


Oral Drug Absorption
The absorption of drugs via the oral route is a subject of intense and continuous investigation in the pharmaceutical industry since good bioavailability implies that the drug is able to reach the systemic circulation by mouth. Oral dry absorption is affected by both drug properties and the physiology of the gastrointestinal tract (GIT1), or patient properties, including drug dissolution from the dosage form, the manner in which drug interacts with the aqueous environment and membrane, permeation across membrane, and irreversible removal by first-pass organs such as the intestine, liver, and lung (Martinez and Amidon, 2002). The purpose of this minireview is to highlight the processes governing drug bioavailability when the drug is already in solution, and emphasizes the roles of intestinal transporters and metabolism on oral bioavailability. The description of physical models for drug dissolution on drug absorption (Higuchi, 1967) or hepatic modeling (Pang and Chiba, 1994; Pang et al., 1998; Abu-Zahra and Pang, 2000), however, is beyond the scope of this work.
The intestine, in addition to the liver, is an important tissue that regulates the extent of absorption of orally administered drugs, since the intestine and liver are involved in first-pass removal (Gibaldi et al., 1971; Rowland, 1972). The majority of drug absorption occurs at the small intestine because of the large surface area since the presence of villi and microvilli increases the absorptive area manyfold. The duodenum and jejunum possess the greatest surface areas due to the highest concentration of villi and microvilli in these regions, and surface area is least for the ileum (Magee and Dalley, 1986).
The circulation of the intestine is unique in that the intestine is the anterior or portal tissue that regulates the flow of substrates to the liver. The intestinal venous blood constitutes the majority of the blood supply to the liver, accounting for 75% of total liver blood flow. For drugs that are highly cleared by the intestine, the contribution of the liver or lung to drug metabolism will become reduced, whereas for drugs that are poorly extracted by the intestine, the substrate is able to reach the next organs, the liver and the lung, for removal (Gugler et al., 1975; Xu et al., 1989; Hirayama et al., 1990). The concentration of drug entering the intestine (Xu et al., 1989; Hirayama and Pang, 1990) and the intestinal flow rate (Chen and Pang, 1997) alter the rate of drug delivery and affect the degree of saturability of intestinal enzymes. These ultimately affect the rates of intestinal and hepatic first-pass metabolism. Additionally, variables such as other drugs or food that alter the transit times within the gastrointestinal tract further modulate the absorption of substrates by the intestine (Welling, 1984; Kimura and Higaki, 2002).
Drug Properties
Many efforts exist to interrelate the physicochemical properties of the drug with absorption. The drug, whether a weak acid or a weak base, and its pKa determine the extent of ionization according to the pH partition hypothesis at various pH values (pH 1.3 for stomach and 6 for intestine) of the gastrointestinal tract (Hogben et al., 1957, 1959; Schanker et al., 1957a,b). Deviations from the predictions were found, and these had been explained by the presence of an unstirred water layer (USWL) (Suzuki et al., 1970a,b), or a microclimate pH (Winne, 1977). The concept of absorption potential was then utilized to describe drug absorbability based on the partition coefficient, the solubility, dose, and fraction un-ionized (Dressman et al., 1985; Macheras and Symillides, 1989; Yu et al., 1996). Drugs that are un-ionized or that undergo hydrogen bonding exhibit a much greater lipophilicity toward membrane permeation than their ionic counterparts. Too many hydrogen donor or acceptor groups, however, is not good (Lipinski et al., 1997). Lipophilicity, a major determinant for predicting the extent of membrane permeation, is often correlated with the partition coefficient, when aqueous solubility is not exceeded and when the unstirred water layer is not an imposing barrier (Ungell et al., 1998). For very lipophilic agents, absorption may be rate-limited by the inability of drug traversing the USWL. For very hydrophilic or polar agents, the converse is true, such that membrane resistance is higher than the aqueous layer resistance. However, when drugs possess both hydrophilic and lipophilic qualities and permeate the USWL and membrane well, blood perfusion rate becomes the overall rate-limiting step for absorption. Predictive models based on Lipinski's Rule of Five (Lipinski et al., 1997), the quantitative structure-bioavailability relationship (Andrews et al., 2000), or other quantitative structure-activity relationship models (Norinder et al., 1999; Zhao et al., 2001, 2002; Klopman et al., 2002) have been developed to forecast the drug permeation potentials. The Rule of Five predicts that oral availability will be poor when there are more than 5 H-bond donors, 10 H-bond acceptors, the logP value is greater than 5, and the molecular weight exceeds 500 (Lipinski et al., 1997).
Drug permeation across the intestinal membrane is described by the effective permeability coefficient, Peff, or the overall rate of loss of compound per unit area (Elliott et al., 1980; Amidon et al., 1981; Ho et al., 1983; Fagerholm et al., 1996). The coefficient Peff may be estimated from luminal perfusion studies by measurement of drug loss across the lumen of any given segment, where Cout,lumen and Cin,lumen are the concentrations of drug leaving and entering the lumen, respectively; r is the radius, l is the length, and Qlumen is the experimentally determined luminal flow rate.  Since changes in water content in the lumen occur for these studies, the change in water volume may be corrected for by the inclusion of a nonabsorbable substance such as polyethylene glycol 4000. For passively absorbed drugs, Peff is dependent on the physicochemical properties of the substrate, including lipophilicity, molecular size, hydrogen bonding capacity, and polar surface area (Winiwarter et al., 1998), and is related to the permeability in the aqueous unstirred water layer (Paq) and the permeability across the membrane (Pm), and may be estimated from the following equation (Ho et al., 1983).
Since changes in water content in the lumen occur for these studies, the change in water volume may be corrected for by the inclusion of a nonabsorbable substance such as polyethylene glycol 4000. For passively absorbed drugs, Peff is dependent on the physicochemical properties of the substrate, including lipophilicity, molecular size, hydrogen bonding capacity, and polar surface area (Winiwarter et al., 1998), and is related to the permeability in the aqueous unstirred water layer (Paq) and the permeability across the membrane (Pm), and may be estimated from the following equation (Ho et al., 1983).  The fraction of dose absorbed (Fa or Css,out,lumen/Css,in,lumen) is estimated as that portion of the dose which disappeared from the intestinal lumen. The fraction of dose absorbed will depend on the physicochemical properties of the compound, the presence of carrier-mediated systems for absorption and exsorption, and intestinal metabolism within the enterocytes.
The fraction of dose absorbed (Fa or Css,out,lumen/Css,in,lumen) is estimated as that portion of the dose which disappeared from the intestinal lumen. The fraction of dose absorbed will depend on the physicochemical properties of the compound, the presence of carrier-mediated systems for absorption and exsorption, and intestinal metabolism within the enterocytes.
The Intestine, a Drug Metabolizing and Excretion Tissue
The intestine is noted for its absorptive function because of the presence of villi and microvilli and transporters for organic anions and cations (Fei et al., 1994; Sadee et al., 1995; Tsuji and Tamai, 1996; Arimori and Nakano, 1998; Craddock et al., 1998; Koepsell 1998; Ito et al., 1998; Zhang et al., 1998). Additionally, there exist drug-metabolizing enzymes for oxidation due to the appreciable cytochrome P450 3A (CYP3A4 in humans) (Hoensch et al., 1975; Watkins et al., 1987; Dubey and Singh, 1988a; Kolars et al., 1992; Thummel et al., 1996; Paine et al., 1996, 1997) and conjugation enzymes (Dubey and Singh, 1988b; Ilett et al., 1990), and efflux transporters at the apical and basolateral membranes (Lin et al., 1999; Suzuki and Sugiyama, 2000; Wacher et al., 2001) (Fig. 1). Drug exsorption occurs at the villous tips of the enterocytes at the apical membrane via the 170-kDa P-glycoprotein [Pgp; or the multidrug resistance gene product (MDR1)] (Thiebault et al., 1987; Hsing et al., 1992; Smit et al., 1998a,b) and multidrug resistance-associated protein 2 (MRP2) (Paulusma et al., 1996; Gotoh et al., 2000; Mottino et al., 2000) that cause drug efflux into the lumen, effectively reducing the sojourn of drug within the enterocyte (Saitoh et al., 1996; Lown et al., 1997b; Kim et al., 1998). Many drugs are substrates of both Pgp and cytochrome P450 3A (Terao et al., 1996; Wacher et al., 1998; Benet and Cummins, 2001). These are exemplified by verapamil (Saitoh and Aungst, 1995; Sandström et al., 1998); anticancer drugs such as vincristine, etoposide, daunorubicin, and paclitaxel (Leu and Huang, 1995; Sonnichsen et al., 1995; Nakayama et al., 2000; Chico et al., 2001; Wacher et al., 2001; Abraham et al., 2002); digoxin (Cavet et al., 1996; Greiner et al., 1999); indinavir, the human immunodeficiency virus protease inhibitor (Hochman et al., 2000, 2001; Li et al., 2002); and immunosuppressive agents cyclosporin (Gan et al., 1996; Lown et al., 1997b), tacrolimus (Lampen et al., 1995; Hashimoto et al., 1998; Hashida et al., 2001), and sirolimus (Lampen et al., 1998; Paine et al., 2002).

Schematic diagram of the enterocyte of the intestine showing absorptive and efflux transporters at the apical and basolateral membranes, and enzymes for intracellular metabolism.
Gastric Emptying and Intestinal Motility
The rate of gastric emptying strongly impacts the rate and extent of intestinal drug metabolism and drug absorption. Various disease conditions and food intake affect stomach emptying and/or intestinal transit (Welling, 1984). Double peak absorption has been correlated with antral gastric motility (Oberle and Amidon,1987; Plusquellec et al., 1987; Suttle et al., 1992; Langguth et al., 1994; Lipka et al., 1995; Suttle and Brouwer, 1995; Wang et al., 1999; Takamatsu et al., 2002; Kimura and Higaki, 2002; Yin et al., 2003) as well as other factors including the presence of adjuvants (Basit et al., 2002) or bile salts (Lennernäs and Regardh, 1993). The data showing double peaks during absorption have been modeled as the discontinuous oral absorption model (Witcher and Boudinot, 1996). Inevitably, a delay in stomach emptying reduces the rate of drug absorption since the rate of delivery to the site of absorption, the small intestine, is prolonged (Table 1). With regard to gastric emptying, drugs may be classified as 1) acid-labile compounds, 1) relatively insoluble compounds, 3) drugs with good water and lipid solubility, and 4) drugs absorbed by carriers. For the acid-labile drugs such as penicillin and ampicillin, a greater degradation occurs with prolongation of stay in the stomach, diminishing the extent of absorption (Terry et al., 1982; Ali, 1985). By contrast, increasing the transit time of relatively insoluble compounds in the stomach favors drug dissolution and improves the extent of the absorption of griseofulvin and phenytoin (Aoyagi et al., 1984; Hamaguchi et al., 1993), whereas no change in extent exists for well absorbed drugs such as acetaminophen and fadrozole, compounds of sound water and lipid solubility (Terry et al., 1982; Choi et al., 1993). For drugs whose transport into the intestine is via apical transporters, however, it is envisaged that the reduced intermittent release of drug from the stomach to the intestine brings about a desaturation of the transporter system, rendering an increase in the extent of drug absorption (Table 1).
Effect of delayed gastric emptying and effect of metabolism and secretion on drug absorption
Methods to Study Intestinal Transport and Metabolism
Because of the significance of the intestine as an important firstpass organ, high-throughput in vitro systems have been developed to assess the importance of intestinal absorption, metabolism, and excretion for the prediction of drug-drug interactions. Gene expression systems (Smit et al., 1998b; Cvetkovic et al., 1999; Gotoh et al., 2000; Shitara et al., 2002) provide direct information on the involvement of individual transporters or enzymes. Then there are the intestinal membrane segments/preparations (Wilson and Treanor, 1975; Hopfer et al., 1976; Lasker and Rickert, 1978; Johnson et al., 2001), cells (Koster and Noordhoek, 1983; Traber et al., 1991), everted sacs (Munck, 1965; Barr and Riegelman, 1970; Kaplan and Cotler, 1972), and the Ussing chamber (Fiddian-Green and Silen, 1975; Rogers et al., 1987; Lampen et al., 1995). For flux measurements, a donor compartment is used for drug administration and a receiving compartment is used for sampling. With drug given to the mucosal side, sampling allows the examination of drug absorption, metabolism, and efflux as well as entry into the basolateral compartment. Moreover, drug may be given at the serosal compartment to ascertain the net flux from the basolateral side to the mucosal lumen. Drug efflux and exsorption has been studied with vesicles prepared from the basolateral and apical membranes (Weinberg et al., 1986; Tsuji and Tamai, 1989; Bair et al., 1991).
A popular in vitro system is the Caco-2 cell line, derived from human colon carcinoma cells (Hidalgo et al., 1989; Hunter et al., 1990), that contains the Pgp (Hunter et al., 1993a,b). A slight drawback may be the existence of a USWL that may pose as a barrier for lipophilic drug transport (Hidalgo et al., 1991). The development of the Caco-2 cell has greatly facilitated progress and led to the testing of diverse drug classes as Pgp substrates (Burton et al., 1993; Hosoya et al., 1996; Tanaka et al., 1996; Terao et al., 1996). The differentiated Caco-2 cell monolayers were initially used to study drug efflux by the Pgp, or the multidrug resistance gene product MDR1. Involvement of Pgp is inferred when the basolateral to apical flux (B to A) exceeds that of A to B. It was further found that, upon culture in 1α-25-dihyroxy vitamin D3 for 2 weeks postconfluence, cytochrome P450 3A activities were up-regulated (Crespi et al., 1996, 2000; Schmiedlin-Ren et al., 1997; Thummel et al., 2001). More recent development involved transfection with the cytochrome P450 gene and stimulation by butyrate (Cummins et al., 2001) to provide the added P450 activity. The incubation system, the donor and receiving compartments separated by the cell monolayer, is an efficient, high-throughput system for examination of whether newly developed pharmaceuticals are substrates of cytochrome P450 3A4 and/or P-glycoprotein such that interactions with other drugs may be predicted.
The in situ vascularly perfused rat small intestine preparation is a useful preparation for studying the disposition of both orally and systemically administered agents (Windmueller and Spaeth, 1977, 1981; Pang et al., 1986; Hirayama et al., 1989; Doherty and Pang, 2000). In this preparation, the native architecture of the small intestine is maintained with respect to the circulation such that the extents of metabolism, absorption, and secretion can be studied simultaneously (Pang et al., 1985; Xu et al., 1989; Hirayama and Pang, 1990). The technique allows for single-pass or recirculating experiments involving systemic or luminal drug administration, including luminal administration in closed loops or segments (Pang et al., 1986; Cong et al., 2001). In other instances, the intestine-liver preparation may be used for the study of first-pass metabolism and for examination of the role of the intestine on regulation of hepatic metabolism (Pang et al., 1985; Chen and Pang, 1997).
In vivo techniques exist for the study of intestinal drug absorption. The Doluisio method entails use of an in situ rat gut technique for drug administration into the lumen (Doluisio et al., 1969; Sim and Back, 1988). In some rat preparations, the inflow and outflow of a select segment were monitored for drug disappearance, and arterial blood was sampled and the volume of blood was replenished by transfusion (Barr and Riegelman, 1970). Some studies involve luminal instillation of drug to select or closed segments of rats (duodenum, jejunum, or ileum) (Hirayama et al., 1990; Cong et al., 2001) or humans (Gramatté and Richter, 1994; Gramatté, 1996; Gramatté et al., 1994, 1996). Gene knockout mice (Kim et al., 1998; Smit et al., 1998; Greiner et al., 1999) and mutant animal models (Gotoh et al., 2000) have also been utilized to examine intestinal transport. The surgical manipulation of portacaval transposition allowed the direct assessment of systemic intestinal removal in the absence of the liver (Gugler et al., 1975; Effeney et al., 1982; Lo et al., 1982). The method is almost analogous to the oral and systemic administrations of the test drug, midazolam, to anhepatic patients for the estimation of intestinal drug metabolism (Paine et al., 1996). Through varying sites of drug administration, namely, orally, intraportally, and intravenously, comparison of the area under the curves under first-order conditions yields the available fraction of the intestine and liver (Mistry and Houston, 1985, 1987; Hirayama et al., 1990).
Transporters for Absorption And Efflux at Apical Membrane
Much attention is given to the presence of transporters at the apical or brush-border membrane of the intestine, not only for drug absorption but also for exsorption (Table 2). The various apical transporters for organic anions and cations have been reviewed (Tsuji and Tamai, 1996; Koepsell, 1998; Zhang et al., 1998). The ABC (ATP-binding cassette proteins) efflux transporters, Pgp for lipophilic entities and MRP2 for drug conjugates, are both present on the intestinal membrane. Moreover, the newly characterized breast cancer resistance protein (BCRP) multidrug transporter confers resistance to mitoxantrone, topotecan, the anthracyclines, and related drugs in cell lines (Doyle et al., 1998; Allen et al., 1999; 2002; Miyake et al., 1999; Jonker et al., 2000; Zamber et al., 2003). These efflux transporters are known to delimit drug absorption.
Involvement of intestinal transporters for drug absorption (see also references contained in Tsuji and Tamai, 1996 )
Heterogeneity of Intestinal Transporters. Analogous to that found for the liver, there exists an increasing body of literature on the heterogeneity of intestinal transporters (Table 3). The absorption of salicylate (Ungell et al., 1998), antipyrine (Raoof et al., 1998), acetaminophen (Gramatté and Richter, 1994), griseofulvin (Gramatté, 1996), and (-)-carbovir (Soria and Zimmerman, 1994) was found to be the same among all segments. Metoprolol absorption is the same among segments in humans (Vidon et al., 1985). Preferential absorption in segmental regions has also been noted for the intestinal absorption of ranitidine (Suttle and Brouwer, 1995) and diltiazem (Homsy et al., 1995). In like fashion, the absorption of ranitidine (Gramatté et al., 1994) and talinolol (Gramatté et al., 1996) is higher in the proximal intestine in humans. For hydrochlorothiazide, atenolol, furosemide, and cimetidine (Lennernäs, 1998), the net mucosal to serosal absorption was greater for the jejunum than for the ileum. For verapamil (Hunter et al., 1990), phenytoin, almokalant, gemifibrozil, metoprolol, omeprazol, propranolol, foscarnet, erythritol, dDVAP (Ungell et al., 1998), and etoposide (Makhey et al., 1998), net mucosal to serosal absorption was greater for the ileum over the jejunum.
Segmental distribution of transporters and enzymes among the intestinal segments
Recent advances in expression cloning of intestinal transporters have provided more definitive tools for the examination of regional distribution of the transporters (Fei et al., 1994; Schneider et al., 1995; Mottino et al., 2000; Walters et al., 2000; Ngo et al., 2001). There are consequences of heterogeneously distributed apical absorptive and secretory transporters. Varying rate constants for benzoic acid intestinal uptake were observed among the segments, and a higher absorption rate constant was shown for the jejunal segment versus the duodenal or ileal segment in the perfused rat intestine preparation. The higher jejunal distribution of Mct1 was confirmed with Western blotting along the length of the rat small intestine (Cong et al., 2001). The proton-coupled oligopeptide transporter of the rabbit was found more abundantly in the proximal intestine (duodenum and jejunum) (Fei et al., 1994); the rat organic anion transporter 3, Oatp3, is higher in the jejunum (Walters et al., 2000); and the apical bile salt transporter (abst) predominates in the distal ileum of the hamster and rat (Wong et al., 1994; Schneider et al., 1995). Gotoh et al. (2000) demonstrated the greater mRNA expression of Mrp2 in the rat jejunum, followed by the duodenum and ileum, with very little in the colon, as confirmed by Mottino et al. (2000). The excretion of the glutathione conjugate 2,4-dinitrophenyl-S-glutathione by Mrp2 was greatest in the jejunum, as expected by mRNA expression (Gotoh et al., 2000). The efflux transporter rat Mrp2 is higher in the proximal region (Gotoh et al., 2000; Mottino et al., 2000), whereas the Pgp efflux pump is higher distally at the jejunum and ileum (Thiebault et al., 1987; Hunter et al., 1990; Chianale et al., 1995; Lown et al., 1997b; Collett et al., 1999; Nakayama et al., 2000; Stephens et al., 2001; Li et al., 2002). The rat basolateral Mrp3 that transports drug out of the cell is higher toward the distal ileum and colon (Rost et al., 2002). The nonhomogeneous distributions of the transporters (Table 3) are expected to affect drug absorption and bioavailability.
Heterogeneity of Intestinal Enzymes for Metabolism. The intestinal tissue is endowed with phase I and II enzymes, although at lower levels than those for the liver. Drug metabolizing enzymes: UDP-glucuronosyltransferases (UGTs), sulfotransferases (PSTs), and glutathione S-transferases (GSTs) exhibit a decreasing gradient along the intestinal wall, from duodenum to ileum (Clifton and Kaplowitz, 1977; Pinkus et al., 1977; Schwarz and Schwenk, 1984; Dubey and Singh, 1988a). The human intestinal CYP3A4 shows a slightly lower level at the duodenum before levels rise again at the jejunum, then finally decreasing toward the ileum (Paine et al., 1996; Thummel et al., 1996). The same was found in animals (Hoensch et al., 1975; Li et al., 2002).
Route-Dependent Intestinal Metabolism/Excretion
Route-dependent intestinal metabolism has been observed (Table 4). “Route-dependent” intestinal metabolism describes a greater intestinal metabolism/extraction of drug upon oral or luminal dosing versus “systemic” dosing. In studies pertaining to the perfused, rat small intestine preparations, greater extents of metabolism were noted for acetaminophen (Pang et al., 1986), enalapril (Pang et al., 1985) morphine (Doherty and Pang, 2000) and (-)-aminocarbovir, the prodrug that was converted to (-)-carbovir (Wen et al., 1999), when given luminally, whereas metabolism was either absent or negligible when the drug dose was given into the reservoir for systemic delivery. These results from the vascular intestinal perfusion model mirrored the observations on midazolam hydroxylation in humans. The drug exhibits a low intestinal extraction ratio (0.09) in anhepatic patients undergoing liver transplantation with systemic administration, but extensive first-pass metabolism was noted orally (extraction ratio of 0.43), with much of the intestinally formed primary metabolite, 1-hydroxy midazolam, reaching the hepatic portal blood (Paine et al., 1996; Thummel et al., 1996, 1997). Interestingly, intestinal CYP3A4 levels in humans correlated well with rates of midazolam 1′- and 4-hydroxylation (Thummel et al., 1996) but failed to show a relation with the erythromycin breath test (Lown et al., 1994, 1997b) that is ordinarily used to correlate with liver CYP3A4 levels (Watkins et al., 1989). The exclusivity of the intravenous erythromycin test to liver function only translates to the inaccessibility of the systemically administered substrate to the intestinal mucosal (CYP3A4) enzymes. The overall findings infer that the enzymes for preabsorptive intestinal metabolism are present on enterocytes facing the lumen and are unavailable to drugs in the circulation. A general hypothesis was put forth to explain route-dependent intestinal metabolism: intestinal drug metabolism behaves as if it were a preabsorptive event occurring predominantly during absorption, but little or no intestinal removal occurs for drug in the systemic circulation due to the inaccessibility of enzymes (Doherty and Pang, 1997). Analogously, pretreatment of humans with rifampin, a pregnane X receptor ligand, on Pgp secretion exerted an effect only on the oral but not the intravenous kinetics of digoxin (Greiner et al., 1999), suggesting that the intestinal Pgp is accessible for digoxin administered into the lumen.
Route-dependent intestinal metabolism
Intestinal Modeling
The dynamic interactions of metabolism and secretion, and the role of transporters on drug absorption have been under considerable debate. The thorough understanding of intestinal processes would be learnt through modeling of intestinal data only in the absence of the contribution from the liver. Recent studies on the interactions between the P450s and Pgp in vitro have led to the conclusion that intestinal metabolism is enhanced by the secretory action of P-glycoprotein due to an increase in the mean residence time (MRT) of drug in the intestine (Benet and Cummins, 2001; Cummins et al., 2001; Johnson et al., 2001). Theoretical examinations on Caco-2 cells and intestinal vascular perfusion systems support the notion that the mean residence time was increased, but metabolism under linear conditions was in fact decreased in the presence of secretion, even though the mean residence time has increased; under nonlinear metabolism, instances exist whereby the rate of metabolite accrual may increase, even though the ultimate amount of metabolite formed remained equal to the dose (Tam et al., 2003a). However, under cases of nonlinear metabolism, increased secretion together with rapid re-entry of drug from the apical compartment to the cell evokes increased rates of drug metabolism. The reason for this anomaly is attributed to the desaturation of intestinal enzymes (Tam et al., 2003a).
The next question is, what about the intact intestine? How should data arising from studies pertaining to intestinal metabolism, efflux, absorption, and gastric emptying/intestinal transit be handled? Modeling efforts have included the gastrointestinal absorption and kinetic models that examine absorption of drug from the stomach, duodenum, upper and lower jejunum and ileum, cecum, and large intestine with varying transit times and absorption (Kimura and Higaki, 2002). There had been theoretical investigations on the compartmental absorption and transit (CAT) model that considered the transit and absorption of drug in the small intestine (seven compartments), and there was no absorption within the stomach and colon compartments. Absorption within the seven mixing-tanks in series compartments for the small intestine was governed by the gastric emptying rate and different intestinal transit times (Yu et al., 1996; Yu and Amidon, 1998, 1999). Since the CAT model did not include the physical modeling of drug dissolution from dosage solid forms or controlled release formulations, degradation in lumen, changes in absorptive surface area, absorption in the stomach and colon, metabolism in liver, and transporter densities for absorption and efflux, a refined and expanded model, known as the advanced CAT or ACAT model was developed, with implementation of software, for the prediction of drug absorption (Agoram et al., 2001). Details such as particle size, pH, particle density, and diffusion coefficient were included for consideration of drug dissolution and absorption. In another model, Ito et al. (1999) described intestinal metabolism and secretion, intracellular drug diffusion, and permeation through the basolateral membrane; however, the model seemed to lack description of intestinal flow. Moreover, these models do not relate to blood flow, drug partitioning, and the phenomenon of route-dependent intestinal metabolism. A greater understanding of the interplay will allow sound interpretations on intestinal drug metabolism and transport with respect to other drugs or the intake of fruit juices (Bailey et al., 1993; Lown et al., 1997a; Dresser et al., 2002).
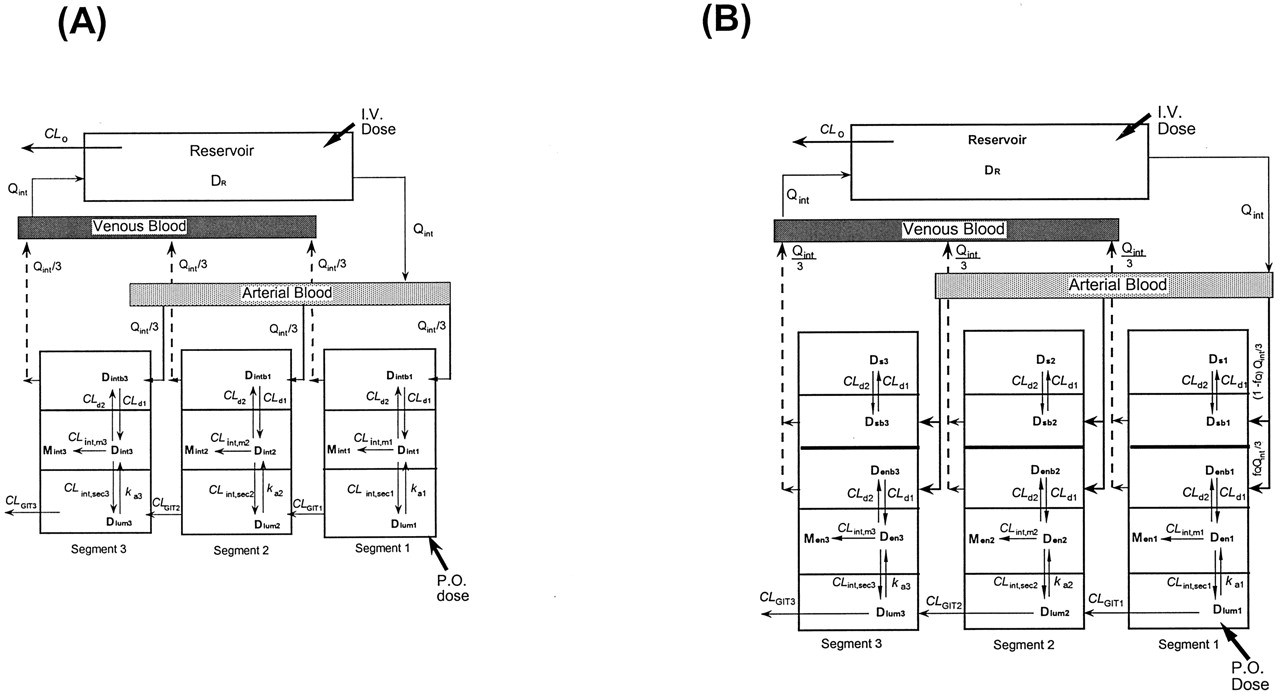
Physiological Models: Traditional Model (TM) and Segregated Flow Model (SFM). Thus, the strategy is to turn to physiologically based models that encompass all of the salient variables of transport, metabolism, efflux, gastrointestinal transit, and absorption (Doherty and Pang, 2000) (Fig. 2). The TM (Fig. 2A) was developed to describe data arising from recirculation of tracer morphine in the perfused rat small intestine preparation (Doherty and Pang, 2000). In this preparation, morphine glucuronidation was observed with dosing of morphine into the duodenal lumen and not with administration into the reservoir that mimicked intravenous administration (Figs. 3 and 4). To address the apparent “inaccessibility” of the intestinal enzymes to the drug borne in the circulation but not lumen, or route-dependent metabolism, the segregated flow model, based on modifications of the model of Klippert and Noordhoek (1985) (Fig. 2B), was developed. SFM describes a partial and low blood flow to the “active” enterocyte region where the absorptive and exsorptive carriers and metabolic enzymes reside, and a much higher bulk flow to a nonabsorptive and nonmetabolizing region that included the serosa, submucosa, and mucosa, excluding the enterocyte region (Cong et al., 2000). Thus, the SFM recognizes the subtle demarcation of tissue layers and distributions in blood supply (Svanvik, 1973; Granger et al., 1980). The literature values for the blood flow to the absorptive enterocyte layer of the mucosa vary greatly, ranging from 5% to 30% (Micflikier et al., 1976; MacFerran and Mailman, 1977; Mailman 1978; Granger et al., 1980). Drug in the lumen must enter via the enterocyte region before reaching the circulation, whereas drug already in circulation is primarily channeled to other tissular regions. The condition conduces to a greater intestinal metabolism with oral versus intravenous dosing. For data on tracer morphine glucuronidation in the recirculating, vascular perfused rat small intestine, the SFM was superior to describe the phenomenon of route-dependent intestinal metabolism (Cong et al., 2000).
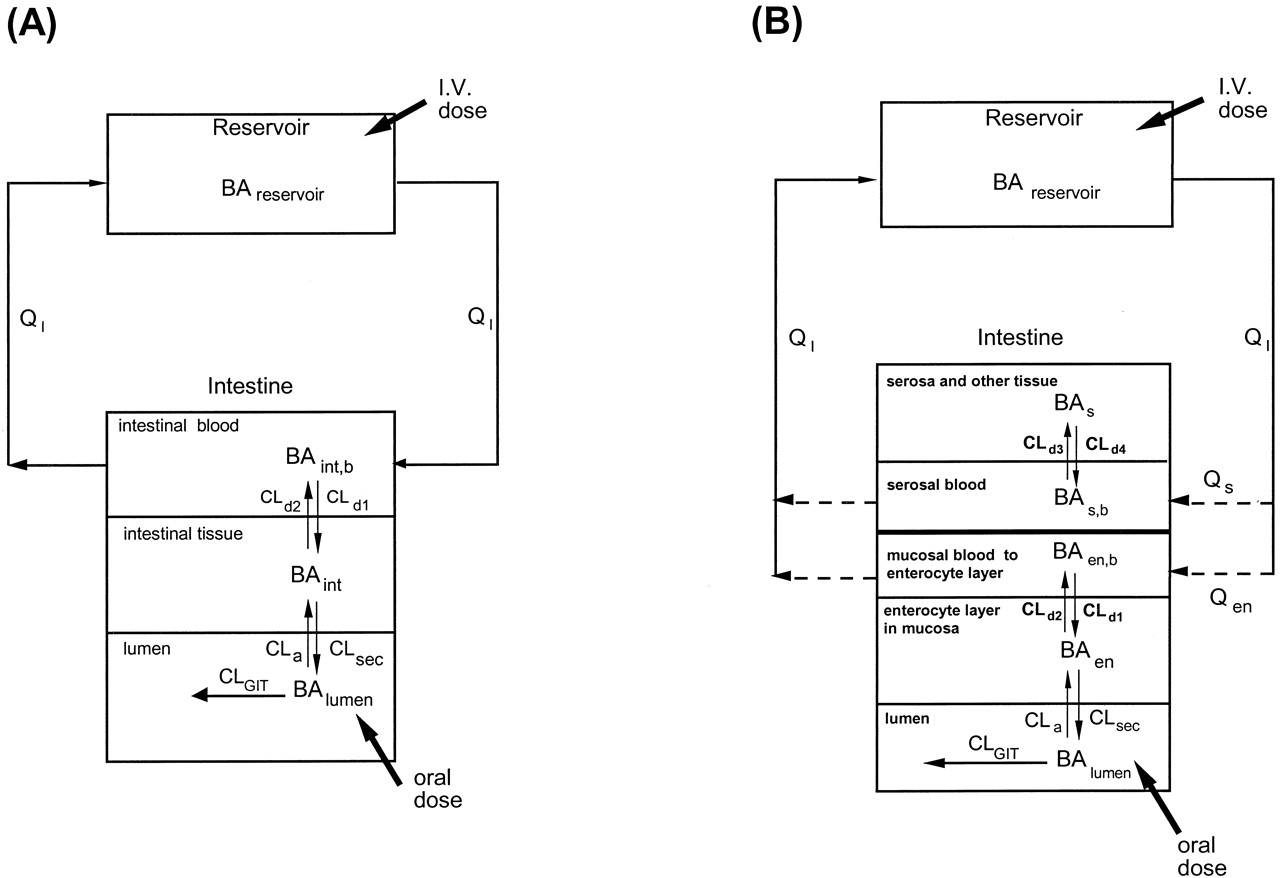
Fates of drug and metabolite in the TM (A) and SFM (B).
Note that the entire oral dose passes through the enterocyte region for TM and SFM, whereas only partial intravenous dose reaches the enterocyte region for the SFM. For TM, the intestinal blood (QI) perfuses the entire intestinal tissue, the site of metabolism and absorption from the lumen. For SFM, intestinal blood is segregated to perfuse the nonmetabolizing (Qs) and enterocyte-mucosal (Qen) regions. Drug equilibrates with those in the corresponding tissue layers with intrinsic transfer clearances CLd1 and CLd2 for TM, or CLd1 and CLd2, CLd3 and CLd4 for SFM. The absorptive, metabolic, and efflux activities within the villus tips of the mucosal layer are represented by the rate constant, ka, and metabolic and secretory intrinsic clearances, CLint,m and CLint,sec, respectively. Gastrointestinal transit clearance is denoted by CLGIT. Adapted from Cong et al. (2000), with permission.

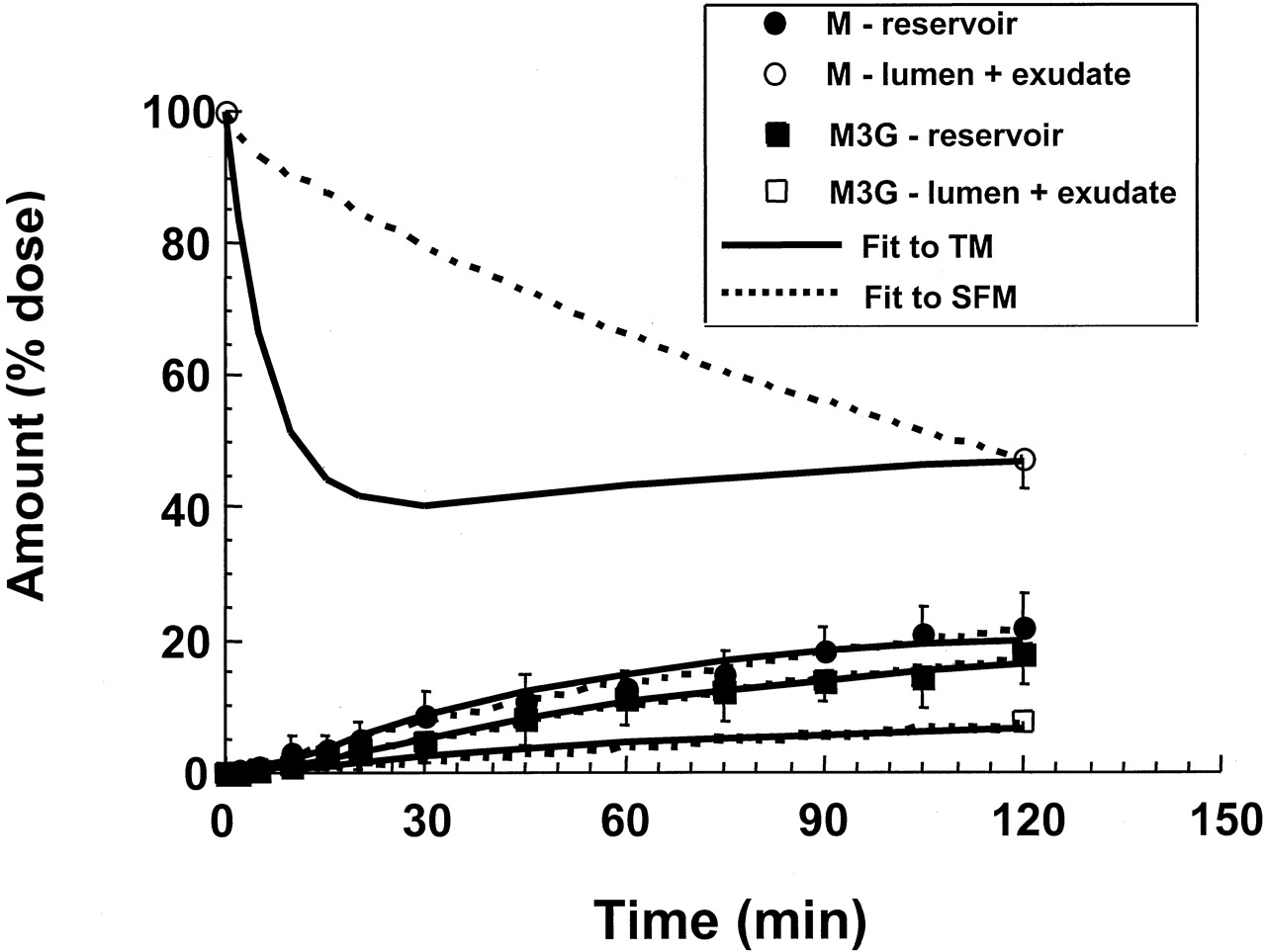
Fits of tracer [3H]morphine data from the vascularly perfused, recirculating rat small intestine preparation to the TM and SFM, after systemic administration of dose into the reservoir (data fromCong et al., 2000).
There was a total lack of morphine glucuronide formed. Only morphine was excreted into the lumen (and luminal fluid that was collected as exudates). Note the superior fit of the data to the SFM. Adapted from Cong et al. (2000), with permission.

Fits of tracer [3H]morphine data from the vascularly perfused, recirculating rat small intestine preparation to the TM and SFM, after duodenal administration of dose into the intestinal lumen (data fromCong et al., 2000).
Note that morphine glucuronide was formed with luminal administration of morphine. Both morphine and morphine glucuronide were found in the lumen and luminal fluids that were collected as exudates. Note the superior fit of the data to the SFM. Adapted from Cong et al. (2000), with permission.
These physiological-based models developed for the intestine provide estimations of metabolism and MRT. Under linear conditions, intestinal secretion results in reduced intestinal metabolite formation, as confirmed in a recent theoretical examination of TM and SFM (unpublished data of K. S. Pang, E. Tseng, and D. Tam). The conclusion also agrees with intuitive and deductive reasoning since reduction of intracellular substrate concentration in the intestine accompanies drug efflux at the apical membrane, yielding lower rates of intestinal metabolism (Sirianni and Pang, 1997; Schuetz and Schinkel, 1999). This was also found in a simulation study based on Caco-2 cell efflux and metabolism (Tam et al., 2003a). Increased absorption neutralizes the effect of intestinal secretion and increased bioavailability (F), whereas increased secretion and metabolism reduce the bioavailability (Table 1).
Segmental, Traditional Model (STM) and Segmental Segregated Flow Model (SSFM). To accommodate the attendant heterogeneities of metabolic and transporter activities and to examine their impact on intestinal clearance and availability, more advanced models have been developed (Tam et al., 2003b). The single intestinal compartments of the TM and SFM were expanded into three equal segmental compartments, as for the zonal modeling of the liver (Abu-Zahra and Pang, 2000), in the development of the segmental, traditional model (Fig. 5A) and the segmental, segregated flow model (Fig. 5B) (Tam et al., 2003b). The varying distributions of absorptive and secretory transporters and metabolic enzymes that evoked high and low availabilities were found from simulations based on intestinal modeling for the rat small intestine (Fig. 6). Of note is the strong influence of the distribution of the metabolic enzymes along the intestinal length on F. The predicted trends were generally similar for both STM and SSFM, although the magnitudes differed. When segments of equal activities for the transporters and metabolic enzymes were examined in a simulation study that viewed the intestine as the only eliminating tissue, a fast absorption, reflected by high absorption rate constants within the segments, counteracted the depth of the virtual, peripheral compartment and reduced the mean residence time of drug in intestinal tissue, MRTcell. Rapid reabsorption of the secreted species canceled the effect of intestinal secretion and increased the F. However, high metabolic and secretion activities within the enterocytes greatly reduced F. A greater metabolic intrinsic clearance within the segments decreased MRTcell, whereas a greater secretory intrinsic clearance within the segments increased the MRTcell (Figs. 7 and 8) (Tam et al., 2003b).
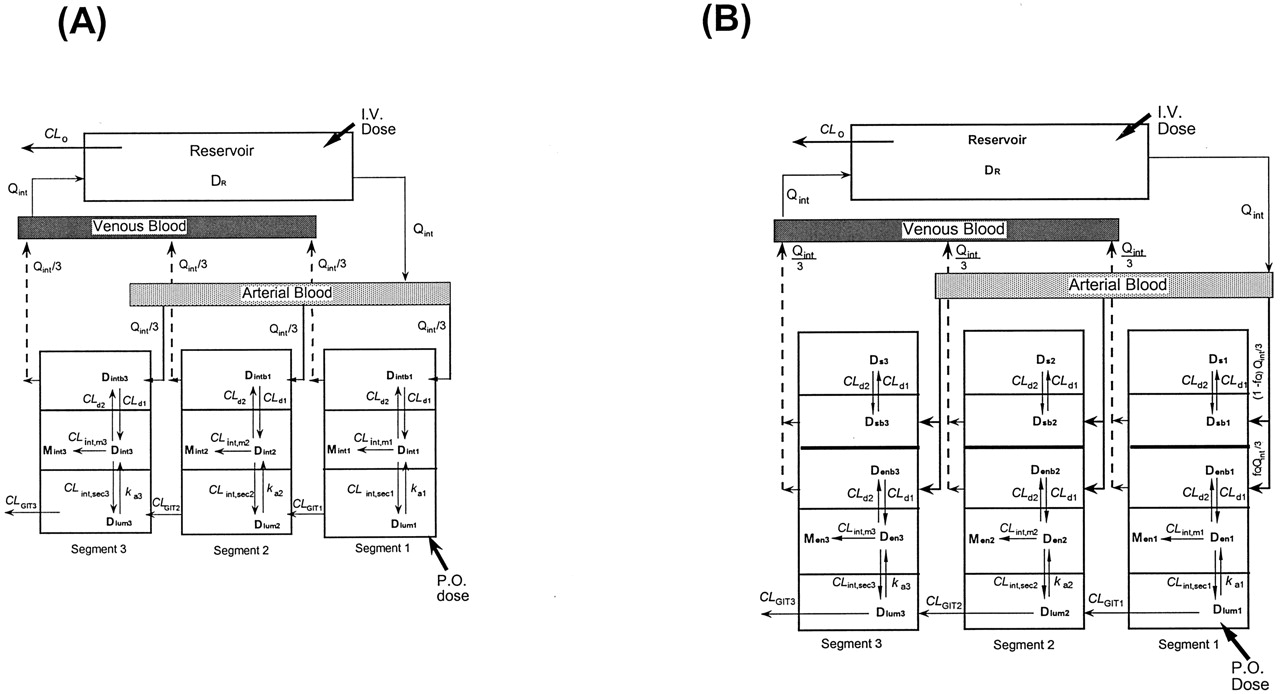
The STM, which views the intestine as three equal segments (or expanded ith compartments where i = 1, 2, or 3) of TM (Fig. 2A) with metabolism taking place to form the metabolite (M) in the intestinal tissue (A), and the SSFM, an expansion of the SFM (Fig. 2B), which views the intestine as three segments (or expanded ith compartments where i = 1, 2, or 3) with low and partial flow to the enterocyte regions (fQQint/3), where fQ is the fraction of total intestinal flow (Qint) perfusing the enterocyte region, where the enzymes and efflux transporters reside and bulk flow [(1 - fQ) Qint/3] to the “other” noneliminating region (B).
Drug (D) in blood equilibrates with that in the corresponding tissue layers with intrinsic transfer clearances CLd1 and CLd2 for both STM and SSFM. The absorptive, metabolic, and efflux activities within the villus tips of the mucosal layer are represented by the rate constant, kai, and metabolic and secretory intrinsic clearances, CLint,mi and CLint,seci, respectively, for each ith segment. The gastrointestinal transit clearance is denoted by CLGITi. Adapted from Tam et al. (2003b), with permission.

Simulations based on intestinal modeling for the rat small intestine, to determine the varying distributions of absorptive and secretory transporters and metabolic enzymes that evoke high and low bioavailabilities.
Segmental distributions (designated as ith segments where i = 1, 2, and 3) of the metabolic intrinsic clearance, CLint,mi (0.9, 0.5, and 0.1 ml/min), the absorption rate constant, kai (5, 3, and 1 min-1), and the secretory intrinsic clearance, CLint,seci (2, 4, and 6 ml/min) furnished the lowest bioavailability (F) (left panel) and the highest F (right panel; CLint,seci values were 2, 4, and 6 ml/min; CLint,mi values were 0.1, 0.5, and 0.9 ml/min, and kai values were 5, 3, and 1 min-1) according to the predictions of the STM and SSFM. The predicted difference existed in the descending versus ascending, segmental distributions of the metabolic intrinsic clearance, CLint,mi within the segments 1, 2, and 3. Taken from Tam et al. (2003b), with permission.
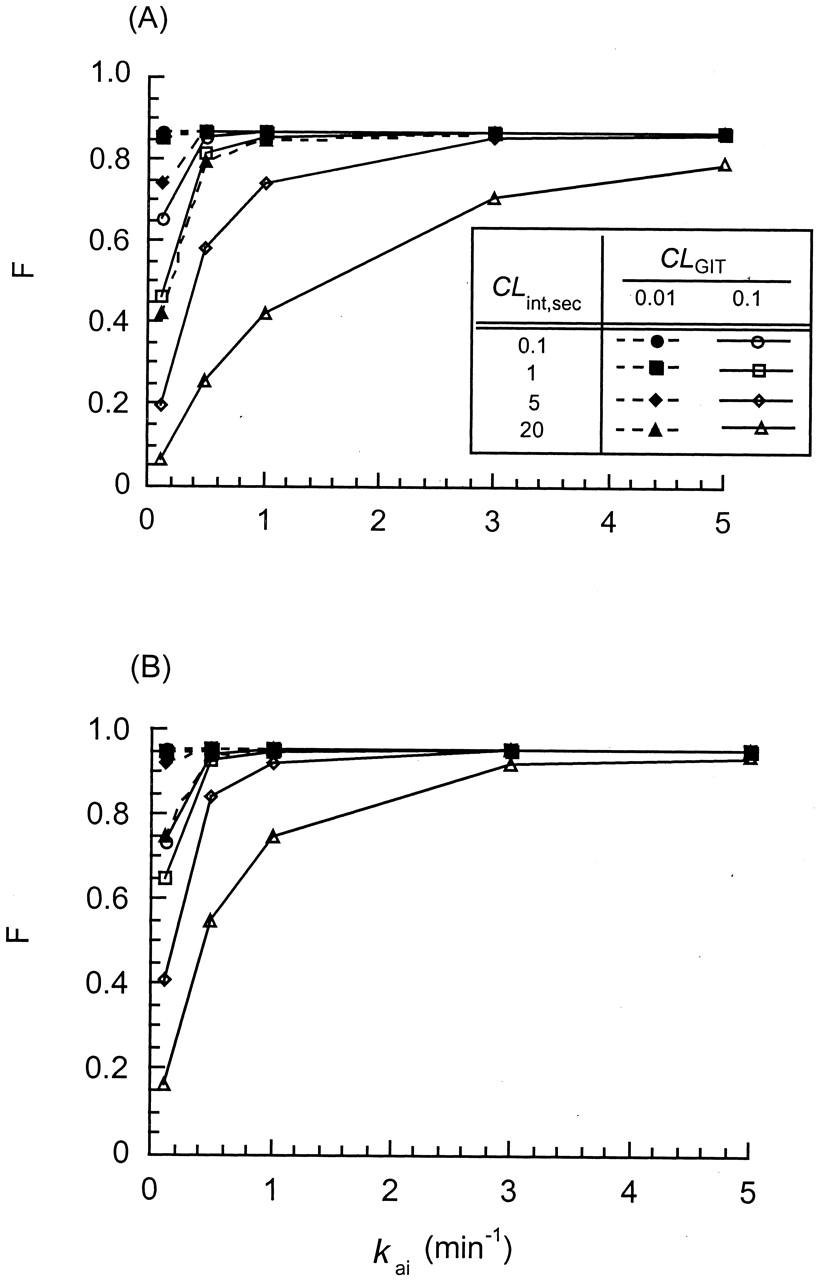
Simulations performed for the STM (A) and SSFM (B) to demonstrate the effect of the segmental, gastrointestinal transit clearance (CLGITi = 0.01 or 0.1 ml/min), the secretory intrinsic clearance (CLint,seci), and the absorption rate constant (kai) on the intestinal (systemic) bioavailability, F.
The metabolic intrinsic clearance, CLint,mi, was kept at 0.1 ml/min; the partitioning clearance of drugs, CLd1 and CLd2, was 0.9 ml/min. All the intestinal segments (i = 1, 2, and 3) were assigned to be equal and contained equal amounts of transporter, metabolic, and absorptive activities. Adapted from Tam et al. (2003), with permission.
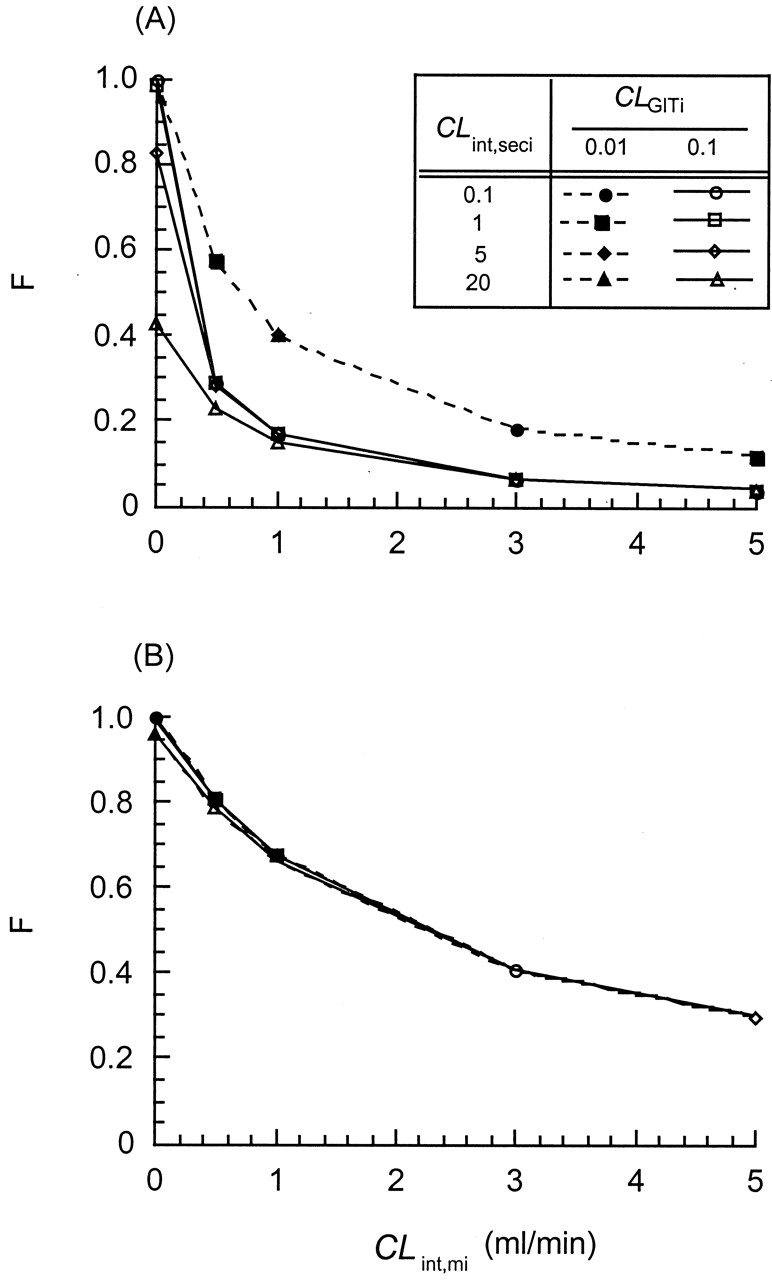
Simulations performed for the STM (A) and SSFM (B) to demonstrate the effect of the gastrointestinal transit clearance (CLGITi = 0.01 or 0.1 ml/min), the secretory intrinsic clearance (CLint,seci), and the metabolic intrinsic clearance (CLint,mi) on the intestinal (systemic) bioavailability, F.
The absorption rate constant for the each segment, kai, was kept at 3 min-1; the partitioning clearance for drugs, CLd1 and CLd2, was 0.9 ml/min. All the intestinal segments (i = 1, 2, and 3) were assigned to be equal and contained equal amounts of transporter, metabolic, and absorptive activities. Adapted from Tam et al. (2003), with permission.
Concluding Remarks and Future Modeling
Another question that remains is whether in vitro data would reflect intestinal metabolism in vivo. But data interpretation and correlation between in vitro and in vivo intestinal drug metabolism/removal is not straightforward. In addition to many of the usual problems often encountered for correlating in vitro-in vivo data on drug metabolism (Pang and Chiba, 1994), other variables are present, and these may further complicate the relationship. One reason is the efflux transporters at the apical membrane and the presence of intestinal motility that work in unison to remove drug out to the lumen, thereby preventing metabolism, although drug may be re-exposed to the enzymes upon reabsorption. Thus, not all of the effluxed drug is subject to intestinal metabolism, even though absorption is rapid. The second is route-dependent intestinal metabolism due to the peculiarity in intestinal blood flow to the “active” metabolic and absorptive region (Cong et al., 2000). The overall estimate of bioavailability is, therefore, complex, and is the consequence of membrane permeation by different arrays of transporters operating in the same or opposite directions, and cellular enzymatic activities that reduce the intracellular drug concentration.
The newly proposed physiologically based models that emphasize metabolism and secretion by the enterocytes have engendered many of the physiological processes governing the overall drug bioavailability and absorption. These models have provided tremendous insight into route-dependent intestinal metabolism/excretion, and the dynamic interplay among intestinal transit times, the absorptive carriers, and metabolic enzymes on intestinal clearance and bioavailability in the absence of a contribution by the liver. For application of the conceptual frameworks developed within the SFM and SSFM, the physiological parameters for the flow rate and volume, as well as more appropriate intestinal transit times, need to be properly scaled-up for modeling of human absorption.
The models, however, considered only drug already in solution. The complexities of drug dissolution and the physiochemical attributes of the various classes of drugs have yet to be integrated to the models. Mass balance with respect to ionization and absorption of the nonionic species may be further accommodated (Ishizaki et al., 1997). The disparate surface area in drug absorption (Agoram et al., 2001) and the intermittent release of drugs in various aggregated and de-aggregated forms from the stomach, gastric emptying, bile salt effects, and the presence of mucus may need to be considered. Improvement in prediction of systemic availability will result upon coupling of the drug-release phase of the ACAT model (Agoram et al., 2001). Sloughed off enterocytes that contribute to intestinal drug metabolism in lumen may need to be modeled (Glaeser et al., 2002). Nonlinearity issues, in terms of intestinal drug-absorptive transporters, metabolic enzymes, and drug efflux transporters, must be considered in drug absorption, especially when the drug is absorbed rapidly to evoke saturation (Tamai et al., 1997; Tam et al., 2003a).
The modeling of drug metabolism and transport by the intestine is at the early stages of development. Proof of the model relies not only on drug but also metabolite disposition during both oral and systemic dosing. Needless to say, the liver should be properly considered for first-pass removal and systemic bioavailability. It is expected that the consolidation of physiologically sound intestinal and liver models will greatly improve our predictiveness on drug absorption bioavailability.
Footnotes
-
↵1 Abbreviations used are: GIT, gastrointestinal tract; USWL, unstirred water layer; Pgp, P-glycoprotein; MDR1, multidrug resistance gene product 1; MRP2, multidrug resistance-associated protein 2; BCRP, breast cancer resistance protein; CAT, compartmental absorption and transit; ACAT, advanced CAT; Mct1, monocarboxylic acid transporter 1; Oatp3, organic anion-transporting polypeptide 3; UGT, UDP-glucuronosyltransferase; PST, sulfotransferase; GST, glutathione S-transferase; MRT, mean residence time; P450, cytochrome P450; TM, traditional model; SFM, segregated flow model; STM, segmental, traditional model; SSFM, segmental segregated flow model.
-
This work was supported by the Canadian Institute for Health Research, Grant MOP36457 and MOP117793.
-

-
Dr. K. Sandy Pang received the B.Sc. degree from the University of Toronto and Ph.D. degree from University of California at San Francisco and was a Fogarty fellow at the National Institutes of Health. She is presently Professor of Pharmacy and Pharmacology at the Faculties of Pharmacy and Medicine at the University of Toronto. Dr. Pang is known for her work in the fields of pharmacokinetics, drug metabolism, and liver physiology. Her research is aimed toward a mechanistic-based understanding of the handling of drugs and their metabolites within eliminating organs, namely the liver, intestine, and kidney. The approach is via development and formulation of physiologically relevant processes of organ clearances through theoretical treatises and the validation by experimental approaches in vitro (expression systems, cells, and subcellular preparations) and in perfused organs. Her work on metabolic kinetics emphasizes the need for consideration of heterogeneity and the order of sequential processing and has led to the finding that differences exist in the kinetics of formed versus preformed metabolites. Her more recent focus is on extension of the principles learned on hepatic clearances to examine heterogeneity in intestinal absorption and clearance with respect to the first-pass effect. Her recent interests include drug discovery programs in examination of hemoglobin conjugates and effective new drugs in a metastatic rat liver tumor model. She was the recipient of the National Institutes of Health Research Career Development Award, Faculty Development Award from the Medical Research Council of Canada, the McNeil Award from the Faculties of Pharmacies in Canada, and the Research Achievement Award in Pharmacokinetics, Pharmacodynamics, and Drug Metabolism from the American Association of Pharmaceutical Scientists (AAPS).
- Received March 3, 2003.
- Accepted July 14, 2003.

- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Oral Drug Absorption
- Drug Properties
- The Intestine, a Drug Metabolizing and Excretion Tissue
- Gastric Emptying and Intestinal Motility
- Methods to Study Intestinal Transport and Metabolism
- Transporters for Absorption And Efflux at Apical Membrane
- Route-Dependent Intestinal Metabolism/Excretion
- Intestinal Modeling
- Concluding Remarks and Future Modeling
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters