order of potency of irbesartan > valsartan > losartan in one study (Belz et al., 1999) and of candesartan > telmisartan > valsartan > irbesartan > losartan (apparent Ki doses at 24-hour time point: 6, 54, 93.5, and 123 mg, respectively; Ki dose for losartan could not be determined due to little remaining antagonism at that time point) (Belz et al., 2000); the reason for the apparently contradictory order of potency reported from a single group of investigators remains unclear, but given the large variance of reported affinities in in vitro studies (Fig. 2) is not surprising. A similar apparent Ki dose for candesartan (1.9 mg) had been determined by the same investigators in an earlier study (Malerczyk et al., 1998). A direct comparative study from another investigator group found that irbesartan produced greater inhibition of vasoconstriction and greater occupancy in the radioreceptor assay than losartan or valsartan when recommended starting doses of all three ARBs were compared (Mazzolai et al., 1999), indicating that standard doses of three ARBs fall on different parts of the relative dose-response curve. Moreover, analysis of the latter study indicated that the correlation between receptor occupancy as measured in the radioreceptor assay and inhibition of vasoconstriction is best at the 4-hour time point (Maillard et al., 1999), indicating a possible hysteresis between the two assays.
In vivo antagonism of AT1R has also been quantified in humans for other responses such as renin release for candesartan, irbesartan, losartan and valsartan (Munafo et al., 1992; Müller et al., 1994; Maillard et al., 2002b), aldosterone release for candesartan (Ogihara et al., 1995) and ANG-induced increase in plasma NO for valsartan (Gossmann et al., 2000).
E. Inverse Agonism, Biased Agonism, Surmountability, and Reversibility of Antagonism
From early on in ARB research it became clear that the antagonist properties of this drug class in many cases are not easily explained by classic competitive antagonism (Robertson et al., 1994a). Therefore, various studies have explored the contribution of more recently discovered drug properties such as inverse agonism or biased agonism and have also explored the basis for insurmountable and poorly reversible antagonism by some ARBs.
1. Inverse Agonism.
For a long time it had been assumed that agonists activate receptors whereas antagonists block such activation but in the absence of agonist are without effects. This concept has undergone fundamental change in the last decade, and it is now generally accepted that many, perhaps even most, antagonists at G-protein-coupled receptors will also reduce receptor activity in the absence of agonist, a feature called inverse agonism (Kenakin, 2004). Not surprisingly, this concept also applies to AT1R (van Liefde and Vauquelin, 2009).
For technical reasons inverse agonism is easiest to detect with constitutively active mutants of receptors, i.e., those exhibiting considerable signaling even in the absence of agonist. Such mutants have not been reported to occur naturally for AT1R but have been created by site-directed mutagenesis in the studies of the next paragraph. Such mutated receptors do not only technically facilitate the detection of inverse agonism but have also helped to understand the drug-receptor interactions leading to this phenomenon at the molecular level (Miura et al., 2011, 2013).
On the basis of such approaches, inverse agonism has been shown for azilsartan (Ojima et al., 2011; Miura et al., 2013), olmesartan (Miura et al., 2006), telmisartan (Bhuiyan et al., 2009a), and valsartan (Miura et al., 2008; Bhuiyan et al., 2009a). Although several studies have demonstrated inverse agonism for the active metabolite EXP3174 (Noda et al., 1996; Miura et al., 2003; Feng et al., 2005), it is not fully clear whether the parent compound losartan also possesses this property, as it was detected in some (Bhuiyan et al., 2009a) but not other studies (Miura et al., 2003; Feng et al., 2005). Similarly, inverse agonism by candesartan was reported in one study (Feng et al., 2005) but not confirmed in another one (Miura et al., 2013).
The overall clinical relevance of the phenomenon of inverse agonism of ARBs has not been established. An interesting study in this regard compared nephroprotective effects of olmesartan and a closely related analog lacking inverse agonism in Dahl salt-sensitive rats; while neither lowered blood pressure in this hypertension model, only olmesartan significantly reduced urinary protein excretion by ~25%, whereas its analog without inverse agonism did not (Kiya et al., 2010); whether the differential effect of the two compounds on proteinuria is solely attributable to that in inverse agonism remains to be studied.
2. Biased Agonism.
It was recently realized that some compounds may exhibit a property called “biased agonism,” “protean agonism,” or “ligand-directed signaling,” i.e., the ability to selectively stimulate one signaling pathway of a given receptor compared with another; in some cases a compound can be an antagonist for one response and an agonist for a different response mediated by the same receptor (Patel et al., 2010). Although biased agonism has also been reported with experimental ARBs, losartan, telmisartan, and valsartan apparently are unbiased compounds, i.e., antagonists for all ANG responses (Violin et al., 2010).
Upon agonist exposure, GPCRs can typically undergo internalization, a process often involved in desensitization. In line with findings with antagonists with many other GPCR, ARBs do not cause internalization of normal AT1R, but if constitutively active AT1R mutants are studied, several ARBs, including candesartan, losartan, telmisartan, and valsartan can cause considerable internalization (Bhuiyan et al., 2010b), a phenomenon possibly reflecting biased agonism.
3. Surmountability and Reversibility of Antagonism.
Most clinically used antagonists at G-protein-coupled receptors exhibit a competitive, reversible, and surmountable interaction with the receptor. This means that the presence of antagonist shifts the agonist concentration-response curve to the right toward higher concentrations without affecting its maximum. In other words, in the presence of antagonist more agonist is needed to achieve the same response but, at least in an experimental setting, a very high concentration of agonist can overcome the antagonist effect and still yield a full response. Most clinically used ARBs do not adhere to this rule (van Liefde and Vauquelin, 2009). However, a mechanistic understanding of this is hampered by a lack of adherence to internationally recommended terminology (Jenkinson et al., 1995; Neubig et al., 2003). Thus, insurmountable antagonism is a rather descriptive term indicating that the maximum effect of an antagonist is reduced by pretreatment with or presence of the antagonist. Although surmountable antagonism is generally associated with reversible competitive antagonism, insurmountable antagonism can have several molecular or cellular reasons. Competitive antagonism implies that agonist and antagonist bind to mutually exclusive, i.e., identical or at least overlapping, sites in the receptor molecule. If both agonist and antagonist form only short-lasting associations with the receptor, competitive antagonism typically is also surmountable. Reversibility again is a rather descriptive term implying that the ligand-receptor association is short-lived; irreversible antagonism may be caused by covalent modification of the receptor, but a noncovalent interaction with a very slow dissociation rate may also appear as being irreversible under some assay conditions.
The surmountability and reversibility of antagonism by ARBs has largely been tested for their ability to inhibit contraction of isolated rabbit aorta and other blood vessels in vitro. In most such studies losartan exhibited surmountable antagonism (Wienen et al., 1992; Cazaubon et al., 1993; Zhang et al., 1993; Dickinson et al., 1994; Mizuno et al., 1995; Jin et al., 1997; Garcha et al., 1999; Morsing et al., 1999; Li et al., 2001), whereas the active losartan metabolite EXP3174 (Wienen et al., 1992; Shibouta et al., 1993; Mizuno et al., 1995; Garcha et al., 1999), azilsartan (Ojima et al., 2011), candesartan (Noda et al., 1993; Shibouta et al., 1993; Jin et al., 1997; Ojima et al., 1997; Garcha et al., 1999; Morsing et al., 1999), eprosartan (Keiser et al., 1995), irbesartan (Cazaubon et al., 1993; Morsing et al., 1999; Li et al., 2001), olmesartan (Mizuno et al., 1995; Ojima et al., 2011), and telmisartan (Wienen et al., 1993) mostly exhibited insurmountable antagonism, although to different degrees. Similar findings were also obtained for other functional responses such as inhibition of ANG-stimulated inositol phosphate formation (Dickinson et al., 1994; Vanderheyden et al., 1999; Vauquelin et al., 2001; van Liefde and Vauquelin, 2009). Insurmountable antagonism of ANG-induced vasoconstriction has also been demonstrated in rats in vivo (Champion et al., 1998). In one study, losartan exhibited surmountable antagonism in low and insurmountable antagonism in high concentrations (Zhang et al., 1994), and the surmountability of losartan antagonism may differ between vascular beds within a given species (Li et al., 2001).
Insurmountable antagonism could theoretically result from irreversible or pseudo-irreversible competitive antagonism, noncompetitive antagonism, and functional antagonism (Neubig et al., 2003), and these possibilities have been explored for various ARBs. The possibility of noncompetitive antagonism has been ruled out by saturation binding experiments in the presence of ARB, e.g., candesartan (Noda et al., 1993; Shibouta et al., 1993) or telmisartan (Wienen et al., 1993), or studies in which the surmountable losartan prevented insurmountable antagonism, e.g., by candesartan (Ojima et al., 1997), EXP3174 (Wienen et al., 1992), or by telmisartan (Wienen et al., 1993). Two types of studies have been instrumental in positively identifying the mechanism underlying insurmountable antagonism by most ARBs: first, experiments in which the functional antagonism by a given ARB was determined depending on the time of preincubation or of washout and, second, studies in which dissociation rates of the various ARBs from the AT1R were determined.
Experiments with candesartan (Noda et al., 1993; Ojima et al., 1997), EXP3174 (Mizuno et al., 1995), or olmesartan (Mizuno et al., 1995) have demonstrated that their insurmountable antagonism increased with duration of preincubation. Conversely, antagonism remained detectable for hours upon washout of azilsartan (Ojima et al., 2011), candesartan (Noda et al., 1993; Shibouta et al., 1993; Ojima et al., 1997), EXP3174 (Wienen et al., 1992; Noda et al., 1993; Shibouta et al., 1993; Mizuno et al., 1995), olmesartan (Mizuno et al., 1995; Ojima et al., 2011), or telmisartan (Wienen et al., 1993), although the degree of remaining antagonism differed between drugs in direct and indirect comparisons. In radioligand binding studies, candesartan also continued to inhibit binding after washout (Hara et al., 1995). These functional data pointed to an important role of slow dissociation rates from the receptor in insurmountable antagonism.
Several studies directly determined dissociation rates (t1/2 values; not to be confused with terminal elimination half-life in pharmacokinetic studies, which is also referred to as t1/2) from AT1R in kinetic radioligand binding studies, mostly based on a radiolabeled version of the ARB. In such studies t1/2 values were reported for candesartan of 66–133 minutes (Ojima et al., 1997; Fierens et al., 1999; Maillard et al., 2002a; Kakuta et al., 2005), losartan of 67 minutes (Kakuta et al., 2005), EXP3174 of 81 minutes (Kakuta et al., 2005), olmesartan of 65–166 minutes (Kakuta et al., 2005; Le et al., 2007), telmisartan of 29–354 minutes (Wienen et al., 2000; Maillard et al., 2002a; Kakuta et al., 2005; Le et al., 2007), and valsartan of 70 minutes (Kakuta et al., 2005). Because of major differences for a given ARB between studies, indirect comparison of these values is difficult. In the only direct comparative study using cloned human AT1R, the dissociation half-lives from the receptor exhibited an order of telmisartan > olmesartan ≥ candesartan > valsartan ≈ losartan (213, 166, 133, 70, and 67 minutes, respectively) (Kakuta et al., 2005).
On the basis of a comparison of dissociation rates from the receptor and degree of insurmountability of ANG-stimulated inositol phosphate formation it was proposed that a direct hyperbolic relationship exists between the two (van Liefde and Vauquelin, 2009). This would indicate that the surmountability of antagonism by ARBs is not a yes/no but rather a quantitative question. Accordingly, it was reported that vascular antagonism by candesartan and EXP3174 involves surmountable and insurmountable components (Shibouta et al., 1993). Moreover, out of four candesartan isomers, two exhibited insurmountable and two mixed antagonism (Noda et al., 1993), indicating that rather subtle chemical properties of an ARB can determine its type of antagonism, possibly by altering receptor dissociation rates. A more direct investigation was reported based on four pairs of closely related ARBs with surmountable versus insurmountable antagonism, including losartan/EXP3174 and telmisartan and a closely related analog thereof (Schambye et al., 1994). In these studies, mutations of the AT1R affected binding of the surmountable analog to a considerably greater degree than that of the insurmountable analog. Specifically, the nonconserved Asn295 in transmembrane (TM) segment VII of the AT1R appears to play a critical role in determining half-life at the receptor and hence surmountability of antagonism.
In conclusion, insurmountable antagonism is a property that almost all ARBs have to some degree. The degree of insurmountable antagonism appears to be a direct function of dissociation rate from the receptor, and molecular determinants of this have been established. Although insurmountability is an interesting in vitro feature of drugs, its in vivo role is not fully clear as competitive surmountable and insurmountable antagonism have very similar consequences if the concentration of agonist cannot be increased to a major extent, as is typically the case with endogenous neurotransmitters and hormones. On the other hand, the underlying slow dissociation from the receptor may well have therapeutic importance, because this can lead to a longer presence of antagonist in the microenvironment of the receptor and hence contribute to a long duration of action irrespective of its pharmacokinetic properties at the organism level.
F. Modeling of the AT1R Binding Pocket
Binding of ARBs to AT1R has been explored by structure activity relationships (Mire et al., 2005), modeling (Buhlmayer et al., 1991; Berellini et al., 2005; Miura et al., 2006; Bhuiyan et al., 2010a, 2009b), conformational analysis (Masek et al., 1993; Polevaya et al., 2001; Zoumpoulakis et al., 2002), and receptor mutation studies (Ji et al., 1994; Schambye et al., 1995; Miura et al., 2008; Bhuiyan et al., 2010a). The latter approach was partly based on systematic screening efforts but more often hypothesis driven, i.e., exchanging specific amino acids known to be important in the binding properties of other receptors; of note, site-directed mutagenesis studies have become an important way to experimentally test the predictions from the molecular modeling studies. In some cases, the binding site has been explored using mutants that also occur naturally in people (Arsenault et al., 2010). From early receptor mutation studies on it became clear that a given receptor mutation may differentially affect the affinity of various ARBs (Schambye et al., 1995). Nevertheless these studies are in reasonable agreement, with a few key amino acids, such as Tyr113, Lys199, His256, Ser109, and Gln257 being identified as important components of the binding site by in multiple studies, although individual ARBs may bind at additional sites as well (Bhuiyan et al., 2009b). However, modeling and mutagenesis studies are sometimes contradictory. For example, modeling indicated that His256 forms salt bridges with most ARBs, including telmisartan (Ohno et al., 2011), but a mutagenesis study (Schambye et al., 1995) by contrast showed that changing His256 to alanine or other neutral amino acids had no effect on telmisartan binding, although it did affect binding of a nonclinical ARB, SKF-105,866.
Ohno et al. (2011) used modeling to compare binding of seven ARBs, including the newest entry, azilsartan. They concluded that all the ARBs share a binding site containing 5–7 amino acids, not adjacent to each other in the linear molecular sequence, that develop ionic, hydrophobic, or hydrogen bonding interactions with the ARBs. The biphenyl-tetrazole and imidazole regions of the ARBs bind in this binding site. Because the ARBs are quite similar in the biphenyl-tetrazole region (see Section II), major differences in binding are due to the structure in the imidazole region. Thus, modeling and mutagenesis studies further suggest that the cyclopentyl ring in irbesartan may bind in a hydrophobic pocket of the receptor not accessible to other ARBs. This was referred to as “pentagon attachment,” and may explain high binding affinity (Fujino et al., 2010; Miura et al., 2011). Telmisartan, due to the second benzimidazole substituent, binds in a unique third site, an additional hydrophobic pocket, which may contribute to its high affinity and slowly reversible binding. This three-point binding had been labeled “delta lock” (Ohno et al., 2011).
Studies of the binding site have contributed to our understanding of the mechanism of inverse agonism (see Section IV.E.1). Specific binding interactions of ARBs have been suggested to be involved in inverse agonism because these interactions stabilize the receptor in an inactive conformation. Mutation studies demonstrated that Tyr113, Lys199, Gln257, and His256 were important to olmesartan binding but that interactions between olmesartan and Tyr113, Lys199, and His256 but not Gln257 were required for inverse agonist activity (Miura et al., 2006). Modeling further suggested that inverse agonism was related to a conformational interaction between Tyr113 in TM III and His256 in TM VI, termed the “double chain domain,” and this could relate to clinical potency of olmesartan (Kiya et al., 2010). Irbesartan, another potent inverse agonist, also requires Tyr113 for this activity (Fujino et al., 2010). However, vasartan’s inverse agonist activity may be primarily mediated through Lys199 of TM V (Miura et al., 2008).
V. Receptor Selectivity and Ancillary Effects
All clinically used ARBs display marked, i.e., >1000-fold, selectivity for AT1R over AT2R [azilsartan (Ojima et al., 2011), candesartan (Shibouta et al., 1993), eprosartan (Keiser et al., 1995), irbesartan (Cazaubon et al., 1993), losartan (Chiu et al., 1990a), olmesartan (Mizuno et al., 1995), telmisartan (Wienen et al., 1993), valsartan (Criscione et al., 1993)], but inhibition of AT1R may also affect AT2R function as there can be cross-talk between the two receptor types (Miura et al., 2010). The clinically used ARBs typically also lack cross-reactivity with other G-protein-coupled receptors. Some reports to the contrary most likely reflect that ANG may be involved as a mediator of responses elicited via other receptors (Madwed and Winquist, 1994). In support of this, candesartan and losartan were found to lack effects in AT1aR/AT1bR double knockout mice (Tsuchida et al., 1998).
Nevertheless, in many cases ARB effects have been reported that apparently are not AT1R mediated. For some ARBs, notably telmisartan, many of these AT1R-independent effects are mediated by PPAR, specifically PPAR-γ, but other PPARs as well as a range of other molecular targets have been implicated. Direct effects on some ion channels as well as on unidentified targets have also been reported. Some ARBs are substrates or inhibitors of certain transporters, these effects are described in Section III.B and consequences of such effects are covered in Section VI.
A. Peroxisome Proliferator-Activated Receptors
PPARs are a family of ligand-activated transcription factors that belongs to the superfamily of nuclear receptors and have three known members, PPAR-α, PPAR-γ, and PPAR-δ. PPAR-α is the molecular target of fibrates in lipid-lowering treatment, PPAR-γ is the target of thiazolidinediones in metabolic regulation; a clinical use for PPAR-δ agonists has not been firmly established.
PPAR-γ is an important mediator in the pathogenesis of insulin resistance (Kurtz and Pravenec, 2004; Guo and Tabrizchi, 2006), a condition frequently coexisting with hypertension, but PPAR-γ may also have effects more directly related to hypertension, e.g., by affecting endothelial function (Storka et al., 2008). A possible role for PPAR-γ in some ARB responses has been studied by multiple approaches. These include direct measurement of binding to and activation of PPAR-γ (Fujimoto et al., 2004; Schupp et al., 2004) and inhibition of ARB responses by PPAR-γ antagonists such as GW9662 (Willson et al., 2000), AT1R knock-out mice (Rong et al., 2010), and PPAR-γ knockdown by siRNA approaches (Nakaya et al., 2007; Scalera et al., 2008; Walcher et al., 2008). More indirect evidence for a role of PPAR-γ in ARB effects comes from comparisons of ARBs with and without alleged PPAR-γ agonism (Benson et al., 2008; Scalera et al., 2008) and comparison of ARB effects with those of established PPAR-γ agonists such as pioglitazone or rosiglitazone (Zanchi et al., 2007; Walcher et al., 2008).
The most direct biochemical evidence for an interaction of ARBs with PPAR-γ comes from binding studies in a cell-free system in which telmisartan and valsartan had EC50 values of 463 nM and 6.2 µM, respectively (Storka et al., 2008). A similar study reporting ARB binding affinities to PPAR-γ reported values of 3–5 µM for candesartan, irbesartan, losartan, and telmisartan (but not for olmesartan or valsartan) (Erbe et al., 2006). A direct molecular interaction with PPAR-γ was also demonstrated for irbesartan and telmisartan in protease protection and in fluorescence resonance energy transfer assays, in which the two ARBs apparently induced a different PPAR-γ conformation compared with rosiglitazone (Schupp et al., 2005); this alternative conformation recruits intracellular cofactors to PPAR-γ that are not recruited by glitazones, which may represent the cellular basis why PPAR-γ-activating ARBs in contrast to glitazones do not promote weight gain. Therefore, some authors refer to such ARBs as selective PPAR-γ modulators. Molecular modeling studies have attempted to define the binding site of telmisartan within PPAR-γ (Benson et al., 2004). More recently crystallography studies at a resolution of 2.18 Å have identified differences in binding site that explain why telmisartan is a partial PPAR-γ agonist compared with, e.g., rosiglitazone (Amano et al., 2012).
Some ARBs not only bind to but also can activate PPAR-γ, e.g., in 3T3-L1 cells where irbesartan and telmisartan induced transcriptional activity of PPAR-γ, whereas losartan mimicked that only at a very high concentration and eprosartan was without effect (Schupp et al., 2004).Within this study, activation of a Gal4-DBD-h PPAR-γ-LBD fusion protein to stimulate a Gal4-dependent luciferase reporter gene was observed with an order of potency of telmisartan > irbesartan > losartan > eprosartan (EC50 5, 27, >50 µM, and nondetectable, respectively). Importantly, PPAR-γ activity was also induced by irbesartan and telmisartan in the AT1R-deficient cell line PC12W, demonstrating that it does not occur secondary to AT1R activation. PPAR-γ activation by irbesartan, losartan, and telmisartan but not eprosartan was also shown in human preadipocytes (Janke et al., 2006) and by telmisartan in murine macrophages (Blessing et al., 2008). In PPAR-γ-transfected CV-1 cells significant activation was only found for telmisartan, whereas candesartan, eprosartan, irbesartan, EXP3174, olmesartan, and valsartan (all tested at 10 µM) lacked such effects; however, even telmisartan was only a partial agonist compared with pioglitazone and rosiglitazone (Benson et al., 2004). Similar findings were reported from another study in PPAR-γ-transfected CV-1 cells in which telmisartan and, to a lesser extent, irbesartan, activated the receptor, whereas candesartan and losartan did not cause activation despite binding to the receptor (Erbe et al., 2006). In similar experiments azilsartan did not activate PPAR-γ (Kajiya et al., 2011). In murine macrophages telmisartan but not losartan, olmesartan, or valsartan activated PPAR-γ (Matsumura et al., 2011). PPAR-γ activation by telmisartan was also shown in human macrophages (Pang et al., 2012). Site-directed mutagenesis studies using reporter gene readouts have indicated that the binding of telmisartan to PPAR-γ has distinct structural requirements compared with that of the thiazolidinediones (Tagami et al., 2009). Upon exposure to irbesartan and telmisartan a downregulation of PPAR-γ mRNA was observed that was quantitatively similar to that induced by pioglitazone (Schupp et al., 2005).
Accordingly, it was found that some ARBs can induce known PPAR-γ target genes. In 3T3-L1 cells, irbesartan, losartan, and telmisartan promoted differentiation into an adipocyte-like phenotype, which was accompanied by markedly increased expression of adipose protein-2 (Schupp et al., 2004). Corresponding EC50 values were 0.13 µM for telmisartan and 3.5 µM for irbesartan, whereas losartan only had detectable effects at 100 µM, and eprosartan was ineffective even at this very high concentration. The effects of 100 µM irbesartan, losartan, and telmisartan were comparable to those of the PPAR-γ agonist pioglitazone (10 µM). Concomitantly other investigators have reported similar data (Benson et al., 2004). Comparing telmisartan and valsartan, a similar induction of adipose protein-2 and other PPAR-γ target genes was also reported in 3T3-L1 cells by other investigators, with telmisartan being active at 1–10 µM and valsartan being inactive in these concentrations (Fujimoto et al., 2004). Similar induction of PPAR-γ target genes was also shown in human primary preadipocytes (Janke et al., 2006) and murine macrophages (Blessing et al., 2008). In rat adipose tissue azilsartan did not alter expression of adipose protein-2 or other PPAR-γ target genes (Zhao et al., 2011). In isolated human monocytes, irbesartan, the losartan metabolite EXP3179, and telmisartan induced the PPAR-γ target gene CD36, whereas losartan itself and its metabolite EXP3174 were inactive (Kappert et al., 2009). In murine macrophages, telmisartan induced expression of ATP binding cassette transporters A1 and G1 and of CD36, an effect blocked by PPAR-γ siRNA (Matsumura et al., 2011). However, it should be noted that some ARBs may affect the expression of PPAR-γ target genes in mice in vivo (Iwai et al., 2007) or in human macrophages in vitro (Pang et al., 2012) without being agonists at the receptor. Taken together these findings demonstrate that some ARBs can bind to and activate PPAR-γ. In this regard telmisartan is most potent, irbesartan is less potent, and losartan even less; for valsartan a moderate binding affinity was reported in at least some studies but it lacked agonism, and azilsartan, eprosartan, and olmesartan were also reported as inactive.
The more important question is whether PPAR-γ activation by some ARBs contributes to their overall pharmacological profile. The most frequently used approach in this regard is the use of PPAR-γ antagonists such as GW9662. This compound is a potent and irreversible PPAR-γ antagonist with 10- and 600-fold selectivity over PPAR-α and PPAR-δ, respectively; it covalently attaches to the ligand binding pocket of PPAR-γ at Cys285 (Willson et al., 2000; Leesnitzer et al., 2002). A wide range of telmisartan effects have been shown to be sensitive to inhibition by GW9662. At the cellular level this includes the activation of PPAR-γ target genes in human preadipocytes (Schupp et al., 2004; Janke et al., 2006), the downregulation of AT1R mRNA and protein in rat vascular smooth muscle cells (Imayama et al., 2006) and in endothelial cells upon repeated passaging (Scalera et al., 2008), the proliferation of human endothelial progenitor cells (Honda et al., 2009; Steinmetz et al., 2010), the IGF-1 receptor expression in human skeletal muscle cells (Storka et al., 2008), the improvement of insulin sensitivity in Hep3B cells (Yoshida et al., 2008), and the enhanced fatty acid oxidation in rat skeletal muscle (Sugimoto et al., 2008). With regard to inflammation, GW9662-sensitive telmisartan in vitro effects include the regulation of inflammatory markers in mouse macrophages and endothelial cells (Nagai et al., 2006), the inhibition of TNF-induced IL-6 expression in rat vascular smooth muscle cells (Tian et al., 2009), the inhibition of TGF-stimulated accumulation of extracellular matrix (Yao et al., 2008), and the increase in permeability in human umbilical vein endothelial cells by downregulation of ZO-1 (Bian et al., 2009). Related to atherosclerosis, this includes the downregulation of advanced glycosylation end product (AGE) receptor in Hep3B cells not shared by candesartan (Yoshida et al., 2006), the downregulation of AGE receptor and cellular damage in human endothelial cells (Yamagishi et al., 2008), and inhibition of AGE-induced MCP-1 expression and downregulation of AGE receptor in human mesangial cells (Matsui et al., 2007).
At the in vivo level, GW9662-sensitive telmisartan effects include the inhibition of choroidal neovascularization in mice (Nagai et al., 2006), the improvement of endothelial NO release and reduction of atherosclerotic plaques in Watanabe hyperlipidemic rabbits (poorly mimicked by candesartan) (Ikejima et al., 2008), the restoration of endothelial NO synthase and PPAR-γ expression in Dahl salt-sensitive rats (Kobayashi et al., 2008b), the improvement of vascular dysfunction in subtotally nephrectomized rats (partly mimicked by losartan) (Toba et al., 2012), and the prevention of hydronephrosis after unilateral ureteral obstruction in AT1R knockout mice (partly mimicked by losartan) (Kusunoki et al., 2012). Metabolic parameters affected by telmisartan in a GW9662-sensitive manner include fatty acid oxidation in rat skeletal muscle (Sugimoto et al., 2008), improvement of glucose levels in Cohen-Rosenthal hypertensive nonobese diabetic rats (Younis et al., 2010), and protection against diabetic complications in a mouse model (Toyama et al., 2011). Telmisartan effects on the size of isoprenaline-induced heart infarcts in diabetic rats (Goyal et al., 2010) and on infarct size in mouse stroke models (Haraguchi et al., 2009; Iwanami et al., 2010) were also GW9662 sensitive. Finally, the telmisartan-induced attenuation of cognitive decline in Alzheimer’s models such as the ddY mouse (Mogi et al., 2008), the amyloid-β-injected mouse (Tsukuda et al., 2009), and upon chronic cerebral hypoperfusion (Washida et al., 2010) were also GW9662 sensitive.
Evidence for an involvement of PPAR-γ in telmisartan effects was obtained by using systemic receptor knockout or siRNA-induced knockdown. Thus, telmisartan prevented hydronephrosis development after unilateral ureteral obstruction in AT1R knockout mouse, an effect that was partly mimicked by losartan and blocked by GW9662 (Kusunoki et al., 2012). The telmisartan-induced expression of ATP binding cassette transporters A1 and G1 and of the scavenger receptor class B type 1 in THP-1 was abolished by PPAR-γ siRNA in murine (Nakaya et al., 2007; Matsumura et al., 2011) and human macrophages (Pang et al., 2012). siRNA-induced PPAR-γ knockdown abolished the ability of telmisartan to delay endothelial senescence during repeated passaging (Scalera et al., 2008). Such PPAR-γ knockdown also abolished the ability of telmisartan to inhibit SDF-1-induced ICAM3 translocation (Walcher et al., 2008).
More circumstantial evidence for an involvement of PPAR-γ in cellular or tissue responses to PPAR-γ-activating ARBs comes from studies in which the effects of ARBs with and without PPAR-γ agonistic properties were directly compared. For example, telmisartan prevented progression of liver steatosis in rats, whereas valsartan did not (Fujita et al., 2007). Other telmisartan effects not shared by other ARBs include induction of apoptosis of renal cancer cells (not mimicked by the weaker PPAR-γ activators irbesartan and losartan nor the inactive candesartan or valsartan) (Funao et al., 2009); insulin sensitization in Hep3B cells (not mimicked by candesartan) (Yoshida et al., 2008); downregulation of AGE receptor in Hep3B cells (not shared by candesartan) (Yoshida et al., 2006); inhibition of human vascular smooth muscle proliferation (partly mimicked by irbesartan, not mimicked by eprosartan or valsartan) (Yamamoto et al., 2009); inhibition of TGF-stimulated accumulation of extracellular matrix (not mimicked by valsartan) (Yao et al., 2008); stimulation of adiponectin expression in 3T3-L1 cells (not mimicked by eprosartan) (Clasen et al., 2005); improvement of insulin sensitivity in Hep3B cells (not mimicked by candesartan) (Yoshida et al., 2008); increase in permeability in human umbilical vein endothelial cell layers (not mimicked by valsartan) (Bian et al., 2009); reduction of serum glucose, insulin, and triglyceride levels and of weight gain in rats fed a high-fat diet (not mimicked by losartan) (Benson et al., 2004); or improvement of glucose levels in Cohen-Rosenthal hypertensive nonobese diabetic rats (not mimicked by valsartan despite both similarly lowering blood pressure) (Younis et al., 2010).
Similar but even more circumstantial evidence comes from studies in which effects of telmisartan were compared with those of established PPAR-γ activators from the thiazolidinedione group such as pioglitazone or rosiglitazone. Telmisartan mimicked thiazolidinedione-induced signal transduction in human skeletal muscle and umbilical vein endothelial cells (Storka et al., 2008), adiponectin expression in 3T3-L1 cells (also stimulated by irbesartan) (Clasen et al., 2005), monocyte migration (Kappert et al., 2009), and prevention of progression of liver steatosis in rats (Fujita et al., 2007).
Only limited evidence is available for a possible involvement of PPAR-α or PPAR-δ in ARB effects. Thus, even high concentrations of several ARBs, including azilsartan (Kajiya et al., 2011) or telmisartan (Benson et al., 2004; Erbe et al., 2006), did not activate PPAR-α or PPAR-δ receptors in biochemical assays. On the other hand, it was reported that telmisartan treatment activates PPAR-α target genes in mouse liver but not skeletal muscle; this was replicated in vitro in Hep2G only in high concentrations, where it was markedly attenuated by PPAR-α siRNA (Clemenz et al., 2008). Telmisartan but not candesartan or losartan also upregulated PPAR-δ expression in 3T3-L1 preadipocytes and reduced visceral fat and prevented obesity development in wild-type mice and spontaneously hypertensive rats but not in PPAR-δ knockout mice (He et al., 2010). Thus, a possible role of PPAR-α or PPAR-δ in ARB effects remains equivocal.
In conclusion, numerous studies have demonstrated that telmisartan binds to and activates PPAR-γ; irbesartan causes PPAR-γ activation only in higher concentrations, and losartan in even higher concentrations, whereas other ARBs do not activate PPAR-γ. Studies against a large variety of endpoints in isolated cells and more importantly in experimental animals in vivo demonstrate that PPAR-γ mediates or at least contributes to known telmisartan effects. However, the role of PPAR-γ in in vivo effects of therapeutic telmisartan doses in patients remains under discussion (Ernsberger and Koletsky, 2007; Rizos et al., 2009). Evidence in favor of this hypothesis includes the findings from many animal models in which typical antihypertensive doses of telmisartan exerted at least some of their effects via PPAR-γ, but these animal models may not be predictive for patients. Unfortunately, only very few clinical studies have been reported that are suitable to address this. In one study, telmisartan reduced expression of some inflammatory markers in cultured human lymphocytes in an AT1R-independent manner, and in a small, placebo-controlled, double-blind in vivo study in patients with hypertension and coronary heart disease, telmisartan treatment also reduced expression of these inflammatory markers (Link et al., 2006). In a small, open-label, parallel group study with losartan treatment of patients with hypertension, drug plasma levels reached values compatible with PPAR-γ activation; concomitantly, PPAR-γ target genes were found to be upregulated in monocytes from losartan-treated patients (Kappert et al., 2009). In a follow-up study, high doses of telmisartan (80 and 160 mg/day) in patients with metabolic syndrome also upregulated the PPAR-γ target genes CD36 and CD136 in monocytes (Bähr et al., 2011). In small crossover study, overweight patients with hypertension were randomized to eprosartan versus telmisartan treatment; some metabolic parameters, which in animal studies involved PPAR-γ, were significantly more improved by telmisartan than by eprosartan (Fogari et al., 2009). However, in another small crossover study between irbesartan and placebo in obese men, irbesartan lowered blood pressure but did not affect adiponectin or lipoprotein lipase expression (Janke et al., 2006). In another placebo-controlled study in patients with obesity but not those with diabetes or hypertension, telmisartan did not improve insulin sensitivity during 16 weeks of treatment (Hsueh et al., 2010). Therefore, due to a scarcity of adequately powered and designed dedicated studies, it remains unclear whether and to which degree PPAR-γ activation by telmisartan and other ARBs contributes to their well established beneficial clinical effects.
B. Effects on Ion Channels
Several ARBs can affect currents through various ion channels, mostly K+ channels, but such effects were typically observed at supratherapeutic concentrations. Candesartan (100 nM) and eprosartan (1 µM) inhibited hKv1.5, hERG (human ether-a-go-go-related gene), and KvLQT11minK channels cloned from human heart and Kv4.3 channels cloned from rat heart and expressed in mammalian cell lines (Caballero et al., 2001). Losartan and EXP3174 (1 µM) transiently increased the hKv1.5 current followed by a voltage-dependent block in guinea pig heart; in the same preparation losartan reduced HERG currents, whereas EXP3174 increased them (Caballero et al., 2000). Telmisartan was reported to inhibit cloned Kv1.3 and Kv1.5 voltage-gated potassium channels expressed in X. laevis oocytes with IC50 values in the low micromolar range (Tu et al., 2008; Li et al., 2009a). Kv1.3 inhibition was also reported from rat lymphocytes at 10–100 µM telmisartan (Luo et al., 2010). Telmisartan was also reported to inhibit HERG channels with an IC50 of 24 µM (Tu et al., 2008). Of note, in line with such direct ion channel effects typically occurring in the micromolar concentrations, clinical correlates have been reported for none of them, and specifically QT prolongation, which can be caused by HERG channel inhibition, has not been reported.
C. Other Ancillary Effects
Some studies have hinted at AT1R-independent effects of ARBs but have not positively identified the underlying molecular mechanism. For example, losartan was reported to have a qualitatively different profile of inhibiting blood pressure responses in rats compared with enalapril, eprosartan, or a peptide AT1R antagonist (Ohlstein et al., 1992); similarly, effects of eprosartan differed from those of losartan, irbesartan, and valsartan in pithed rats in the hands of the same investigators (Ohlstein et al., 1997). Telmisartan lowered systolic and diastolic blood pressure and body weight, increased high-density lipoprotein cholesterol and reduced and stabilized plaques in ApoE1/AT1R double-knockout mice (Fukuda et al., 2010). Similarly, telmisartan reduced oxidative stress equally well in mesangial cells obtained from wild-type or AT1R knockout mice, but based upon lack of inhibition by GW9662 this effect was not attributable to PPAR-γ (Shao et al., 2007). Telmisartan also had free radical scavenging effects in mouse mesangial cells, which, based on receptor knockout studies, were not mediated by AT1R or by PPAR-γ (Shao et al., 2007). Telmisartan also reduced TNF-induced vascular cell adhesion molecule expression in human umbilical vein endothelial cells that was not mimicked by losartan or EXP3174 or a PPAR-γ agonist and also not inhibited by a PPAR-γ antagonist (Cianchetti et al., 2008; Nakano et al., 2009). Beneficial metabolic effects of telmisartan against diet-induced obesity, insulin resistance, and fatty liver have also been reported in AT1R knockout mice, but an involvement of PPAR-γ has not been excluded in those studies (Rong et al., 2010).
Some ARBs, particularly irbesartan and losartan, in high concentrations (>1 µM) can inhibit 9,11-dideoxy-9α,11α-methanoepoxy prostaglandin F2α (U46619)-induced thromboxane A2 receptor-mediated vasoconstriction of isolated canine coronary arteries and aggregation of human platelets (Li et al., 2000) or rat hindlimb vasoconstriction in vivo, the latter effect not being mimicked by candesartan (Fukuhara et al., 2001). The inhibitory effect on platelet aggregation has been reported by other investigators and, based on radioligand binding studies, is based on a low potency but direct interaction with the thromboxane receptor (Liu et al., 1992a; Monton et al., 2000). At concentrations of ≥40 ng/ml (approximately 80 nM), telmisartan caused vasodilatation against thromboxane in cannulated rat mesenteric arteries (Asano et al., 2006). In contrast, vasoconstriction by U46619 in feline hindquarters was not sensitive to candesartan (Lambert et al., 1998; Champion et al., 1999). Although not strictly an ancillary effect, candesartan and valsartan blocked aldosterone-induced vasoconstriction that occurs nongenomically via AT1R (Yamada et al., 2008). The molecular mechanisms underlying all of these effects remain to be established.
VI. Pharmacokinetic Properties
To avoid bias in reporting, this section is primarily based on the prescribing information approved by regulatory authorities as assessed in June 2012; in this regard, we primarily used information from the U.S. Food and Drug Administration (www.fda.gov), but for azilsartan, the Japanese label information was used additionally because, in the U.S., azilsartan medoxomil is marketed, whereas in Japan, azilsartan is used. Where available, published studies were used to reference statements from the label information; where no such reference is given, the statement has directly been taken from the corresponding prescribing information. Unless specifically stated otherwise, all pharmacokinetic information relates to human use. A summary of key pharmacokinetic parameters of all ARBs is presented in Table 3. Of note, many reported pharmacokinetic studies unfortunately do not indicate the ethnicity of the study subjects, but they are most likely Caucasian subjects. Studies in non-Caucasian populations are specifically mentioned below.
Key pharmacokinetic parameters of ARBs
Data were obtained from the regulatory authority approved U.S. prescribing information except for azilsartan where the corresponding Japanese document was used. For valsartan information from the prescribing information for both the capsule and tablet formulation were used.
A. Absorption and First-Pass Metabolism
Depending on country, both azilsartan medoxomil (e.g., U.S. and European Union) and azilsartan (e.g., Japan) are available for clinical use, although an early publication from the manufacturer (Kohara et al., 1996) concluded that azilsartan would not need a prodrug. Azilsartan medoxomil and azilsartan are rapidly absorbed from the gastrointestinal tract (tmax 1.5–3 hours), and during this process, azilsartan medoxomil is effectively hydrolyzed to its active metabolite azilsartan. Accordingly, no azilsartan medoxomil is detectable in plasma after oral administration. The pharmacokinetics of orally administered azilsartan are rather similar, but when azilsartan rather than azilsartan medoxomil is administered, bioavailability, Vd, and elimination half-life are slightly greater (Table 3); whether this reflects a true difference in pharmacokinetic profile or normal variation between studies remains unclear because a greater bioavailability may on technical grounds yield longer elimination half-life estimates. In a range of 20–320 mg, exposure to azilsartan is dose proportional upon single-dose oral administration. The estimated bioavailability of azilsartan following administration of azilsartan medoxomil is approximately 60% and not affected by concomitant food intake.
Candesartan cilexetil is rapidly absorbed from the gastrointestinal tract (tmax 3–4 hours) and completely converted to candesartan by ester hydrolysis during this process (Hübner et al., 1997; vanLier et al., 1997). The bioavailability of candesartan upon oral candesartan cilexetil administration is 15% and not affected by concomitant food intake (Riddell, 1997). For comparison, the bioavailability of candesartan upon oral candesartan cilexetil administration in rats and dogs is 28 and 5%, respectively (Kondo et al., 1996). Radioreceptor assay techniques have yielded very similar estimates of pharmacokinetic parameters of candesartan as those obtained by classic analytical techniques (Malerczyk et al., 1998).
Eprosartan is rapidly absorbed from the gastointestinal tract (tmax 1–2 hours) with an absolute bioavailability of 13% (Tenero et al., 1998b). Concomitant food intake delays absorption, as with most drugs, but its effects on Cmax and AUC are not deemed clinically relevant (Tenero et al., 1998b). Increases in eprosartan plasma concentrations are slightly less than dose proportional upon oral administration of 100–800 mg.
Irbesartan is rapidly absorbed from the gastrointestinal tract (tmax 1.5–2 hours) with an absolute bioavailability of 60–80% (Chando et al., 1998; Vachharajani et al., 1998a), independent of concomitant food intake (Vachharajani et al., 1998b). Steady-state concentrations are achieved within 3 days and limited accumulation of irbesartan (<20%) is observed in plasma upon repeated once daily dosing (Brunner, 1997).
Losartan is well absorbed from the human gastrointestinal tract (tmax 1 hour for losartan and 3–4 hours for the active metabolite EXP3174) but undergoes substantial first-pass metabolism by P450 enzymes (Lo et al., 1995; Stearns et al., 1995; Yun et al., 1995). Although only 14% of the administered dose is converted to EXP3174, this active metabolite has an AUC that is about four times as great as that of losartan, although plasma concentrations of losartan and EXP3174 are approximately equal (Ohtawa et al., 1993). Similar findings were also obtained in Japanese subjects (Nakashima and Umemura, 1996), but recent studies suggest that there may be relevant ethnic differences in the pharmacokinetics of losartan, e.g., between ethnic groups within China (Yang et al., 2012). A meal slows absorption of losartan and decreases its Cmax but has only minor effects on losartan AUC or on the AUC of the metabolite (about 10% decreased). In rats the absorption of losartan exhibits distinct differences from humans (Wong et al., 1996), i.e., absorption in the upper (but not lower) gastrointestinal tract is accompanied by glucuronidation rather than generation of EXP3174 (Krieter et al., 1995). Although the bioavailability of losartan in dogs is rather similar to that in humans (Christ et al., 1994), this species forms only little EXP3174 (see Section VI.C). In pigs, only 2% of an administered losartan dose becomes detectable in the circulation as nonmetabolized EXP3174 (Lankford et al., 1997). Such interspecies differences need to be taken into consideration when interpreting pharmacological data from experimental animals.
Olmesartan medoxomil is rapidly and completely converted by ester hydrolysis to olmesartan during absorption by breakdown of the medoxomil ester moiety via the enzyme paraoxonase I/arylesterase in the intestinal wall and plasma (Mire et al., 2005; Ishizuka et al., 2012;). More recent studies have identified a role for carboxymethylenebutenolidase in the conversion of olmesartan medoxomil to olmesartan (Ishizuka et al., 2010). Uptake into human hepatocytes is mediated by the OATP1B1 and OATP1B3 transporters (Nakagomi-Hagihara et al., 2006; Yamada et al., 2007). Peak olmesartan plasma concentrations are reached after 1–2 hours, and the absolute bioavailability following administration of olmesartan medoxomil is approximately 26% (Schwocho and Masonson, 2001) compared with <5% for administration of olmesartan (Laeis et al., 2001). The bioavailability of olmesartan medoxomil is not affected by concomitant food intake. Plasma concentrations of olmesartan reached steady state after 5 days of once daily administration of olmesartan medoxomil (Schwocho and Masonson, 2001). The pharmacokinetics of olmesartan have also been reported based on population pharmacokinetic analysis in patients with hypertension (Tanigawara et al., 2009).
Telmisartan is rapidly absorbed from the gastrointestinal tract (tmax 0.5–1 hour) (Stangier et al., 2000a). The bioavailability is dose-dependent with 42 and 58% at the 40- and 160-mg dose, respectively. Accordingly, the pharmacokinetics of telmisartan are nonlinear at the dose range of 40–160 mg, with greater than dose-proportional increases in plasma concentrations with higher doses. The hepatic uptake of i.v. [11C]telmisartan is limited only by hepatic blood flow and apparently mediated by the transporter OATP1B3 (Takashima et al., 2011). Accordingly, [11C]telmisartan has been proposed to be a biomarker for OATP1B3 activity in rats and humans in vivo (Shimizu et al., 2012). Concomitant food intake slightly reduces the bioavailability of telmisartan with a reduction of AUC of about 6% for the 40-mg dose and ~20% for the 160-mg dose, which are not considered clinically relevant. The pharmacokinetics of telmisartan in Chinese subjects were rather similar except for a slightly later tmax (Zhang et al., 2006). In a pharmacokinetic comparison of orally and intravenously administered telmisartan between mice, rats, rabbits, dogs, and humans, distinct profiles were observed for each species (Wienen et al., 2000), which need to be taken into account when interpreting data from experimental animals.
Valsartan is concomitantly available as a tablet and a capsule, but the two formulations exhibit a similar pharmacokinetic profile (Sechaud et al., 2002). Valsartan is rapidly absorbed from the gastrointestinal tract (tmax 2–4 hours), and the absolute bioavailability is 25% (Flesch et al., 1997; Müller et al., 1997), but higher values were reported when a neutrally buffered solution of radiolabeled valsartan was orally administered, which may not be representative for the valsartan formulation used therapeutically (Waldmeier et al., 1997). Within the clinical dosing range, valsartan exhibits dose-linear pharmacokinetics. Concomitant food intake reduced AUC and Cmax by approximately 40 and 50%, respectively. The pharmacokinetics of valsartan have also been reported in children and adolescents with hypertension (Blumer et al., 2009).
In conclusion, all ARBs are rapidly absorbed, with azilsartan, azilsartan medoxomil, irbesartan, and telmisartan having higher bioavailability than candesartan cilexetil, eprosartan, losartan, olmesartan medoxomil, or valsartan. During this process the prodrugs azilsartan medoxomil, candesartan cilexetil, and olmesartan medoxomil are fully converted into their respective active metabolites; i.e., these prodrugs are not detectable in circulating plasma upon oral administration. In contrast, losartan and its active metabolite are present in similar concentrations in human plasma. Concomitant food intake affects only the absorption of valsartan to a major extent.
B. Distribution
Azilsartan is highly bound to plasma proteins (>99%), mainly albumin, resulting in a Vd of approximately 16 or 21 liters for studies with azilsartan medoxomil and azilsartan, respectively. In rats, minimal azilsartan-associated radioactivity crossed the blood-brain barrier. In pregnant rats, azilsartan passed the placental barrier and was distributed to the fetus.
Candesartan is highly bound to plasma proteins (>99%), resulting in a Vd of 0.13 l/kg. In rats, candesartan crosses the blood-brain barrier poorly, if at all. Candesartan passed the placental barrier in rats and was distributed to the fetus. There is wide distribution of candesartan in rats and dogs (Kondo et al., 1996).
Eprosartan is highly bound to plasma proteins (98%). In a pooled population, pharmacokinetic analysis from two phase III trials mean Vd for a 65-year-old patient was 308 l, indicating possible distribution into tissues; however, this value represents the Vss for oral administration, whereas the Vd after i.v. administration was reported at 13 liters (Tenero et al., 1998b).
Irbesartan is stated to have 90% plasma protein binding in the label information [but published data indicate a value of 99.5% (Morsing et al., 1999)], primarily to albumin and α1-acid glycoprotein with negligible binding to cellular components of blood, resulting in an average Vd of 53–93 l, indicating possible distribution into tissues. Although Vd, expressed in liters per kilogram, is similar in rats, it appears to be much greater in monkeys, possibly reflecting differences in plasma protein binding (Davi et al., 2000). The combined use of rat, dog, and monkey data has allowed good predictions of human Vd and clearance (Kumar and Srinivas, 2008). Studies in rat indicate that irbesartan weakly crosses the blood-brain barrier and placenta; it is excreted in the milk of lactating rats.
The pharmacokinetics of losartan and its active metabolite EXP3174 were determined after i.v. doses of each component separately in healthy volunteers. Both losartan and its active metabolite are highly bound to plasma proteins, primarily albumin, with plasma-free fractions of 1.3 and 0.2%, respectively (Christ, 1995), resulting in a Vd of losartan and EXP3174 of ~34 and 12 liters, respectively, indicating possible modest distribution into tissues (Lo et al., 1995). Studies in rats indicate that losartan crosses the blood-brain barrier poorly, if at all.
Olmesartan is highly bound to plasma proteins (99%) and does not penetrate red blood cells, resulting in a Vd of 17 liters (Schwocho and Masonson, 2001). In rats, olmesartan crossed the blood-brain barrier poorly, if at all. Olmesartan passed the placental barrier in rats and was distributed to the fetus; it was distributed to milk at low levels.
Telmisartan is highly bound to plasma proteins (>99.5%), mainly albumin and α1-acid glycoprotein (Stangier et al., 2000a), resulting in a Vd of 500 liters, indicating possible distribution into tissues. Tissue penetration despite high-plasma protein binding is indicative of permissive binding. It reversibly distributes into erythrocytes (Stangier et al., 2000a). The uptake of telmisartan into human hepatocytes is predominantly mediated by OATP1B3 (Ishiguro et al., 2006). Upon oral or intravenous administration of radiolabeled telmisartan to rats, the highest levels of radioactivity were detected in liver, kidney, blood, and lung, with no radioactivity being detected in the brain (Wienen et al., 2000). The Vd of telmisartan is smaller in rats than in humans and even smaller in dogs (Wienen et al., 2000).
Valsartan is highly bound to serum proteins (95%), mainly albumin (Colussi et al., 1997), resulting in a Vd of 17 liters (Flesch et al., 1997), indicating that valsartan does not distribute into tissues extensively. In contrast to telmisartan, plasma protein binding here apparently limits tissue penetration. The hepatic uptake of valsartan is mediated by the transporters OATP1B1 and OATP1B3 (Yamashiro et al., 2006).
In conclusion, the ARBs differ considerably in Vd, with candesartan having the lowest values, eprosartan having a high value, and telmisartan having the highest value. This is at least partly explained by the lipophilicity and/or restrictive versus permissive plasma protein binding of the various compounds (Table 1) and may have implications for their overall tissue distribution (see Section III). All ARBs are highly plasma protein bound, and at least for irbesartan and telmisartan this involves α1-acid glycoprotein, which typically provides a tighter binding mode than albumin. In molecular modeling and docking studies, the binding pocket for ARBs to albumin has been characterized in detail (Li et al., 2010). For possible implications of plasma protein binding on drug-drug interactions, see Section VI.G.
C. Metabolism
Azilsartan is metabolized primarily by CYP2C9 and has two primary metabolites, the major one named M-II by O-dealkylation, and the minor one named M-I by decarboxylation. The systematic exposure to M-II and M-I was approximately 50% and <1% of azilsartan exposure. The azilsartan metabolites are not considered to contribute to the pharmacologic activity of orally administered azilsartan medoxomil or azilsartan. In in vitro studies, azilsartan did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4 and did not induce CYP3A.
Candesartan is mainly excreted in unchanged form but can undergo minor O-deethylation, yielding an inactive metabolite (vanLier et al., 1997). Candesartan is not significantly metabolized by the P450 system and at therapeutic concentrations has no effects on P450 enzymes (Taavitsainen et al., 2000). An inhibition of CYP2C9 function has only been observed at very high and probably supratherapeutic concentrations (Kamiyama et al., 2007). In rats and dogs, candesartan is partly metabolized by glucuronidation (Kondo et al., 1996).
Eprosartan was not metabolized by the P450 system and did not inhibit human P450 enzymes CYP1A, CYP2A6, CYP2C9/8, CYP2C19, CYP2D6, CYP2E, and CYP3A in vitro (Taavitsainen et al., 2000). Eprosartan was mainly excreted in unchanged form, and no active metabolites have been detected upon oral or intravenous administration of [14C]eprosartan. Accordingly, eprosartan was the only drug-related compound found in plasma or feces.
Irbesartan was metabolized via glucuronide conjugation (Perrier et al., 1994) and oxidation. After oral or intravenous administration of [14C]irbesartan, >80% of the circulating radioactivity is attributable to unchanged irbesartan (Chando et al., 1998). The primary circulating metabolite is inactive irbesartan glucuronide, accounting for approximately 6% of radiolabel. The remaining oxidative metabolites do not appreciably contribute to irbesartan’s pharmacologic activity. In vitro studies of irbesartan oxidation indicated that irbesartan was oxidized primarily by CYP2C9; metabolism by CYP3A4 was negligible (Bourrie et al., 1999). Irbesartan was neither metabolized by nor did it substantially induce or inhibit other isoenzymes commonly associated with drug metabolism (CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2D6, CYP2E1) (Taavitsainen et al., 2000). There was no induction or inhibition of CYP3A4. However, known CYP2C9 substrates/inhibitors such as sulphenazole, tolbutamide, or nifedipine significantly inhibit formation of the oxidized irbesartan metabolite (see Section VI.G).
Losartan underwent extensive, P450-mediated first-pass metabolism to yield the active carboxylic acid metabolite EXP3174 (Stearns et al., 1991) that is responsible for most of the AT1R antagonism observed with losartan treatment and several inactive metabolites, which can be detected in plasma and urine. Its metabolism is highly species-dependent and differs considerably between primates, including humans and rodents (Stearns et al., 1992). After oral or intravenous administration of [14C]losartan to humans, circulating plasma radioactivity was primarily attributed to losartan and EXP3174. In vitro studies indicate CYP2C9 and CYP3A4 are involved in the biotransformation of losartan, but the former may be more important (Yasar et al., 2001); drug-drug interaction (see Section VI.G) and pharmacogenomic studies (see Section VI.H) also support a greater role for CYP2C9. The extent of this biotransformation can markedly differ between subjects because ~1% of subjects yield only <1% EXP3174 compared with 14% of the dose in normal subjects. Of note, in contrast to humans, dogs apparently do not form EXP3174 in vivo, and hence dogs can be used to assess the differential in vivo effects of losartan and its active metabolite (Suzuki et al., 2001). In in vitro studies, losartan did not inhibit CYP2A6, CYP2D6, or CYP2E1 but did inhibit CYP1A2, CYP2C9, CYP2C19, and CYP3A4 (Taavitsainen et al., 2000), which is at least partly reflected in its drug-drug interaction profile (see Section VI.G). EXP3174 is further metabolized by glucuronidation at the N2 position within the tetrazole ring (Alonen et al., 2008). This glucuronidation step may be species-dependent (Huskey et al., 1993).
After the rapid and complete conversion of olmesartan medoxomil to olmesartan, there was virtually no further metabolism of olmesartan. Even in very high concentrations, it did not inhibit CYP2C9 function (Kamiyama et al., 2007).
After oral or intravenous administration, [14C]telmisartan largely remains unchanged (Stangier et al., 2000a). The very minor component being metabolized is represented by a pharmacologically inactive acyl glucuronide, which is the only metabolite that has been identified in human plasma and urine and accounts for approximately 11% of total radioactivity in plasma. Formation of the glucuronide metabolite in humans can best be predicted preclinically using canine data (Deguchi et al., 2011). The P450 enzymes are not involved in the metabolism of telmisartan, and an inhibition of CYP2C9 function has only been observed at very high and probably supratherapeutic concentrations (Kamiyama et al., 2007). Telmisartan had no effects in vitro on P450 enzymes, except for some inhibition of CYP2C19.
Only ~20% of administered valsartan dose was recovered as metabolites. The primary metabolite, accounting for 9% of administered dose, is valeryl-4-hydroxy valsartan, which is generated by CYP2C9 (Nakashima et al., 2005). Valsartan did not inhibit P450 enzymes to a clinically relevant extent in vitro (Taavitsainen et al., 2000) and, even in very high concentrations, did not inhibit CYP2C9 function (Kamiyama et al., 2007).
In conclusion, despite their pharmacodynamic similarity, the various ARBs differ considerably in their metabolism. With the exception of losartan, pharmacologically active metabolites are of little relevance once the orally administered prodrug has been cleaved during the absorption process where applicable. Therefore, specific differences in the involvement of P450 enzymes in ARB metabolism may largely be relevant in the context of differential drug-drug interactions (see Section VI.G).
D. Excretion
In studies with oral administration of [14C]azilsartan medoxomil, approximately 55 and 45% of radioactivity were recovered in feces and urine, respectively, with ~15% of the latter as azilsartan. The renal clearance of azilsartan is 2.3 ml/min. For healthy Japanese subjects, a total and renal clearance of 21.3–24.2 ml/min and 2.17–2.42 ml/h, respectively, have been reported. The elimination half-life was 11 hours, and steady-state was achieved within 5 days, with no accumulation in plasma upon repeated dosing.
Upon oral administration of [14C]candesartan cilexetil, 67 and 33% of radioactivity were recovered in feces and urine, respectively (vanLier et al., 1997). After i.v. administration of [14C]candesartan, 36 and 59% were recovered in feces and urine, respectively, indicating that biliary excretion contributes to the elimination of candesartan (vanLier et al., 1997). The biliary and urinary excretion of candesartan occurs largely as unchanged drug. The total and renal candesartan clearances are 0.37 and 0.19 ml/min/kg, respectively. The elimination half-life was 9 hours, with no accumulation in plasma upon repeated dosing (Hübner et al., 1997). In rats and dogs, candesartan is predominantly excreted via the bile; the retention of candesartan in blood vessels is longer than in plasma (Kondo et al., 1996).
Eprosartan was eliminated by biliary and renal excretion, primarily as unchanged compound. After an oral dose of [14C]eprosartan, 90 and 7% of radioactivity were recovered in feces and urine, respectively, reflecting poor oral bioavailability. After an i.v. dose of [14C]eprosartan, 61 and 37% of radioactivity were recovered in feces and urine, respectively. Approximately 20% of radioactivity in urine was an acyl glucuronide (i.e., <2% of an oral dose), with 80% being unchanged eprosartan. The elimination half-life was 20 hours, and no accumulation occurred in plasma upon repeated dosing. On the basis of pooled population pharmacokinetic analysis from two phase III trials, mean oral clearance for an average 65-year-old patient was 48.5 l/h, i.e., approximately 800 ml/min.
Irbesartan and its metabolites are eliminated by biliary and renal excretion. After either oral or intravenous administration of [14C]irbesartan, approximately 20% of radioactivity was recovered in urine and the remainder in feces (Chando et al., 1998; Vachharajani et al., 1998a). Total plasma and renal clearance were 157–176 and 3.0–3.5 ml/min, respectively. The elimination half-life was 11–15 hours, and steady state was reached within 3 days (Marino et al., 1998a). Limited accumulation (<20%) was observed upon repeated daily dosing.
Losartan, EXP3174, and their inactive metabolites were eliminated by biliary and renal excretion. After oral administration of [14C]losartan, approximately 35% of radioactivity was recovered in the urine and ~60% in the feces. After an i.v. dose of [14C]losartan, ~45% of radioactivity was recovered in the urine and 50% in the feces. The total plasma clearance of losartan and EXP3174 was ~600 and 50 ml/min, respectively, with renal clearance of approximately 75 and 25 ml/min, respectively (Lo et al., 1995). After single doses of losartan administered orally, ~4% of the dose was excreted unchanged in the urine and ~6% was excreted in urine as EXP3174. The terminal half-life of losartan was ~2 hours, whereas that of EXP3174 was 6–9 hours (Lo et al., 1995). Neither losartan nor EXP3174 accumulated in plasma upon repeated once-daily dosing because of its rapid elimination.
Olmesartan was eliminated by biliary and renal excretion with 35–50% of the administered dose recovered from urine, and the remainder was eliminated in feces via the bile (Schwocho and Masonson, 2001); a role for P-gp in the biliary clearance has been shown in rat studies (Abe et al., 2008). The total plasma and renal clearance of olmesartan were 1.3 and 0.6 l/h, respectively. The elimination of olmesartan appears to be biphasic with a terminal half-life of approximately 13 hours but may be somewhat shorter in patients with hypertension compared with healthy subjects (Warner and Jarvis, 2002). Steady-state levels of olmesartan occurred within 3–5 days, and no accumulation occurred with repeated dosing (Schwocho and Masonson, 2001). Olmesartan was not cleared by hemodialysis (Tanaka et al., 2009), probably because of its high plasma protein binding.
Telmisartan was largely (>97% of administered oral or intravenous dose) eliminated in feces via biliary excretion; only minute amounts were found in urine (<1%) (Stangier et al., 2000a). The biliary excretion of telmisartan has also been studied using [11C]telmisartan in PET studies (Takashima et al., 2011). Telmisartan had a total plasma clearance of >800 ml/min and showed biexponential decay kinetics with a terminal half-life of approximately 24 hours, which was independent of administered dose. Telmisartan had an accumulation index in plasma of 1.4–2 upon repeated once daily dosing. The elimination half-life in mice, rats, rabbits, and dogs was considerably shorter than in humans, particularly upon i.v. dosing (Wienen et al., 2000).
Upon oral administration, valsartan was largely eliminated in feces (83%), with a smaller component in urine (13%) (Brookman et al., 1997; Müller et al., 1997; Waldmeier et al., 1997; Abe et al., 2008). The efflux transporter responsible for biliary excretion of valsartan is mrp2 (Yamashiro et al., 2006). After i.v. administration, the plasma and renal clearance are 2 and 0.62 l/h, respectively. Valsartan shows biexponential decay kinetics after i.v. administration with an average elimination half-life of ~6 hours (Flesch et al., 1997). Valsartan does not accumulate appreciably in plasma with repeated administration (Müller et al., 1997). In rats, an in vitro assay based upon sandwich-cultured hepatocytes has been found to be a good predictor of valsartan biliary excretion in vivo (Fukuda et al., 2008).
In conclusion, ARBs differ considerably in their routes of elimination, and this needs to be taken into account in patients with renal or hepatic impairment (see Section VI.F). They also differ in half-life, with losartan having the shortest and eprosartan and telmisartan the longest elimination half-life. At least in some cases such as telmisartan, slow dissociation kinetics from the receptor (see Section IV.F) may contribute to slow elimination. Long elimination half-lives may be clinically relevant for 24-hour blood pressure control. A lack of sustained blood pressure control over a full 24-hour period may be associated with early morning blood pressure rises, which in turn may be associated with adverse outcomes in patients with hypertension (Kario et al., 2011).
E. Age, Sex, and Ethnicity
The Cmax and AUC of azilsartan were not significantly different between young and old subjects, males and females, or races. Information on pediatric patients is not available.
The exposure to candesartan is not affected by sex. However, it differs by age with subjects ≥65 years exhibiting a 50 and 80% higher Cmax and AUC, respectively, than younger subjects (Hübner et al., 1997), but this difference does not result in a need for dose adjustments. Children experience a greater exposure upon oral administration of the same dose compared with adults, but the overall pharmacokinetic profiles within a range of 1–17 years were similar to those found in adults (Trachtman et al., 2008; Schaefer et al., 2010).
Eprosartan pharmacokinetics were not influenced by body weight, race, or sex (Xu et al., 2007), but oral clearance was a linear function of age with a decline of 0.62 l/h for every year increase (Tenero et al., 1998c). Thus, the AUC, Cmax, and tmax increased approximately 2-fold in elderly men (68–78 years) compared with younger men (20–39 years) without changes in plasma protein binding. Information on pediatric patients is not available.
Irbesartan pharmacokinetics were not influenced by sex in healthy subjects (Vachharajani et al., 1998c). In patients with hypertension there were no sex differences for half-life or accumulation but somewhat higher plasma levels in females (11–44%); no sex-related dosage adjustment is necessary. Although elimination half-life was not significantly altered in 68- to 80-year-old versus 18- to 40-year-old subjects, AUC and Cmax were ~20 and 50%, respectively, greater in the eldery subjects (Vachharajani et al., 1998c), but no dosage adjustment is necessary. In black subjects, irbesartan AUC values were approximately 25% greater than in whites, but Cmax values did not differ. The pharmacokinetics of irbesartan have also been characterized in children and adolescents aged 6–16 years and were similar to values found in adults (Sakarcan et al., 2001).
Plasma concentrations of losartan were approximately twice as high in female as in male patients with hypertension, but those of EXP3174 were similar in both sexes; no dosage adjustment is necessary. Plasma concentrations of losartan and EXP3174 are similar in elderly and young patients with hypertension. The pharmacokinetic parameters of losartan were generally similar in all age groups of children (aged 6 years and above) to those in adults; the suspension formulation of losartan used in children has a similar bioavailability with regard to both losartan and EXP3174 compared with the tablet formulation used in adults.
Minor differences were seen in the pharmacokinetics of olmesartan in Caucasian men and women, with females exhibiting an approximately 10–15% higher AUC and Cmax than males (Yoshihara et al., 2005), whereas no significant sex difference has been observed in Chinese subjects (Jiang et al., 2009). Although the Cmax values of olmesartan were similar in elderly compared with young adults, modest accumulation of olmesartan was observed in the elderly with repeated dosing (a 33% increase in AUC corresponding to a 30% reduction in renal clearance) (von Bergmann et al., 2001). The clearance of olmesartan in children aged 1–16 years was similar to that in adults when adjusted for body weight; hence, no dosage adjustment is necessary. The suspension formulation of olmesartan used in children is bioequivalent to the tablet formulation used in adults. Population pharmacokinetic analysis suggests that the pharmacokinetics of olmesartan do not differ between Western and Japanese populations (Yoshihara et al., 2005).
Plasma concentrations of telmisartan are generally 2–3 times higher in females than in males, but this does not translate into significant differences in blood pressure response or the incidence of orthostatic hypotension. The pharmacokinetics of telmisartan do not differ between the elderly and those <65 years of age (Stangier et al., 2000f). In population pharmacokinetic analysis, differences were found between exposure in Japanese compared with European subjects, but it appeared that these were less likely to be due to ethnicity but rather to concomitant food intake (studies in Japanese and European subjects conducted in fed and fasting state, respectively) (Tatami et al., 2004)
The pharmacokinetics of valsartan do not differ significantly between males and females. The clearance of the suspension formulation of valsartan, which is used for pediatric patients and has a 1.6-fold greater bioavailability than the tablet formulation, was similar in children aged 1–16 years as in adults receiving the same formulation. In the elderly, the exposure to valsartan is 70% higher and the half-life is 35% longer than in the young (Sioufi et al., 1998), but no dosage adjustment is necessary.
F. Renal and Hepatic Impairment
The Cmax and AUC of azilsartan are not significantly affected by mild to moderate renal impairment or by mild hepatic impairment. Severe renal or moderate hepatic impairment is associated with a 1.5- to 2-fold increase in AUC but not significant increase in Cmax; dose adjustment is not necessary in these populations. Severe hepatic impairment has not been tested.
The exposure to candesartan is mildly increased with mild to moderate renal impairment and approximately doubles in patients with severe renal impairment and those undergoing hemodialysis (de Zeeuw et al., 1997). In line with its lipophilicity, candesartan cannot be removed by hemodialysis, probably because of restrictive plasma protein binding. With mild and moderate hepatic impairment the AUC for candesartan increases by 30 and 145%, respectively (de Zeeuw et al., 1997). However, adjustment of initial dose is not necessary with either renal or hepatic impairment. Severe hepatic impairment has not been tested.
Eprosartan Cmax and AUC increase by 30–50% and by 70–90% with moderate to severe renal impairment, respectively, and the unbound fraction of the drug increases by 35 and 59% in patients with moderate or severe renal impairment, respectively (Martin et al., 1998). Eprosartan was poorly removed by hemodialysis (Kovacs et al., 1999). Eprosartan AUC but not Cmax was increased on average by approximately 40% with decreased hepatic function (Tenero et al., 1998a). However, adjustment of initial dose is not necessary with either renal or hepatic impairment. Severe hepatic impairment has not been tested.
The pharmacokinetics of irbesartan were not altered in patients with renal impairment or in patients on hemodialysis (Sica et al., 1997), and no dosage adjustment is necessary on pharmacokinetic grounds in mild to severe renal impairment. Irbesartan is not removed by hemodialysis (Sica et al., 1997). The pharmacokinetics of irbesartan were not significantly altered upon repeated oral administration in patients with mild to moderate cirrhosis of the liver (Marino et al., 1998b), and no dosage adjustment is required in such patients. Severe hepatic impairment has not been tested.
The plasma concentrations and AUCs of losartan and EXP3174 are increased by 50–90% in patients with mild or moderate renal insufficiency, and the renal clearance was reduced by 55–85% for both losartan and EXP3174 in such patients (Sica et al., 1995); no dosage adjustment is necessary for these patients. Neither losartan nor its active metabolite can be removed by hemodialysis, but the pharmacokinetics of losartan in patients do not differ substantially from those in healthy subjects in patients with end-stage renal disease undergoing hemodialysis (Sica et al., 2000) or continuous ambulatory peritoneal dialysis (Pedro et al., 2000). After oral administration in patients with mild to moderate alcoholic cirrhosis of the liver, plasma concentrations of losartan and EXP3174 were, respectively, 5 and ~1.7 times those in young male volunteers. Compared with normal subjects, the total plasma clearance of losartan in patients with hepatic insufficiency was ~50% lower, and the oral bioavailability was approximately two times higher. Therefore, a lower starting dose is recommended for patients with a history of hepatic impairment. Severe hepatic impairment has not been tested.
The serum concentrations of olmesartan were elevated in patients with renal insufficiency compared with subjects with normal renal function. After repeated dosing, the AUC was approximately tripled in patients with severe renal impairment (von Bergmann et al., 2001). On the basis of population pharmacokinetic analyses, the clearance of olmesartan is reduced by ≥30% in patients with severe renal impairment (Yoshihara et al., 2005). The pharmacokinetics of olmesartan in patients undergoing hemodialysis have been described (Tanaka et al., 2009) but not compared with those in subjects without renal impairment. In patients with moderate hepatic impairment compared with those in matched controls increases in AUC (~60%), and Cmax values were observed (von Bergmann et al., 2001). No adjustment of starting dose is recommended with moderate to marked renal impairment or with moderate to marked hepatic dysfunction. Severe hepatic impairment has not been tested.
In line with the virtual lack of renal elimination of telmisartan, no dose adjustment is necessary in patients with decreased renal function (Stangier et al., 2000c). Telmisartan is not removed from blood by hemofiltration. In patients with hepatic insufficiency plasma concentrations are increased and bioavailability approaches 100% (Stangier et al., 2000g). In such patients telmisartan treatment should be initiated at low doses and titrated slowly. Severe hepatic impairment has not been tested.
The exposure to valsartan (measured by AUC) is apparently not correlated to renal function in patients with different degrees of renal impairment. Consequently, dose adjustment is not required in patients with mild-to-moderate renal dysfunction. No studies have been performed in patients with severe impairment of renal function (creatinine clearance <10 ml/min). Valsartan is not removed from the plasma by hemodialysis. On average, patients with mild-to-moderate chronic liver disease have twice the exposure (measured by AUC values) to valsartan of age-, sex-, and weight-matched healthy volunteers (Brookman et al., 1997). In general, no dosage adjustment is needed in patients with mild-to-moderate liver disease. Severe hepatic impairment has not been tested.
In conclusion, hepatic impairment differentially affects ARB pharmacokinetics, and this needs to be taken into account in the prescribing in such patients. Similarly, renal impairment differentially affects ARB pharmacokinetics, with irbesartan and telmisartan not being affected even by severe renal impairment. As ARBs pharmacodynamically have beneficial effects in patients with impaired renal function and are generally classified as renoprotective, their use in such patients is generally recommended (Sica and Gehr, 2002).
G. Pharmacokinetic Drug-Drug Interactions
Reversible increases in serum lithium concentrations have been observed with multiple ACE inhibitors and ARBs, which are based on the mechanism of action of these drug classes on the renal tubules. Although this interaction has not been incorporated into all ARB labels, it is considered a class effect and will not be discussed specifically below for the individual drugs.
For both azilsartan medoxomil and azilsartan, no clinically significant interactions have been observed with amlodipine, antacids, chlorthalidone, digoxin, fluconazole, glyburide, ketoconazole, metformin, pioglitazone, and warfarin.
In line with the lack of significant interaction of candesartan with the P450 system (see Section VI.C), no significant drug interactions have been reported in studies of candesartan cilexetil given with other drugs such as glyburide, nifedipine, digoxin, warfarin, hydrochlorothiazide, and oral contraceptives in healthy volunteers (Jonkman et al., 1997), or with enalapril to patients with heart failure (New York Heart Association classes II and III), or with mycophenolate mofetil in renal transplant patients (Miura et al., 2009b).
In line with the lack of significant interaction of eprosartan with the P450 system, eprosartan steady-state concentrations were not affected by concomitant administration of ketoconazole or fluconazole (Kazierad et al., 1997), potent inhibitors of CYP3A and 2C9, respectively. They were also not affected by concomitant administration of ranitidine (Tenero et al., 1998d). Reciprocally, eprosartan had no effect on single oral-dose digoxin pharmacokinetics (Martin et al., 1997), steady-state prothrombin time ratios in healthy subjects receiving warfarin (Kazierad et al., 1998), or on 24-hour plasma glucose profiles in patients with diabetes receiving glyburide.
On the basis of in vitro studies with irbesartan, no interaction is expected with drugs whose metabolism is dependent upon P450 isoenzymes CYP1A1, 1A2, 2A6, 2B6, 2D6, 2E1, or 3A4. An inhibition of CYP2C9 function has only been observed at very high and probably supratherapeutic concentrations (Kamiyama et al., 2007). Indeed no pharmacokinetic interactions have been observed with hydrochlorothiazide, digoxin, warfarin, nifedipine, tolbutamide, simvastatin, or antacids; however, a minor increase in exposure was observed upon concomitant fluconazole administration (Marino and Vachharajani, 2001), as expected for a CYP2C9 substrate. In rats, irbesartan did not interfere with the pharmacokinetics of indapamide (Yan et al., 2012).
On the basis of its metabolism, including strong first-pass metabolism involving CYP2C9 and, to a lesser degree, 3A4, losartan exhibits several drug-drug interactions. Coadministration of losartan and cimetidine led to an increase of ~18% in AUC of losartan but did not affect the pharmacokinetics of EXP3174 (Goldberg et al., 1995). Coadministration of losartan and phenobarbital led to a reduction of ~20% in the AUC of losartan and EXP3174 (Goldberg et al., 1996). A somewhat greater interaction (~40% reduction in the AUC of EXP3174 and ~30% reduction in the AUC of losartan) has been reported with rifampin (Williamson et al., 1998). Fluconazole, an inhibitor of CYP2C9 and CYP3A4, decreased the AUC of EXP3174 by approximately 40%, but increased the AUC of losartan by approximately 70% following multiple doses (Kazierad et al., 1997; Kaukonen et al., 1998). Conversely, an inhibition of CYP2C9 function has only been observed at very high and probably supratherapeutic losartan concentrations (Kamiyama et al., 2007). However, fluvastatin, an inhibitor of CYP2C9, but not CYP3A4 mimicked the effect of fluconazole on losartan or EXP3174 pharmacokinetics (Meadowcroft et al., 1999). A reduced formation of EXP3174 was also observed with the CYP2C9 inhibitor bucolome in both rat and human studies (Kobayashi et al., 2008a). Conversion of losartan to its active metabolite after intravenous administration is not affected by ketoconazole or itraconazole (Kaukonen et al., 1998), inhibitors of CYP3A4. The AUC of EXP3174 following oral losartan was also not affected by erythromycin (Williamson et al., 1998), another inhibitor of CYP3A4, but the AUC of losartan was increased by 30%. Although losartan did not affect the pharmacokinetics of phenytoin, phenytoin reduced the conversion of losartan to EXP3174 (Fischer et al., 2002). Taken together, these data suggest that a combined inhibition of CYP2C9 and CYP3A4 may be required to obtain noticeable effects on the pharmacokinetics of losartan (Meadowcroft et al., 1999). On the basis of such data, losartan conversion to EXP3174 has also been used as a probe to study potential inhibition of CYP2C9 by other compounds, e.g., the antimalaria drug amodiaquine (Wennerholm et al., 2006). Losartan did not affect the pharmacokinetics or pharmacodynamics of a single dose of warfarin (Kong et al., 1995) or the pharmacokinetics of oral or intravenous digoxin (De Smet et al., 1995). There also is no pharmacokinetic interaction between losartan and hydrochlorothiazide. Inhibition of P-gp, e.g., by grapefruit juice, may increase cellular net efflux of losartan, as this effect may counteract those on CYP3A4 (Soldner et al., 1999). In vivo grapefruit juice prolonged the lag time to detect losartan in plasma upon oral administration and the half-life of EXP3174 (Zaidenstein et al., 2001).
In line with the virtual lack of olmesartan metabolism, no pharmacokinetic drug-drug interactions have been reported (Laeis et al., 2001). For example, no pharmacokinetic interaction was observed upon the combined administration of olmesartan with glibenclamide (Huber et al., 2006), hydrochlorothiazide (Kreutz et al., 2006), or amlodipine (Rohatagi et al., 2008).
Coadministration of telmisartan can increase median digoxin peak and trough concentrations by 49% and 20%, respectively (Stangier et al., 2000b); mechanistically, this may be related to the potent P-gp inhibition that has been reported for telmisartan (Weiss et al., 2010). Therefore, digoxin levels should be monitored when initiating, adjusting, or discontinuing telmisartan treatment. Coadministration of 80 mg telmisartan with 10 mg of ramipril in healthy subjects increased steady-state Cmax and AUC of ramipril 2.3- and 2.1-fold, respectively; in contrast, such coadministration reduced the Cmax and AUC of telmisartan by 31 and 16%, respectively. Because of the possible additive pharmacokinetic effect, concomitant use of telmisartan and ramipril is not recommended. Although concomitant administration of telmisartan did not affect the pharmacokinetics of nisoldipine, nisoldipine increased the AUC and reduced the clearance of telmisartan in patients with hypertension (Bajcetic et al., 2007). Exposure to mycophenolate mofetil was reported to be lower in renal transplant patients upon concomitant administration of telmisartan, possibly because the metabolism of mycophenolate mofetil involves uridine diphosphate-glucuronosyltransferase 1A9, which has been identified as a PPAR-γ target gene (Miura et al., 2009b). Telmisartan is not expected to interact with drugs that inhibit P450 enzymes; it is also not expected to interact with drugs metabolized by P450 enzymes, except for possible inhibition of the metabolism of drugs metabolized by CYP2C19. Accordingly, coadministration of telmisartan did not result in a clinically significant interaction with acetaminophen or ibuprofen (Stangier et al., 2000d), amlodipine (Stangier and Su, 2000), glyburide, simvastatin, hydrochlorothiazide (Young et al., 2000), or warfarin (Stangier et al., 2000e). In rats, telmisartan did not interfere with the pharmacokinetics of indapamide (Yan et al., 2012).
No clinically significant pharmacokinetic interactions were observed when valsartan was coadministered with amlodipine, atenolol (Czendlik et al., 1997), cimetidine (Schmidt et al., 1998), digoxin, furosemide (Bindschedler et al., 1997), glyburide, hydrochlorothiazide, indomethacin, vildagliptin (He et al., 2008), dapagliflozin (Kasichayanula et al., 2012), or mycophenolate mofetil (Miura et al., 2009b). The valsartan-atenolol combination was more antihypertensive than either component, but it did not lower the heart rate more than atenolol alone. Coadministration of valsartan and warfarin did not change the pharmacokinetics of valsartan or the time course of the anticoagulant properties of warfarin. In rats, valsartan did not interfere with the pharmacokinetics of indapamide (Yan et al., 2012). Upon coadministration of valsartan and simvastatin the plasma concentrations of both drugs and the β-hydroxy metabolite of simvastatin are increased by about 20% (Sunkara et al., 2007).
In conclusion, with the exception of losartan, ARBs do not have clinically relevant drug-drug interactions based on interaction with the P450 system in the overall population; this may be different in patient groups with specific genotypes, e.g., for CYP2C9 (see Section VI.H), but this possibility has been insufficiently explored. Because of interaction with the transporter molecule P-gp, digoxin levels need to be monitored upon coadministration of telmisartan. ARBs as a class can affect serum lithium concentrations based on pharmacodynamic grounds.
H. Pharmacokinetic Pharmacogenomics
Although several gene polymorphisms have been reported in the AT1R and other components of the RAAS, they have not consistently been associated with altered drug responses to ARBs (Rosskopf and Michel, 2008). However, polymorphisms of drug metabolizing enzymes and, perhaps more importantly, drug transporters may affect the pharmacokinetics of several ARBs. Similar to overall pharmacokinetics, potential pharmacogenomic effects also are substance specific and need to be discussed for each drug separately. In our literature search we have not identified pharmacogenomic studies related to azilsartan or eprosartan.
In vitro studies indicated that genotype at CYP2C9 (*3 allele) can affect metabolism of candesartan and change its susceptibility to CYP2C9 inhibitors such as warfarin (Hanatani et al., 2001). The potential relevance of a 2C9 gene polymorphism was later confirmed in a clinical case report, where it was associated with excessive blood pressure lowering (Uchida et al., 2003). This polymorphism was also associated with increased exposure to irbesartan (Choi et al., 2012; Hong et al., 2005).
Polymorphisms in CYP2D6 or CYP2C19 did not affect the conversion of losartan to its active metabolite EXP3174 (Sandwall et al., 1999). Studies with yeast transfected with various CYP2C9 alleles and with human liver microsome preparation from subjects representing the known alleles of the enzyme demonstrated that polymorphisms of the gene encoding this enzyme, particularly the *2 and *3 alleles, affect the metabolism of losartan to EXP3174 (Yasar et al., 2001). In follow up in vivo studies the *3 allele of the CYP2C9 polymorphism was confirmed to be associated with a reduced EXP3174 formation (Yasar et al., 2002b). Later work from this group proposed that the urinary EXP3174/losartan ratio can even be used as in vivo marker for CYP2C9 genotype (Yasar et al., 2002a). Although this was confirmed by other investigators in Caucasian (Lee et al., 2003a,b) and Japanese subjects (Sekino et al., 2003), it was proposed that tolbutamide may be a superior probe for this polymorphism compared with losartan (Lee et al., 2003a). The importance of genotype at the CYP2C9 locus for conversion of losartan to EXP3174 was also confirmed in a study of almost 1800 Korean subjects (Bae et al., 2011). In a smaller Korean study, CYP*1/*3 and *1/*13 similarly reduced EXP3174 generation but were not different from each other (Bae et al., 2012). Whether an altered conversion of losartan to EXP3174 is associated with an altered blood pressure response to losartan remains unclear, as this was observed in Japanese (Sekino et al., 2003) but not Caucasian healthy volunteers (Lee et al., 2003b); however, the subject groups being tested were small, and ARBs typically cause only minor blood pressure reductions in healthy subjects to begin with. Later studies based on black African subjects found that the *5 and *6 alleles of CYP2C9 are also associated with lower enzyme activity for losartan conversion in vivo, whereas the *8 and *11 alleles had little effect (Allabi et al., 2004). The *13 allele of CYP2C9 was also reported to be associated with a reduced conversion of losartan to EXP3174 in healthy volunteers (Li et al., 2009b). In a small sample of Japanese patients with hypertension, the *30 allele of CYP2C9 was associated with a diminished blood pressure lowering effect of losartan (Yin et al., 2008). Genotype at the CYP2C9 locus also affects the drug-drug interaction between losartan and phenytoin (Fischer et al., 2002). The plant extract silymarin inhibited losartan metabolism in healthy Chinese volunteers only in the absence of the *3 allele of CYP2C9 (Han et al., 2009). A common polymorphism in P-gp, 3435C>T, did not affect CYP2C9 genotype-dependent metabolism of losartan (Yasar et al., 2008).
In a study of polymorphisms of the gene for the transporter OATP1B1, SLCO1B1, the mean Cmax and AUC0–24 of olmesartan tended to be higher in *15/*15 subjects than in *1b/*1b subjects, whereas the mean CLt/F in *15/*15 subjects was significantly lower than that in *1b/*1b subjects (Suwannakul et al., 2008). Other investigators did not confirm a role for polymorphisms in the SLCO1B1 gene and did not detect one for those in the gene encoding breast cancer resistance protein, ABCG2, but found that two single-nucleotide polymorphisms in the gene encoding mrp2, ABCC2, were associated with a greater olmesartan exposure (Kim et al., 2012). A later study from the group originally reporting on a role for SLCO1B1 gene polymorphisms in three ethnic groups also failed to confirm a role in olmesartan exposure and also did not detect one for polymorphisms in the genes encoding OATP1B3 or mrp2 (Endo et al., 2012).
OATP1B3 and mrp2 are also involved in the distribution of other ARBs such as telmisartan (Nishino et al., 2000; Ishiguro et al., 2006, 2008). In a study of 12 Japanese renal transplant recipients being treated with telmisartan for at least 6 months, a polymorphism in the gene encoding mrp2 was associated with greater telmisartan exposure; polymorphisms of SLCO1B3, ABCB1, ABCG2, or several uridine diphosphate-glucuronosyltransferases did not affect telmisartan exposure (Miura et al., 2009a). Similarly, the C3435T polymorphism of P-glycoprotein did not influence the pharmacokinetics of telmisartan in healthy Chinese men (Guo et al., 2009). Another study also did not detect a clear effect of polymorphisms in the SLCO1B3 gene, but found that another polymorphism enhanced the expression of UGT1A3 and thereby increased the glucuronidation activity toward telmisartan and lowered telmisartan plasma concentrations (Yamada et al., 2011).
A polymorphism in the gene for OATP1B1 tended to affect the exposure to valsartan in healthy Japanese volunteers but this did not reach significance (Maeda et al., 2006).
In conclusion, the field of pharmacokinetic pharmacogenomics of ARBs is still emerging but already has yielded a number of potentially clinically relevant findings. Polymorphisms in genes encoding CYP2C9 and several transporters may be most relevant in this regard. However, a considerably greater amount of data will be required before robust clinical recommendations can be derived from such information.
VII. Conclusions
The clinically available ARBs differ in chemical structure, which has implications for a wide range of physicochemical, pharmacological, and pharmacokinetic properties. Differences in lipophilicity may contribute to those in tissue penetration and Vd. The ARBs are differentiated by receptor affinity to a limited extent only. However, their dissociation kinetics from the receptor differ more substantially, and this may have relevance for the surmountability of their effects and may also contribute to in vivo duration of action. It is possible that as a consequence of insurmountability, functional affinity determinations for some ARBs in cellular or tissue (vasoconstriction) assays yield greater potency estimates than those observed in radioligand binding assays. All ARBs are highly selective for AT1R versus AT2R and many other G-protein-coupled receptors. However, some ARBs, particularly telmisartan, also bind to and activate PPAR-γ as partial agonists; this property mediates many telmisartan effects in experimental animals, but its contribution to in vivo effects in patients remains to be fully explored. The primary molecular ARB target, the AT1R, appears to be the most important mediator of clinical effects for all members of this drug class. ARBs also differ in a variety of pharmacokinetic parameters of which a difference in elimination half-life, longest for telmisartan, probably is the most important. Other pharmacokinetic parameters such as metabolism or route of elimination may be important for drug-drug interactions or in patients with renal or hepatic impairment. However, the overall relevance of pharmacological differences between ARBs largely needs to be explored in dedicated clinical studies.
Acknowledgments
The authors gratefully acknowledge the assistance of Drs. Erick Young, Rucha Sana, Joachim Stangier, and Donald Tweedie (all affiliated with Boehringer Ingelheim), who provided specialist input into various sections of the article.
Authorship contributions
Performed data analysis: Foster, Michel.
Wrote or contributed to the writing of the manuscript: Brunner, Foster, Li, Michel.
Footnotes
M.C.M. and C.F. are present and past employees of Boehringer Ingelheim, respectively. H.R.B. has been a consultant to practically all ARB manufacturers in the past but reports no present conflict of interest. L.L. reports no conflict of interest.
Abbreviations
- ACE
- angiotensin converting enzyme
- AGE
- advanced glycosylation end product
- ARB
- angiotensin receptor blocker
- ANG
- angiotensin II
- AT1R
- angiotensin II type 1 receptor
- AT2R
- angiotensin II type 2 receptor
- AUC
- area under the curve
- Cmax
- maximum plasma concentration
- P450
- cytochrome P450
- OATP
- organic acid transporting polypeptide
- PET
- positron emission tomography
- P-gp
- P-glycoprotein
- PPAR
- peroxisome proliferator-activated receptor
- RAS
- renin-angiotensin system
- t1/2
- dissociation rates
- TM
- transmembrane
- tmax
- time to reach Cmax
- Vd
- volume of distribution
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





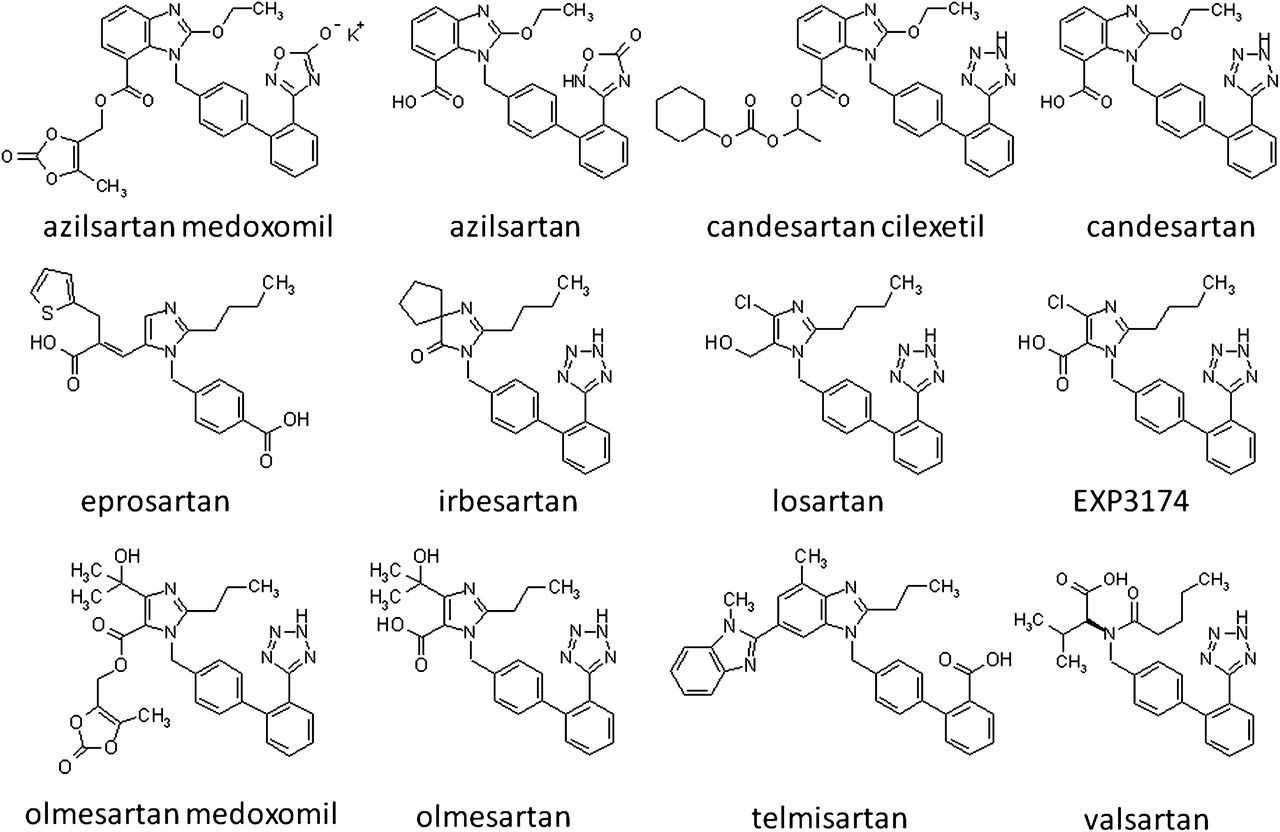{kind=link}
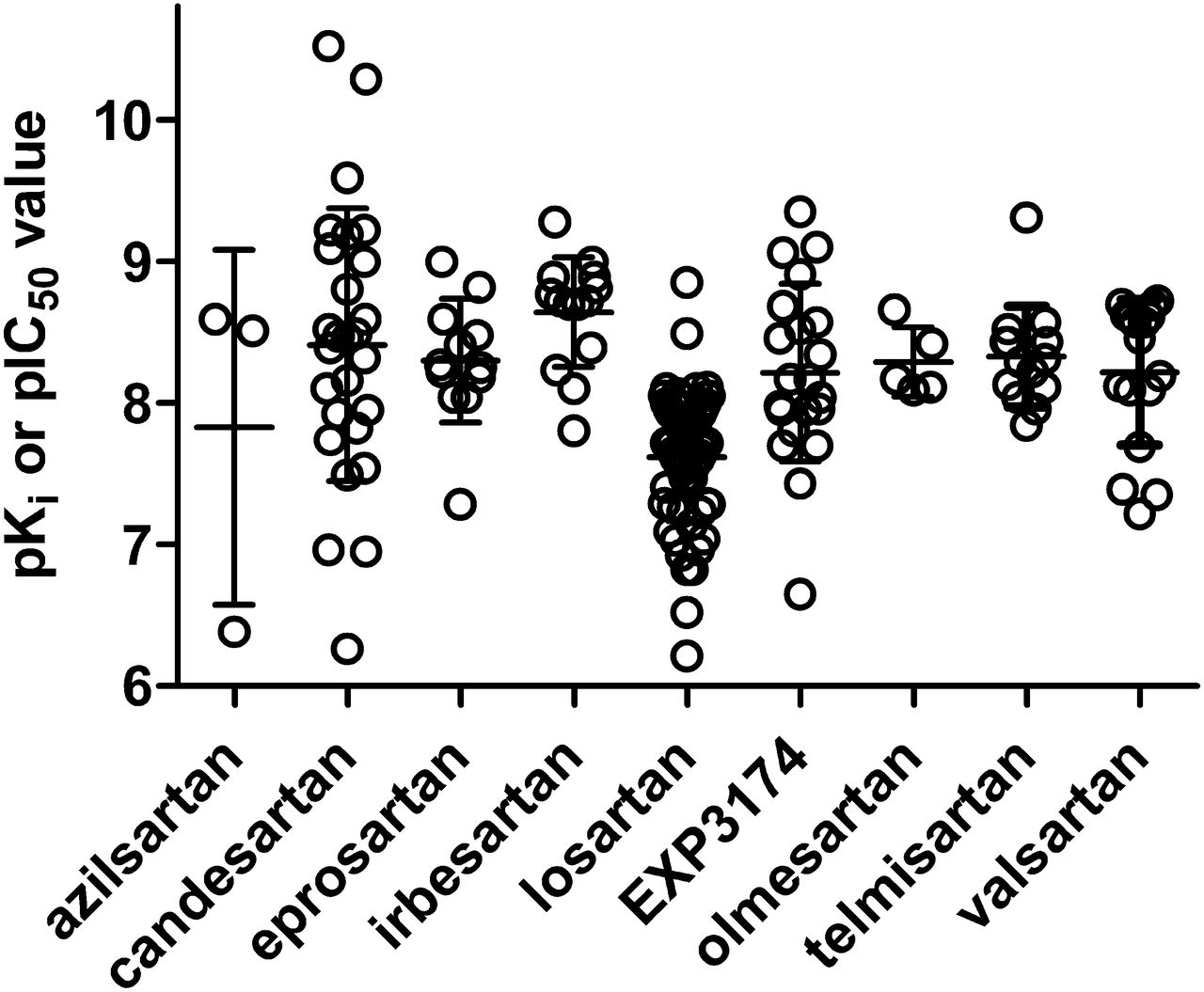{kind=link}
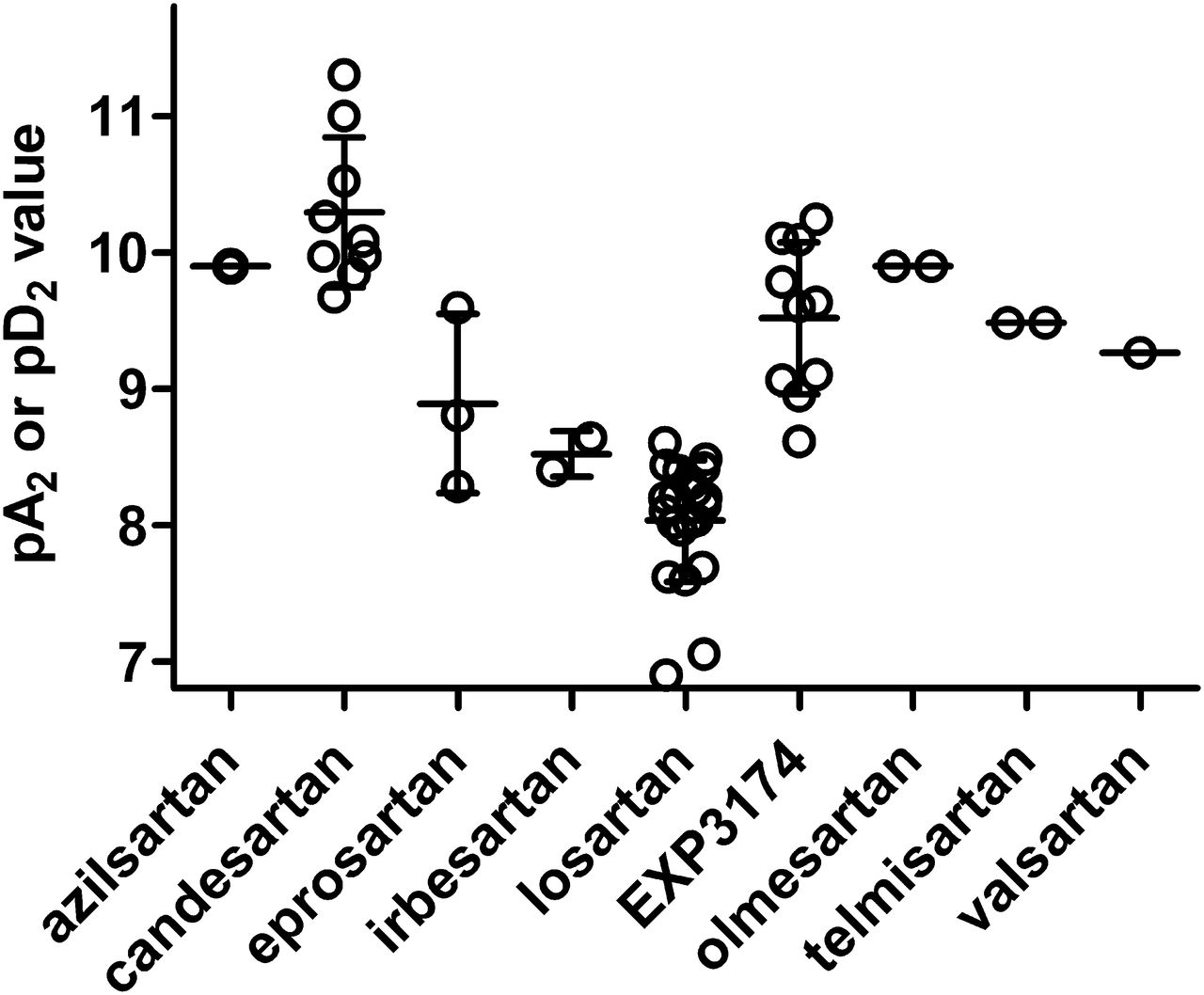{kind=link}