


Abstract
The nonspecific, noncovalent binding of three drugs, imipramine, warfarin, and propranolol, to pooled human and animal liver microsomes has been determined using equilibrium dialysis in conditions where no cofactor (NADPH) was included in the incubation. The binding of warfarin was dependent upon both protein and drug concentration, whereas the binding of propranolol and imipramine was also dependent upon protein concentration but generally independent of drug concentration. At a microsomal protein concentration of 1.0 mg/ml and a warfarin concentration of 10 μM, the free fraction (fu(mic)) was 0.85. The corresponding values for propranolol and imipramine were 0.41 and 0.16, respectively. Thus, although all three drugs exhibit high binding in plasma (fu<0.1) the acidic drug warfarin differs from the basic drugs propranolol and imipramine in the extent to which each binds to microsomal protein. The binding of all three drugs to liver microsomes obtained from commonly studied animal species (rat, dog, and monkey) was almost identical to that observed in human. Additionally, the binding of warfarin and propranolol to microsomes obtained from insect cells used in baculovirus cytochrome P450 expression systems was similar to that exhibited in liver microsomes, when equal protein concentrations were compared. The enzyme kinetics of propranolol, imipramine, and warfarin oxidative metabolism were determined in pooled human liver microsomes, and the intrinsic clearance values obtained were used in scaling up to project human in vivo clearance. The values obtained by incorporating microsomal binding were compared with those in which this factor is ignored. The findings suggest that the parameter fu(mic) is important to obtain when attempting to relate in vitro intrinsic clearance toin vivo clearance. Also, this value is important to consider when comparing substrates with respect to enzyme specificity, since measured apparent KM values should be converted to true “free KM ” values by correcting for the free fraction in the in vitroincubation. Furthermore, the extent of nonspecific binding to microsomes is likely an important parameter to consider when attempting to relate Ki values measured in vitro to observations of drug-drug interactions (or the lack thereof) in vivo.
Over 20 years ago, physiological models of the process of hepatic extraction of drugs were proposed that stated that hepatic clearance was dependent upon three factors: liver blood flow, blood protein binding, and hepatic intrinsic clearance (1, 2). Furthermore, in vivo intrinsic clearance was defined as the ability of the liver to remove drug if the limitations of protein binding and hepatic blood flow were not applied. A bridge between in vivo intrinsic clearance and the enzyme kinetics of drug metabolism reactions measured in vitro was established which stated that, under the conditions of low substrate (drug) concentrations (i.e.[S]≪KM ), in vitrointrinsic clearance (Cl’int) was simply a ratio of the Michaelis-Menten parametersVmax/KM (3). This in vitro intrinsic clearance value, expressed on a per milligram microsomal protein basis, was multiplied up to reflect the total microsomal protein content in a liver. Thus, the examination of the enzyme kinetics of drug metabolism reactions in vitrobecame a powerful tool whereby the in vivo clearance could be predicted provided that several assumptions concerning clearance mechanisms (metabolism vs. renal or biliary), organs responsible for clearance (liver vs. extrahepatic), and type of metabolism (microsomal oxidative vs. other) were applied (4, 5). However, regular application of this approach to predicthuman clearance has only recently become more prevalent with the increase in the availability of human derived materials (6).
In the course of applying this approach to the study of proprietary compounds, we have observed that in some cases values of clearance predicted from in vitro intrinsic clearance determinations using human and animal liver microsomes were substantially lower than those observed in vivo (7). Furthermore, it was known that the metabolic transformations catalyzed by liver microsomes were predominant in the disposition of the compounds in vivo. In an examination of these data in total, it appeared that the compounds exhibiting such a discrepancy were all characterized by high values of plasma protein binding, and furthermore, all were lipophilic amines. Thus, for this class of compounds either intrinsic clearance values were being very poorly measured (i.e. up to 100-fold errors) or one or more of the assumptions described in the previous paragraph were being erroneously applied. When plasma protein binding values were removed from the well-stirred or parallel tube models for these compounds, scaled-up values for clearance were remarkably close to those measured in vivo (7). Thus, the notion that plasma protein binding does not restrict the hepatic extraction of highly bound drugs, because of rapid on-off rates of drug and protein, was also considered.
In an examination of the Ca2+ channel blocker felodipine, such observations were also made (8). If the plasma protein binding of felodipine is included in the expression describing the well-stirred model of hepatic extraction, along with the value obtained from scaling-up in vitro intrinsic clearance without correcting for binding in the in vitro conditions, the in vivo clearance is drastically underpredicted. In this case, the investigators determined that binding of the drug to thein vitro incubation matrix (liver microsomes) equaled the binding of felodipine to plasma proteins (although values were not reported). They then corrected the in vitro intrinsic clearance value observed by dividing this value by the free fraction in the liver microsomal incubation, which resulted in an overall cancelling out of all protein binding terms in the expression describing the well-stirred model of hepatic extraction.
In light of our observations and the data on felodipine, I have undertaken an examination of the microsomal protein binding of three drugs, warfarin, imipramine, and propranolol, that are well-characterized drugs with respect to pharmacokinetic and metabolic properties. They were chosen because all three represent compounds with low free fractions in plasma (fu ≤ 0.1) and each represents an example of an organic acidic compound (warfarin) or a lipophilic amine base (propranolol and imipramine). The results from these microsomal binding experiments are reported and discussed with regard to predicting in vivo clearance from in vitro intrinsic clearance data.
Materials and Methods
Materials.
Pooled human and animal liver microsomes and microsomes from Sf9 insect cells were prepared using standard methods. The human pool consisted of equal amounts of microsomal protein from 19 individual donors. Protein determinations were made using the BCA assay kit (Pierce, Rockford, IL). [4-3H]Propranolol (28 Ci/mmol), [14C]warfarin (58 mCi/mmol) (both as racemates), and [3H]imipramine (20.0 Ci/mmol) were obtained from Amersham (Arlington Heights, IL). Imipramine,rac-propranolol, and rac-warfarin were obtained from Sigma (St. Louis, MO).
Equilibrium Dialysis Procedure.
Radiolabeled drugs (at various concentrations) were mixed with liver microsomes (at various protein concentrations) in 25 mM KH2PO4, pH 7.5 containing 3.3 mM MgCl2. The mixtures (1.0 ml) were delivered to one side of a dialysis cell (Spectra/POR apparatus, Spectrum Medical Industries, Los Angeles, CA) containing a preconditioned dialysis membrane (Spectra-Por #4; molecular weight cutoff 12–14 kDa). To the other side of the membrane was delivered 1.0 ml of the phosphate buffer containing MgCl2. The cells were sealed and the apparatus was incubated, with the cells rotating, in a water bath maintained at 37°C for 4 to 5 hr. After the incubation period, the dialysates were removed. Portions were added to scintillation vials and the volumes were made equal by the addition of buffer to a total volume of 0.7 ml (to ensure equivalent counting efficiencies for microsome and buffer samples). Ultima Gold scintillation fluid (18 ml, Packard, Meriden, CT) was added and the samples were counted. Unbound fraction was calculated as the ratio:
Enzyme Kinetic Experiments.
Before enzyme kinetic experiments were begun, incubation conditions were established to ensure initial reaction velocity linearity.rac-Propranolol (at concentrations of 0.5 to 100 μM), [4-3H]propranolol (as a tracer), pooled human liver microsomes (1.0 mg/ml), MgCl2 (3.3 mM), and NADPH (1.3 mM) were incubated in a total volume of 0.1 ml 25 mM KH2PO4, pH 7.5. The incubations were commenced with addition of NADPH and incubated for 5 min at 37°C, after which acetonitrile (0.2 ml) was added to precipitate the microsomal protein. The precipitated protein was removed by centrifugation (3000g, 5 min), and an aliquot (120 μl) of the supernatant was injected onto an HPLC.
rac-Warfarin (at concentrations of 0.05 to 1000 μM) mixed with [14C]warfarin, pooled human liver microsomes (3.0 mg/ml), MgCl2 (3.3 mM), and NADPH (1.3 mM) were incubated in a total volume of 0.5 ml 100 mM KH2PO4, pH 7.5. The incubations were commenced with the addition of cofactor and incubated for 20 min at 37°C in a shaking water bath open to the air. The reactions were terminated by the addition of HCl (1M, 0.25 ml) and extracted with methyl t-butyl ether (3 ml). The organic fraction was evaporated under N2 and the residue reconstituted in 100 μl H2O/CH3CN (1:1) for HPLC analysis.
Imipramine (at concentrations of 0.1 to 300 μM) mixed with [3H]imipramine, pooled human liver microsomes (1.0 mg/mL), MgCl2 (3.3 mM), and NADPH (1.3 mM) were incubated in a total volume of 0.2 ml 25 mM KH2PO4, pH 7.5. The incubations were commenced by the addition of the NADPH and incubated for 5 min at 37°C in a shaking water bath open to the air. The reactions were terminated by the addition of 0.2 ml CH3CN, the precipitated material removed by centrifugation, and the supernatant analyzed by HPLC as described below.
HPLC Systems.
The HPLC system used in these studies consisted of a Perkin Elmer (Norwalk, CT) ISS 200 autoinjector, Hewlett-Packard (Palo Alto, CA) 1050 gradient pump, LDC Analytical (Riviera Beach, FL) membrane degasser, and Spectromonitor3200 (LDC Analytical, Riviera Beach, FL) variable wavelength ultraviolet detector, and an Inus β-Ram (Tampa, FL) radiometric flow detector. For measurement of propranolol metabolism, a Kromasil C-18 column (3.2 × 150 mm) was used. The mobile phase consisted of H2O/CH3OH/CH3CN/CH3COOH (50:25:25:0.1) at a flow rate of 1.0 ml/min. Tru-Count scintillation fluid (Inus) was used at a flow rate of 5 ml/min. Under these conditions, [3H]propranolol eluted at 4 min and all radiolabeled metabolites eluted between the void volume and 3.6 min. For warfarin metabolism, a Novapak C18 column (3.2 × 150 mm) was used with a mobile phase of 20 mM CH3COOH, pH 4.8 with NH4OH/CH3CN (65:35) at a flow rate of 1.0 ml/min. Scintillation fluid was used as before. Under these conditions, [14C]warfarin eluted at 3.5 min and all radiolabeled metabolites eluted between 1 and 3 min. For imipramine metabolism, a Zorbax Phenyl column (4.6 × 150 mm) was used with a mobile phase of 20 mM HClO4 (adjusted to pH 2.5 with NaOH)/CH3CN (50:50) at a flow rate of 1.0 ml/min. Imipramine eluted at 4.7 min and the two metabolites observed eluted at 3.1 and 4.2 min.
Enzyme Kinetic Calculations.
Apparent enzyme kinetics were determined by fitting reaction velocityvs. substrate concentration data. Initially, the data were examined on double reciprocal and Eadie-Hofstee plots to assess the potential for multiplicity of enzymes. (Curved plots suggested >1 enzyme was involved.) The Michaelis-Menten plots were then fit to expressions containing one or more Michaelis-Menten terms. Residual values for each of the data points were calculated and plottedvs. substrate concentration to ensure a random distribution of residuals.
Results
Binding of Warfarin, Imipramine, and Propranolol to Human Liver Microsomes.
Warfarin, imipramine, and propranolol demonstrated noncovalent binding to pooled human liver microsomes when subjected to equilibrium dialysis. Binding experiments were conducted at 37°C to mimic conditions used in in vitro microsomal metabolism studies but were conducted in the absence of NADPH so that metabolism of the compounds would not occur. Recovery of radiolabeled drugs was consistently 90% or better, thus nonspecific binding to the equilibrium dialysis membranes and/or apparatus was minimal. Initial time course experiments demonstrated that equilibrium was achieved by 4 hr (fig. 1). Adjustments to the determinations of free and bound drugs because of volume shifts during the dialysis experiment (caused by osmotic flow) were not necessary since in almost all cases such shifts were not observed to be significant. (Although volume shifts are commonly observed when conducting equilibrium dialysis experiments with plasma, this was not readily observed in the experiments conducted with microsomes, likely due to the fact that the protein concentration, and hence osmotic force, is substantially greater in plasma than in the drug solutions in microsomes.)

Equilibrium dialysis of warfarin, imipramine, and propranolol vs. pooled human liver microsomes. [14C]Warfarin (•) at 10 μM and [3H]propranolol (▪) and [3H]imipramine (▴) at 1.0 μM were dialyzed vs. human liver microsomes (1.0 mg/ml) for various time periods.
The binding of warfarin to human microsomes was dependent on microsomal protein concentration; the free fraction decreased from a value of 0.99 to 0.47 as the protein concentration was raised from 0.1 to 10 mg/ml (fig. 2). At microsomal protein concentrations typically used in metabolic incubations (1.0 to 3.0 mg/ml), the warfarin free fraction was 0.85 to 0.73. Equilibrium dialysis of microsomes with propranolol and imipramine also showed a dependence on protein concentration (fig. 2). Compared with warfarin, propranolol was more highly bound to microsomes with free fraction values ranging from 0.83 to 0.13 as the protein concentration increased from 0.1 to 10 mg/ml. The binding of imipramine was greater still, with free fractions ranging from 0.34 to 0.05 over the same microsomal protein concentration range. At a microsomal protein concentration of 1.0 mg/ml, which is commonly used in metabolic incubations for propranolol and imipramine, the free fractions were 0.38 and 0.16, respectively, compared to 0.85 for warfarin.

Binding of warfarin, imipramine, and propranolol to pooled human liver microsomes as a function of microsomal protein concentration. [14C]Warfarin (▪) at 10 μM and [3H]propranolol (•) and [3H]imipramine (▴) at 1.0 μM were dialyzedvs. human liver microsomes for 5 hr. Points represent the mean ± SD of triplicate determinations.
The binding of warfarin to pooled human liver microsomes as a function of warfarin concentration is shown in fig.3. With increasing warfarin concentrations from 1.0 to 100 μM, the free fraction increased from 0.72 to 0.95 (at a microsomal protein concentration of 1.0 mg/ml). For propranolol, the extent of binding was independent of the concentration of drug (fu(mic) ∼ 0.40) in the concentration range of 1.0 to 100 μM (fig. 3). Imipramine showed a small dependence on drug concentration (fu(mic) = 0.14 to 0.22) over the 100-fold range in concentration.

Binding of warfarin, imipramine, and propranolol to pooled human liver microsomes as a function of drug concentration. [14C]Warfarin (▪), [3H]imipramine (▴), and [3H]propranolol (•) at 1.0–100 μM were dialyzed vs. human liver microsomes (1.0 mg/ml) for 5 hr. Points represent the mean ± SD of triplicate determinations.
Binding of Warfarin, Imipramine, and Propranolol to Animal Liver and Sf9 Cell Microsomes.
The binding of warfarin, imipramine, and propranolol to liver microsomes from rat, dog, and monkey, three commonly used preclinical species in drug metabolism studies, compared with that measured in human liver microsomes was similar (fig.4).

Binding of propranolol, imipramine, and warfarin to animal liver microsomes. [3H]Propranolol, [3H]imipramine (each at 1.0 μM) and [14C]warfarin (at 10 μM) were dialyzedvs. animal liver microsomes (1.0 mg/ml protein concentration) for 5 hr. Each bar represents the mean (± SD) of triplicate determinations.
Microsomes from Sf9 cells, an insect cell line used in baculovirus expression systems for cytochrome P450 enzymes, were examined for nonspecific binding to warfarin and propranolol, since this in vitro system is becoming a commonplace tool in drug metabolism studies. The binding of warfarin and propranolol to Sf9 cell microsomes that lack P450 enzymes is presented in fig.5. The binding of propranolol (1.0 μM) to these microsomes closely paralleled that observed in pooled human liver microsomes when comparing equal protein concentrations. The free fraction decreased from 0.88 to 0.11 as the protein concentration increased from 0.1 to 10 mg/ml. The binding of warfarin (10 μM) to Sf9 cell microsomes was somewhat weaker than binding to human liver microsomes with free fractions ranging from 0.98 to 0.82 as the protein concentration increased from 0.1 to 10 mg/ml (fig. 5).

Binding of propranolol and warfarin to Sf9 insect cell microsomes. [3H]Propranolol (1.0 μM) and [14C]warfarin (10 μM) were dialyzed vs.Sf9 insect cell microsomes commonly used as a cytochrome P450 expression system for 5 hr. Each point represents the average of duplicate determinations.
Binding of Warfarin, Imipramine, and Propranolol to Serum Proteins and Partitioning Between Serum and Microsomes.
Equilibrium dialysis of warfarin (10 μM), propranolol (1.0 μM), and imipramine (1.0 μM) vs. human serum yielded free fraction values (table 1), consistent with those listed in the literature (9-12). When these values were combined with free fraction values measured in human microsomes (fu(serum)/fu(microsomes)), serum-microsome partitioning values were calculated (table 1). Experimentally determined values for partitioning between serum and microsomes were obtained by dialyzing the three drugs in human serumvs. human liver microsomes on opposing sides of the dialysis membrane. The experimentally determined values for partitioning of propranolol and imipramine between serum and microsomes were similar to those calculated from individually determined free fractions in serum and microsomes (table 1). The corresponding experimentally determined value for warfarin (0.033) was substantially different from that calculated from dividing fu(serum) by fu(microsomes) (0.0094).
Partitioning of propranolol, imipramine, and warfarin between serum and microsomes
Enzyme Kinetics of Warfarin, Propranolol, and Imipramine Metabolism by Pooled Human Liver Microsomes.
The enzyme kinetics of total metabolism of rac propranolol in pooled human liver microsomes were measured (fig.6). An abbreviated HPLC assay was applied using radiolabeled propranolol that provided a separation of parent compound from all metabolites, which were not baseline resolved from each other. This approach was used in this study because the objective of the experiment was to yield enzyme kinetic data for total propranolol consumption and not the formation of individual metabolites. The data fit to a function comprising the sum of two Michaelis-Menten terms. However, the activity of the second enzyme of higher KM
was best described as Cl′int2 · [S]:
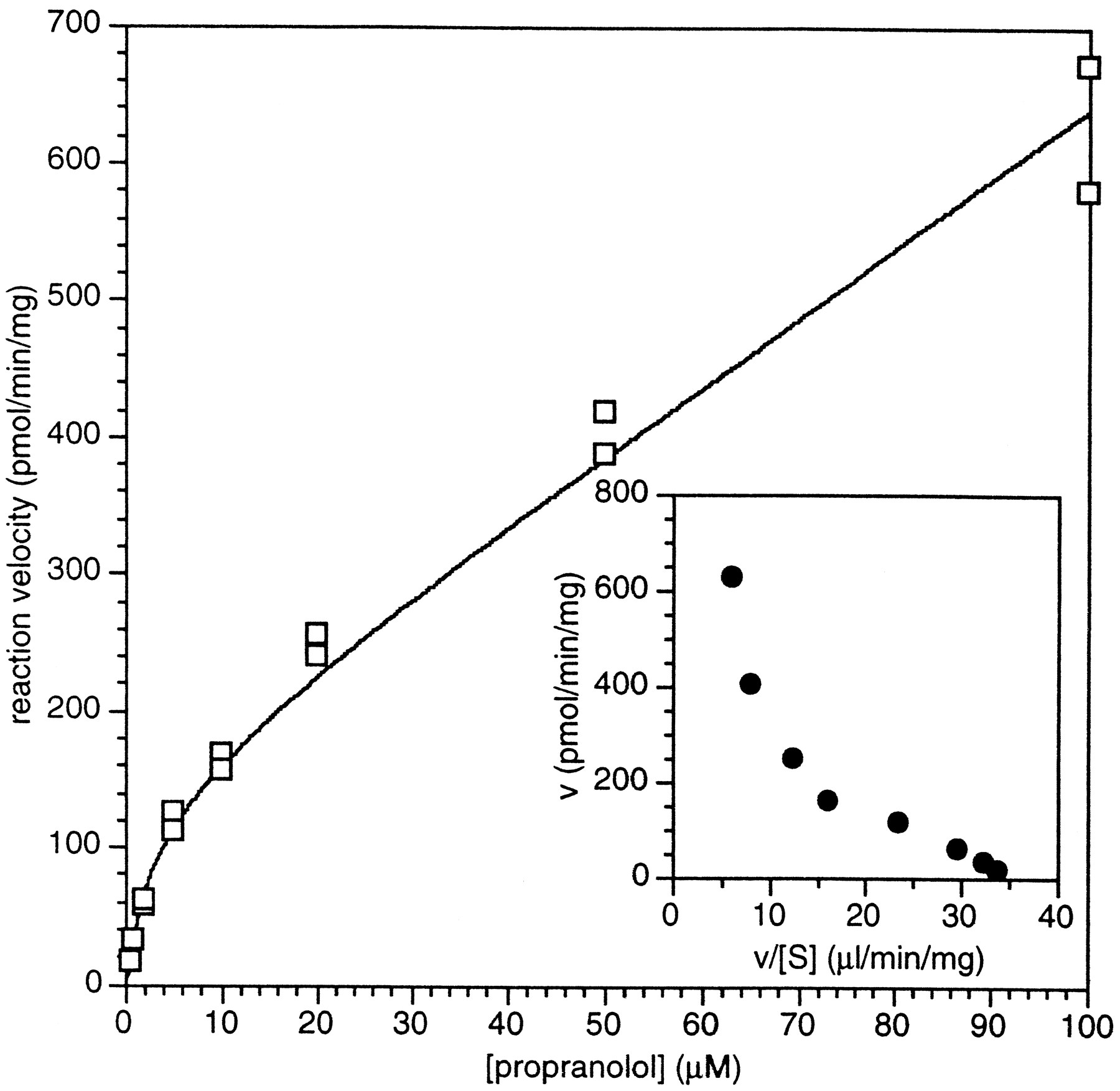
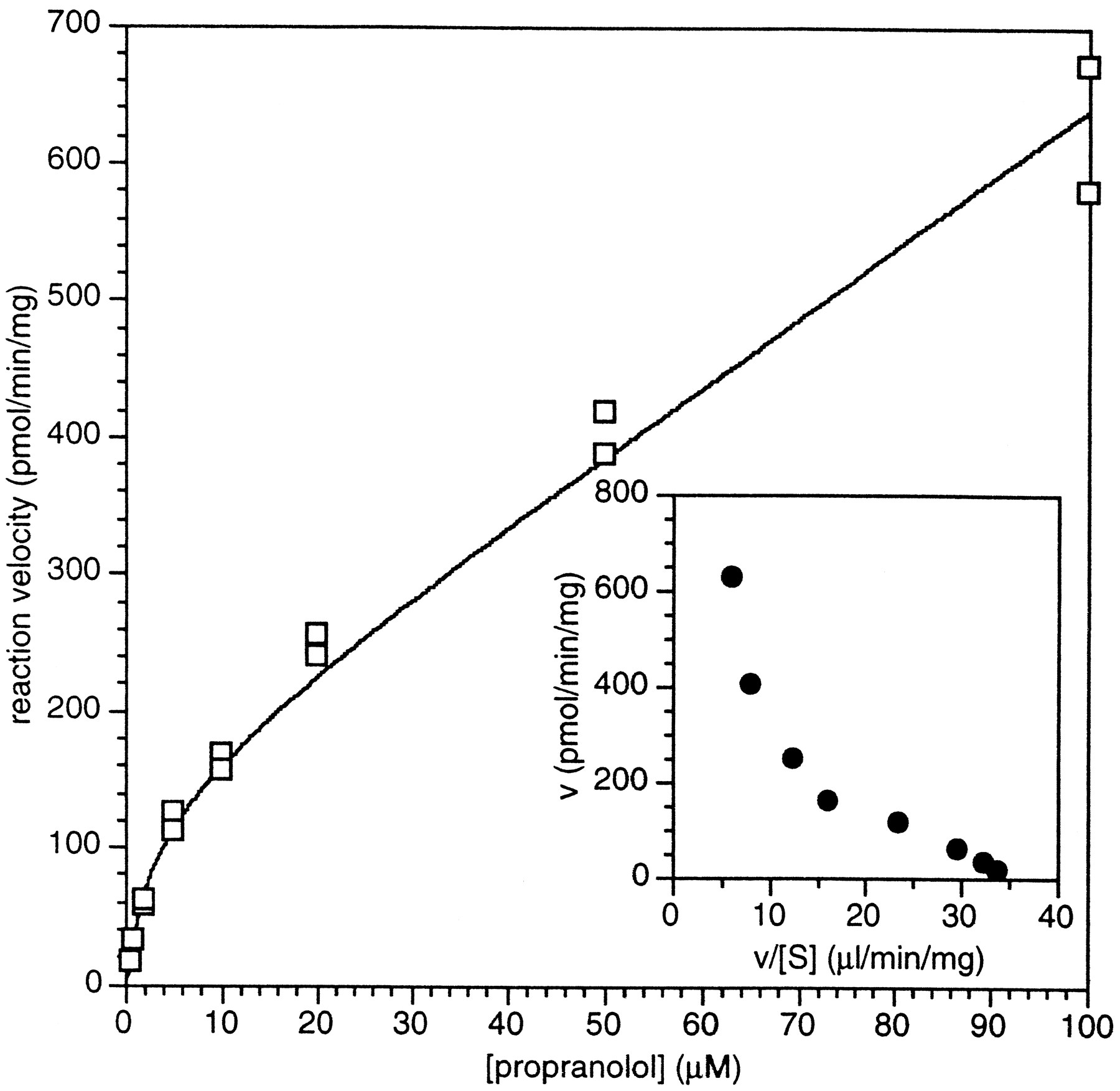
Substrate saturation plot of totalrac-propranolol metabolism by pooled human liver microsomes.
Racemic [3H]propranolol was incubated in the presence of pooled human liver microsomes and NADPH as described inMethods. Incubations were done in duplicate and individual values are presented. The data was fit to the function: representing the sum of two component enzyme activities with low and high KMapp values. Values determined were Vmax1 = 144 pmol/min/mg;KMapp1 = 3.0 μM; Cl’int2 = 0.005 mL/min/mg microsomal protein. Inset figure represents an Eadie-Hofstee plot of averaged data.
representing the sum of two component enzyme activities with low and high KMapp values. Values determined were Vmax1 = 144 pmol/min/mg;KMapp1 = 3.0 μM; Cl’int2 = 0.005 mL/min/mg microsomal protein. Inset figure represents an Eadie-Hofstee plot of averaged data.
Summary of enzyme kinetic data for rac-propranolol, imipramine, and rac-warfarin metabolism in human liver microsomes
The enzyme kinetic data of rac-warfarin metabolism in human liver microsomes are shown in fig. 7. As with propranolol, radiolabeled drug was used with a rapid, isocratic HPLC system designed to distinguish unchanged drug from metabolites. Resolution of individual metabolites was not sought, but rather total overall metabolism of warfarin was measured using this approach. The data were described by the presence of low and highKM
enzyme activities using the equation:

Substrate saturation plot of total rac-warfarin metabolism by pooled human liver microsomes.
Racemic [14C]warfarin was incubated in the presence of pooled human liver microsomes and NADPH as described inMethods. Incubations were done in duplicate and individual values are presented. The data was fit to the function: representing the sum of two component enzyme activities with low and high KM
values. Values determined wereVmax1 = 1.0 pmol/min/mg;KMapp1 = 2.0 μM;Vmax2 = 121 pmol/min/mg;KMapp2 = 150 μM. The inset figure represents an Eadie-Hofstee plot of averaged data.
representing the sum of two component enzyme activities with low and high KM
values. Values determined wereVmax1 = 1.0 pmol/min/mg;KMapp1 = 2.0 μM;Vmax2 = 121 pmol/min/mg;KMapp2 = 150 μM. The inset figure represents an Eadie-Hofstee plot of averaged data.
The substrate saturation plot for [3H]imipramine metabolism by human liver microsomes is in fig. 8. Biphasic kinetics were also observed for this compound withKMapp values of 2.5 and 550 μM (table 2). (The KMapp value of 550 μM represents an estimate, since the highest imipramine concentration examined was 300 μM. However, the data fit better to a sum of two Michaelis-Menten terms as opposed to the sum of a Michaelis-Menten term and an intrinsic clearance term.) The total Cl’int for imipramine was identical to that of propranolol (table 2).
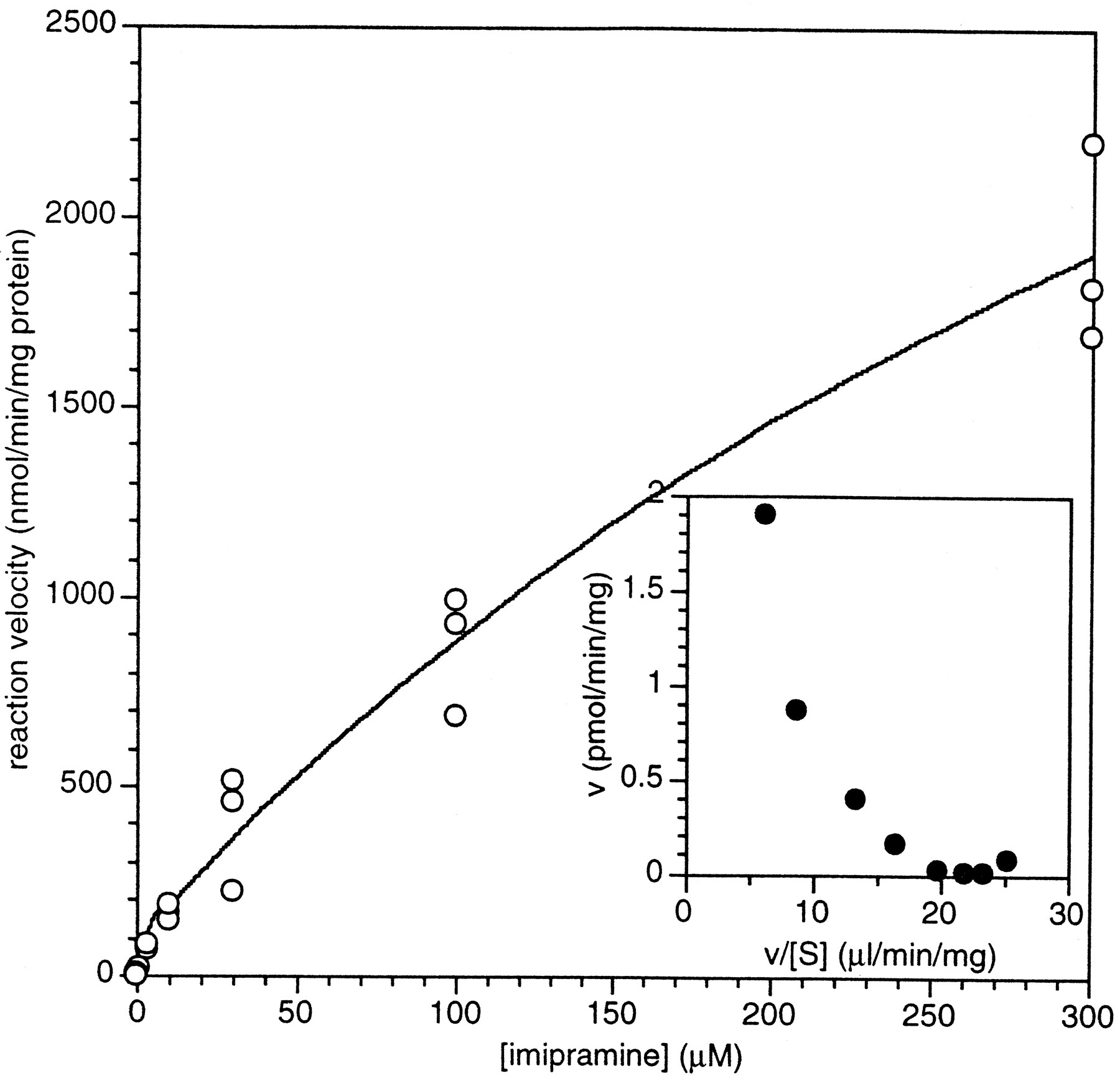
Substrate saturation plot of total imipramine metabolism by pooled human liver microsomes.
[3H]Imipramine was incubated in the presence of pooled human liver microsomes and NADPH as described inMethods. Incubations were done in triplicate and individual values are presented. The data was fit to the function: representing the sum of two component enzyme activities with low and high KM
values. Values determined wereVmax1 = 110 pmol/min/mg;KMapp1 = 2.5 μM;Vmax2 = 5100 pmol/min/mg;KMapp2 = 550 μM. The inset figure represents an Eadie-Hofstee plot of mean data.
representing the sum of two component enzyme activities with low and high KM
values. Values determined wereVmax1 = 110 pmol/min/mg;KMapp1 = 2.5 μM;Vmax2 = 5100 pmol/min/mg;KMapp2 = 550 μM. The inset figure represents an Eadie-Hofstee plot of mean data.
Microsomal Binding Impact on in Vitro-in Vivo Correlation.
For propranolol, scaling-up of the in vitro intrinsic clearance value to reflect intrinsic clearance on a per kilogram body weight basis for human yielded a value of 48 ml/min/kg. Insertion of this value into equations describing hepatic clearance (well-stirred and parallel-tube equations) with and without blood and microsomal binding yielded values listed in table 3. When all protein binding is disregarded (or assuming fu(blood) = fu(microsomes)), values for predicted clearance matched or exceeded the actual blood clearance in vivo(table 3, eqs. 1 and 5). If the value for free fraction in blood was included, but the binding to microsomes was disregarded (table 3, eqs.2 and 6), clearance values estimated from in vitro intrinsic clearance were low (∼ 5 ml/min/kg). Accounting for both free fraction in blood and free fraction in microsomes (at a protein concentration of 1.0 mg/ml that was used in enzyme kinetic experiments) provided predicted blood clearance values equal to or slightly under the actual blood clearance value of 12–16 ml/min/kg (table 3, eqs. 3, 4, 7, and 8). Values ranged from 8–11 ml/min/kg, with the parallel tube equation having higher values than the well-stirred equation.
Human blood clearance values obtained by scaling of in vitrointrinsic clearance data using equations describing the well-stirred and parallel tube models of hepatic clearance and including or excluding serum protein and microsome binding
Scaling the in vitro intrinsic clearance value for imipramine yielded a predicted in vivo intrinsic clearance value of 48 ml/min/kg (table 3). If serum protein binding is included in the relationship between intrinsic clearance, but the binding to microsomes is not included, significant underpredictions of clearance are made (2.2–2.4 ml/min/kg) compared with the reported mean human blood clearance (14 ml/min/kg; reference 13) (table 3). Including the free fraction in microsomal incubations (0.16 at 1.0 mg protein/ml) or using the experimentally determined microsome/serum partitioning value yielded predicted clearance values (8.6 to 11 ml/min/kg).
For warfarin, disregarding all protein binding (table 3, eqs. 1 and 5) yielded a predicted human clearance value well in excess of that measured in vivo. However, including protein binding (blood or blood and microsomal) yielded predicted values below the actual value (eqs. 2, 3, 6, and 7). Using the serum/microsome partitioning data gave the closest prediction of clearance (table 3, eqs. 4 and 8).
Discussion
Methods whereby human clearance can be predicted from in vitro drug metabolism studies are becoming increasingly used in the drug discovery process. The drug metabolism scientist faces the task of selecting compounds for further development that are expected to possess desired human pharmacokinetic properties. In vitro methods using human derived reagents are becoming a regular component of compound selection strategies. While the mathematics of these methods have been in the scientific literature for many years (1-3), they are only becoming of increased importance because, in large part, of the recent increase in the availability of high-quality human tissues suitable for such studies. Two recent comprehensive reviews of the process of scaling-up of in vitro metabolism data to in vivo clearance have described theoretical and practical aspects (4, 5). The conclusion made in these reviews was that clearance in preclinical species and humans can be predicted using liver microsomes (or better still, hepatocytes). Furthermore, whereas the notion of the possible importance of binding of drugs in in vitro incubation matrices was raised (4), it was proposed that this factor would not be expected to have a significant impact on the scale-up of in vitro intrinsic clearance data to in vivo clearance values because of the typically low concentrations of protein used in in vitro metabolism experiments relative to protein concentrations present in plasma (or serum). However, to date, there have been little data in the scientific literature that specifically addresses the potential impact of nonspecific binding inin vitro matrices on the relationship between in vitro Cl’int and in vivoclearance (7). There have been a few instances in which this factor has been addressed, albeit as a minor point (8, 14-17). In a recent report, an alternative approach of including albumin in the microsomal incubation of phenytoin yielded an improved prediction of in vivo pharmacokinetic parameters (18). In work in our laboratory, we have found that many proprietary compounds were highly bound (>90%) to liver microsomes at protein concentrations typically used in enzyme kinetic experiments (1.0–5.0 mg/ml). Including this binding factor provided good agreement between clearance estimated from in vitro Cl’int and clearance measured in vivo. Thus, the purpose of these experiments was to characterize the nonspecific binding of warfarin, propranolol, and imipramine to liver microsomes and to examine the potential impact that such binding has on the prediction of in vivo clearance from in vitro intrinsic clearance data for these well characterized compounds.
Propranolol, imipramine, and warfarin were chosen for this investigation since these represent examples of drugs in which much knowledge exists on the pharmacokinetics and disposition in humans. The method of equilibrium dialysis was chosen for the determination of nonspecific binding to microsomes because this method is least subject to the potential artifact of nonspecific binding to components of the apparatus (common to ultrafiltration and ultracentrifugation techniques). Initial time course experiments established that equilibrium was attained after 4 hr. Dialysis was conducted in the absence of cofactor (NADPH) necessary for P450 catalyzed metabolism to ensure stability of the compounds throughout the dialysis period.
The data described in this report demonstrate that compounds can be extensively and nonspecifically bound to microsomes at microsomal protein concentrations in the range of those used in in vitro drug metabolism studies. Furthermore, the value for fraction unbound under in vitro incubation conditions can have a significant impact on the prediction of clearance from in vitro intrinsic clearance data (see below). The binding of all three compounds increased with increasing microsomal protein concentration through the range of 0.1 to 10 mg/ml microsomal protein. However, whereas the binding of warfarin also demonstrated a dependence on drug concentration, the binding of imipramine and propranolol was mostly independent of drug concentration through the range of 1.0 to 100 μM. Binding was similar across species and sources (i.e. Sf9) of microsomes.
In addition to their utility in the process of relating in vitro Cl’int to in vivoclearance (see below), these binding data are interesting to compare from a physicochemical standpoint, as both amines (propranolol and imipramine) were substantially bound to microsomes while the organic acid (warfarin) did not bind extensively. This is consistent with previous data that demonstrated that lipophilic amines (e.g.chlorpromazine) bound to the lipid fraction of microsomes while organic acids did not (19, 20). However, the fact that warfarin binds poorly while propranolol and imipramine bind well does not support the assumption that microsomal binding phenomena could be ignored for all organic acids but considered important for all neutral compounds and organic bases. Clearly, more data for other compounds are necessary. A more extensive examination of the relationship between physicochemical properties (i.e. lipophilicity, acid vs. basevs. neutral, hydrogen bonding potential) and microsomal binding is in progress.
Enzyme kinetic data for the oxidative metabolism of propranolol, imipramine, and warfarin by pooled human liver microsomes were collected for the purpose of correlating in vivo human clearance values with predictions of human clearance from in vitro intrinsic clearance data. While some enzyme kinetic data already existed for each of the three compounds (21-27), these data were reexamined to obtain data for total substrate consumption using racemic drugs (when applicable) and a pool of human liver microsomal samples from 19 individual donors (more representative of an “average”). Such data are more appropriate to scaling in vitro Cl’int to in vivoclearance since the literature data are primarily focussed only on individual metabolic reactions (e.g. propranolol 4-hydroxylation; warfarin 7-hydroxylation, etc.) for specific enantiomers, using fewer human liver microsomal samples. Also, some disparity exists in different literature reports with regard to enzyme kinetic values, especially for imipramine (25-27). The examination of racemic mixtures (for warfarin and propranolol) in this study was purposely done to best mimic the in vivo situation since these compounds are administered as racemic mixtures, despite information showing some differences in the in vivodisposition of individual enantiomers (28-30). Thus, the potential inhibition of metabolism of one enantiomer by the other (31), which could occur in vivo, would also be accounted for in thein vitro experiments.
The enzyme kinetic data for all three compounds were biphasic, suggesting that multiple enzyme activities (i.e. lowKM /low capacity and highKM /high capacity components) contributed to total metabolism of each compound. Intrinsic clearance values (Vmax/KM ) were summed to yield total in vitro intrinsic clearance values which were then scaled up to reflect the intrinsic clearance value on a per kilogram body weight basis (using scaling factors of 45 mg microsomal protein per gm liver and 20 g liver per kg body weight). Propranolol and imipramine were high intrinsic clearance compounds whereas warfarin was a low intrinsic clearance compound (table 2).
Propranolol, imipramine, and warfarin represent interesting cases whenin vitro metabolism data are scaled-up to in vivoclearance data. All three compounds are highly bound to serum proteins (fu ≤ 0.1). For propranolol, disregarding all binding values led to good predictions of in vivo clearance (table 3, eqs. 1 and 5). However, disregarding protein binding is inappropriate and challenges well-established relationships between serum protein binding and clearance (1, 2). Including only plasma protein binding resulted in a substantial underestimation of clearance. Inclusion of both serum and microsomal binding values yielded a projection of human blood clearance of 9.2 to 11 ml/min/kg (table 3, eqs. 3 and 7), representing a slight underestimate of actual clearance (12–16 ml/min/kg; refs. 9, 28, 32). However, an underestimate should be observed for the projection of propranolol clearance when using liver microsomes since other contributions to clearance (e.g. 10–20% of metabolism as direct glucuronidation), exist for this compound (33, 34) and the potential exists for inhibition of metabolism by the formed metabolites (4, 35, 36). Imipramine represents a case similar to propranolol. Inclusion of the free fraction of imipramine in the microsomal incubation resulted in a predicted value for human clearance of 8.6 to 11 ml/min/kg (table 3, eqs. 3 and 7): a slight underestimate of reported mean human blood clearance values (14–15 ml/min/kg; (13)). Disregarding the binding in microsomes and including only the value for protein binding in blood yielded substantial underestimates of in vivo clearance (2 ml/min/kg; table 3, eqs. 2 and 6). Thus the case of imipramine, along with the data for propranolol, supports the notion that nonspecific binding in in vitro incubations has an impact on the projection of in vivo clearance from in vitrointrinsic clearance data.
For warfarin, completely ignoring all protein binding in the projection of in vivo clearance from in vitro intrinsic clearance clearly provided a poor agreement (1.1 vs. 0.081 ml/min/kg; table 3, eqs. 1 and 5). However, including only blood protein binding yielded an underprediction of nearly 5-fold (table 3, eqs. 2 and 6). Warfarin was not extensively bound to liver microsomes at protein concentrations (3.0 mg/ml) used in the determination of enzyme kinetic values; thus the in vitro data still provided an underprojection (3.5-fold) of in vivo blood clearance, even when binding to microsomes was included (table 3, eqs. 3 and 7). While some of the considerations analogous to those described above for propranolol could account for some underprediction (e.g.alternative non-P450 mediated route of warfarin metabolism (24, 37)), it does not seem that an underprediction of such magnitude could be accounted for by these factors. Interestingly, the closest projection of in vivo clearance comes from using the warfarin serum-microsome partition value obtained from direct dialysis of these two matrices (table 3, eqs. 4 and 8). However, this observation is strictly empirical at this point, and this experimental approach may be fraught with artifacts. The general applicability of determining values in such a manner requires further investigation.
In summary, this work demonstrates the importance of nonspecific binding to liver microsomes in understanding the relationship betweenin vitro metabolism data and in vivo clearance. It is believed that the principles explored for the prototype compounds imipramine, propranolol, and warfarin are generally applicable to other drugs and inhibitors. It is advocated that the free fraction inin vitro metabolism experiments be determined and included in the process of scaling-up in vitro drug metabolism data to in vivo clearance data. The phenomenon of nonspecific binding to microsomes may also contribute to disparities betweenKM (as well asKi ) values measured in different in vitro systems (e.g. liver microsomes vs.heterologously expressed recombinant CYP) and reported by different laboratories, since the extent of nonspecific binding may vary with different experimental conditions. This notion merits further investigation.
Acknowledgments
The author extends gratitude to Drs. Larry Tremaine, Robert Ronfeld, and Donald Tweedie for helpful discussions concerning this work.
Footnotes
-
Send reprint requests to: Dr. R. Scott Obach, Drug Metabolism Department, Central Research, Pfizer, Inc., Groton, CT 06340. E-mail: obachr{at}pfizer.com.
- Received April 25, 1997.
- Accepted August 19, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}