


Abstract
The involvement of flavin-containing monooxygenases (FMOs) in the formation of xanomeline N-oxide was examined in various human and rat tissues. Expressed FMOs formed xanomelineN-oxide at a significantly greater rate than did expressed cytochromes P-450. Consistent with the involvement of FMO in the formation of xanomeline N-oxide in human liver, human kidney, rat liver, and rat kidney microsomes, this biotransformation was sensitive to heat treatment, increased at pH 8.3, and inhibited by methimazole. The latter two characteristics were effected to a lesser extent in human kidney, rat liver, and rat kidney microsomes than were observed in human liver microsomes, suggesting the involvement of a different FMO family member in this reaction in these tissues. As additional proof of the involvement of FMO in the formation of xanomeline N-oxide, the formation of this metabolite by a characterized human liver microsomal bank correlated with FMO activity. The FMO forming xanomeline N-oxide by human kidney microsomes exhibited a 20-fold lowerKM (averageKM = 5.5 μM) than that observed by the FMO present in human liver microsomes (averageKM of 107 μM). The involvement of an FMO in the formation of xanomeline N-oxide in rat lung could not be unequivocally demonstrated. These data and those in the literature suggest that the increased prevalence ofN-oxidized metabolites of xanomeline after s.c. dosing as compared with oral dosing may be due to differences in the affinity of various FMO family members for xanomeline or to differences in exposure to xanomeline that these enzymes receive under different dosing regimens.
Xanomeline is a selective M1-muscarinic agonist that was being developed for the symptomatic treatment of Alzheimer’s disease. After oral administration to human volunteers (healthy males), xanomeline was rapidly absorbed and exhibited significant first pass metabolism withN-oxidation of the tetrahydropyridine ring as one of the major routes of metabolism (Shipley et al., 1993) (Fig.1). Similar metabolism of xanomeline was observed after a single oral dose to rats (Cornpropst et al., 1993). Interestingly, rats and monkeys dosed s.c. had an altered pharmacokinetic profile, with xanomeline exhibiting a slow sustained release. This was felt to be due to a depot effect caused by dosing xanomeline in a peanut oil vehicle. Furthermore, a greater prevalence of N-oxidized metabolites was observed after s.c. dosing as compared with the metabolite profile observed after oral exposure to xanomeline (Shipley et al., 1995).
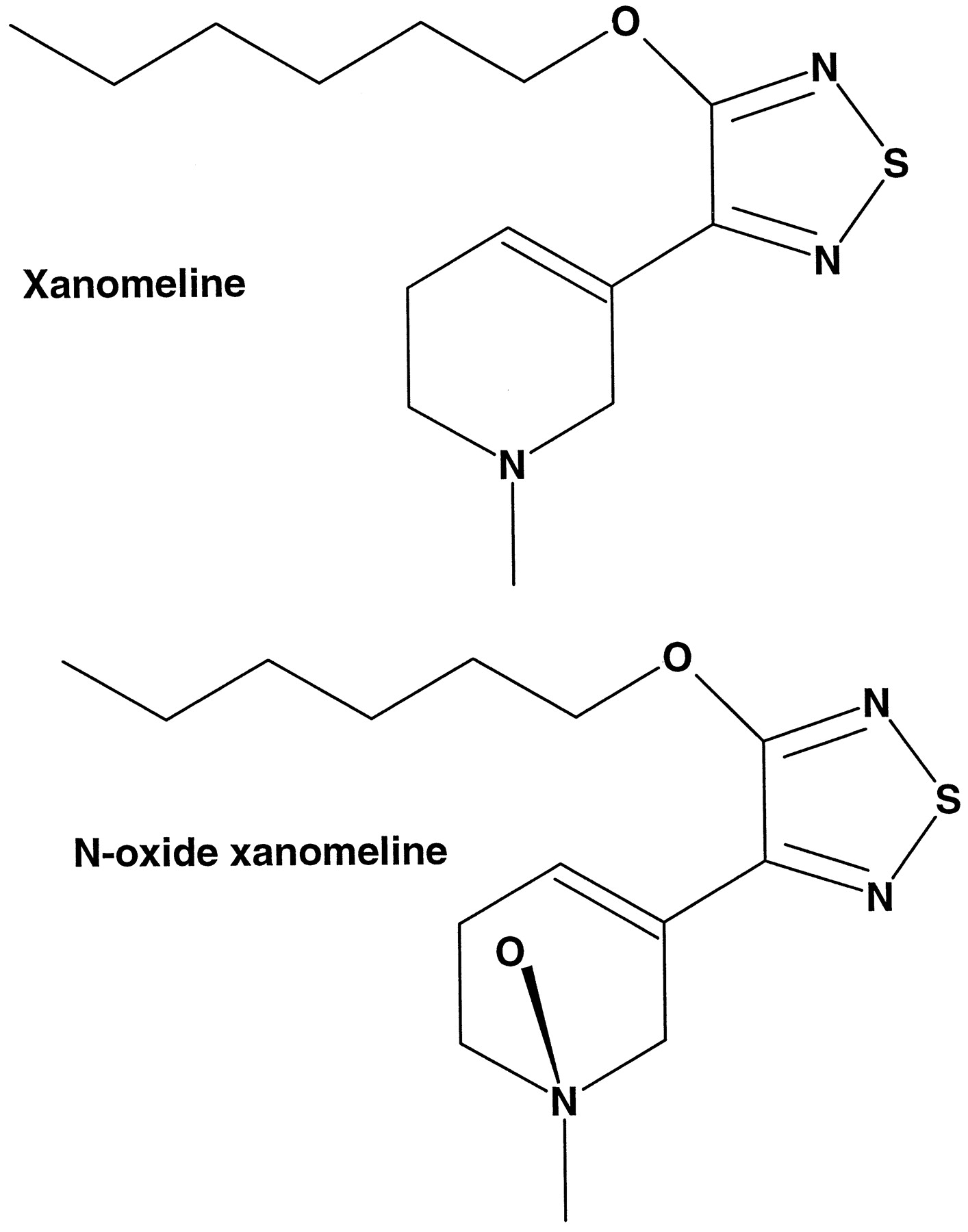
Structure of xanomeline and xanomeline N-oxide.
The flavin-containing monooxygenases (FMOs)1 are enzymes that catalyze NADPH-dependent oxygenation of nucleophilic nitrogen, sulfur, or phosphorus containing functional groups of xenobiotics. Five distinct forms of FMO have been identified (FMO1 through FMO5) that are expressed in a tissue- and species-specific manner (Cashman, 1995; Phillips et al., 1995). Human FMOs exhibit 82 to 88% sequence identity with their orthologs from other species, but only 51 to 57% sequence identity with each other (Phillips et al., 1995). Although FMOs often exhibit broad and overlapping substrate specificity, there are substrate specificities (Hines et al., 1994) and enzymatic properties among the different FMO families that distinguish them from each other (Burnett et al., 1994;Itagaki et al., 1996). The goal of this project was to determine whether the FMOs were responsible for the formation of xanomelineN-oxide. Furthermore, due to the greater prevalence ofN-oxidized metabolites of xanomeline after s.c. dosing as compared with oral dosing, metabolism of xanomeline in both hepatic and extrahepatic tissues was examined.
Materials and Methods
Chemicals.
Xanomeline and xanomeline N-oxide were synthesized at Eli Lilly and Co. (Indianapolis, IN) (Fig. 1). Furafylline andS-mephenytoin were obtained from Ultrafine Chemicals (Manchester, England). Methimazole, quinidine, sulfaphenazole, and NADPH were obtained from Sigma Chemical Co. (St. Louis, MO). Ketoconazole was obtained from Janssen Pharmaceutica, Inc. (Beerse, Belgium). Coumarin and n-octylamine were obtained from Aldrich (Milwaukee, WI). Troleandomycin was obtained from Pfizer (Groton, CT).
Microsomal Samples.
Human liver samples designated HLA through HLN were obtained from either the liver transplant unit at the Medical College of Wisconsin (Milwaukee, WI) or the Medical College of Virginia (Richmond, VA) under protocols approved by the appropriate committee for the conduct of human research. Liver, kidney, and lung tissue from Fisher 344 rats were either immediately processed into microsomes or snap frozen in liquid nitrogen and stored at −70°C before microsomal preparation. Microsomes in these tissues were prepared by differential centrifugation (van der Hoeven and Coon, 1974). The microsomes prepared from the human liver samples HLA through HLN (n = 14) were previously characterized for their relative levels of cytochromes P-450 (CYPs) and FMO through the use of form-selective catalytic activities (CYP1A2, phenacetin O-deethylase activity; CYP2A6, coumarin 7-hydroxylase; CYP2C9, tolbutamide 4-hydroxylase; CYP2C19, S-mephenytoin 4′-hydroxylase; CYP2D6, bufuralol 1′-hydroxylase; CYP3A4, midazolam 1′-hydroxylase; CYP2E1, chlorzoxazone 6-hydroxylase; FMO, nicotine N′-oxidation) (Wrighton et al., 1993b; Hall et al., 1994). Human kidney samples designated IUK2, IUK4, IUK5, and IUK7 were obtained at Indiana University Hospital (Indianapolis, IN) under a protocol approved by the Institutional Review Board. Microsomes were prepared by using differential centrifugation (Haehner et al., 1996). A human lung microsomal sample was obtained from IIAM (Exton, PA). Microsomes made from a human B-lymphoblastoid cell line engineered to express CYPs or a baculovirus cell line engineered to express FMO1 or FMO3 were obtained from Gentest Corp. (Woburn, MA).
HPLC Analysis.
For analysis of xanomeline N-oxide, a 150 × 4.6 mm YMC Basic column (YMC, Inc., Wilmington, NC) with a column heater set at 35°C and UV detection at 295 nm was used. For the analysis of samples generated after incubations with microsomes made from human tissue or cDNA-expressed enzymes, a gradient was used for chromatography in which mobile phase was delivered at 1 ml/min, with variations of the following conditions: 0 to 6 min, 0.05 M ammonium acetate, pH 6.5: acetonitrile (65:35, v/v); 5 to 12 min, 0.05 M ammonium acetate, pH 6.5: acetonitrile/methanol (20:70:10, v/v/v). Samples generated after incubations with microsomes prepared from rat tissues were detected with a chromatographic method that used isocratic mobile phase conditions where mobile phase, delivered at 1 ml/min, consisted of 0.05 M ammonium acetate buffer (pH 6.5)/methanol/acetonitrile (15:15:70, v/v/v).
Kinetic Studies.
Incubations with microsomes prepared from human tissue for studies examining the kinetics of formation of xanomeline N-oxide were performed under initial rate conditions for the formation of this metabolite. In these studies, microsomes were preincubated at 37°C for 3 min in 0.1 M sodium phosphate buffer (pH 7.4 or 8.3) and 1 mM NADPH before the initiation of the reaction with the addition of 0.59 to 585 μM xanomeline (for incubations with microsomes from HLC, HLM, IYK2, IYK4, and IYK7) or 1.17 to 585 μM xanomeline (for incubations with microsomal sample HLB). The reactions were stopped with the addition of an equal volume of solvent, and the denatured protein was removed by centrifugation. The resulting supernatant fraction was subjected to HPLC analysis. Kinetic analyses of the formation of xanomeline N-oxide were initially evaluated by visual examination of Eadie-Hofstee plots to assess whether one or more enzymes were involved in its formation. The kinetic parameters for the formation of xanomeline N-oxide were then determined after fitting the data (Ring et al., 1996) to the appropriate kinetic model (Segel, 1975) by using NONLIN (version VO2-G-VAX, Statistical Consultants, Inc., Lexington, KY).
Correlation Studies.
For correlation studies, incubations were performed as outlined above with xanomeline concentrations of 2.3, 5.9, or 234 μM with microsomes from a human liver microsomal bank which had been previously characterized for the CYPs and FMO (see above). Univariate correlation analyses were performed (JMP, version 3.2.1, SAS Institute, Inc., Cary, NC) between the rate of xanomeline N-oxide formation and these enzymatic activities.
Incubations Examining the Effect of pH, Enzyme Inhibitors, or Heat on Xanomeline N-Oxide Formation.
Microsomal incubations were performed as described previously with a mixture of human or rat microsomes and 293 or 250 μM xanomeline, respectively, at pH 7.4 and pH 8.3. Similar incubations were performed at pH 7.4 in the presence or absence of 2.5 mM methimazole or 4 mM octylamine. To examine the effect of heating at 55°C with or without NADPH present, microsomes were heated at 55°C for 1 min, cooled on ice for approximately 2 min, and incubated with 250 or 293 μM xanomeline as described above. Incubations with 5.9 or 234 μM xanomeline and three different sources of human liver microsomes were also performed either in the presence or absence of CYP-specific inhibitors [CYP2C9, 25 μM sulfaphenazole; CYP2D6, 10 μM quinidine; CYP3A, 2 μM ketaconazole (Newton et al., 1995)] or substrates [CYP2A6, 500 μM coumarin (Yun et al., 1991); CYP2C19, 250 μMS-mephenytoin (Wrighton et al., 1993a)]. In addition, mechanism-based inhibitors of CYP1A2 (10 μM furafylline) and CYP3A (20 μM triacetyloleandomycin) (Newton et al., 1995), preincubated at 37°C with the microsomes and NADPH for 15 min before the initiation of the incubations with xanomeline, were examined for their effect on xanomeline N-oxide formation.
Incubations with Human Lung Microsomes or Microsomes Containing cDNA-Expressed Enzymes.
Human lung microsomes were incubated with 468 μM xanomeline at 1 mg/ml protein for 60 min. Incubations with cDNA-expressed human FMO1 or FMO3 (0.02 mg/ml) with 1 mM NADPH and 5.9 or 59 μM xanomeline were performed for 15 min. Microsomes prepared from cells engineered to express human CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 (Gentest) were incubated for 90 min with 1 mg/ml protein with 2 mM NADPH and 5.9 or 59 μM xanomeline.
Results
Using three different human hepatic microsomal samples (HLB, HLC, and HLM), a single-enzyme Michaelis-Menten model described the kinetics of formation of xanomeline N-oxide with an averageKM (apparent) of 107 μM (n = 3) (Table 1). A single-enzyme Michaelis-Menten model also described the kinetics of formation of xanomeline N-oxide by three different human kidney microsomal samples (IUK2, IUK4, and IUK7) (Table 1). This enzyme exhibited an average KM (apparent) of 5.5 μM (n = 3).
Kinetic analysis of xanomeline N-oxide formation1-a by microsomes prepared from human liver and human kidney
After univariate correlation analysis, the rate of formation of xanomeline N-oxide (Table 2) was found to correlate to the form-selective activity for FMO (ranger = 0.66–0.77) (Table3). Furthermore, there was cocorrelation between the formation rate of xanomeline N-oxide at the three xanomeline concentrations examined (Table 3).
Formation of xanomeline N-oxide2-a in a bank of human liver samples after incubation with 2.3, 5.9, or 234 μM xanomeline
Significant correlation between the formation of xanomeline N-oxide and CYP or FMO form-selective activities or formation between different substrate concentrations of xanomeline in a bank of characterized human liver microsomes (n = 14)
Expressed FMO1 and FMO3 were able to form this metabolite at a high rate of turnover when incubated with either 5.9 or 58.5 μM xanomeline (Fig. 2A). Multiple expressed CYPs were also able to form this metabolite (Fig. 2B), but at only a small fraction (≤2%) of the rate observed for the expressed FMOs.

Formation of xanomeline N-oxide by expressed FMO enzymes (A) or CYPs (B) after incubation with 5.9 and 58.5 μM xanomeline.
Most inhibitors of the CYPs only slightly inhibited the formation of this xanomeline N-oxide (Table4) when incubated with 5.8 μM xanomeline. There was a 60 to 70% inhibition of xanomelineN-oxide formation observed in microsomes incubated with 234 μM xanomeline and the CYP2A6 inhibitor coumarin. The formation of xanomeline N-oxide was significantly inhibited (88%) by the addition of methimazole (2.5 mM) to human liver microsomes. Moderate inhibition by methimazole was observed with human kidney microsomes and all rat microsomal samples examined (inhibition ranged from 32 to 57%) (Fig. 3). A similar pattern of inhibition was observed at pH 8.3 (data not shown). There was little effect byn-octylamine (4 mM) on the formation of xanomelineN-oxide by human liver or kidney microsomes (120 and 103% of control, respectively) at pH 7.4. Although human kidney microsomal formation of xanomeline N-oxide was unaffected byn-octylamine at pH 7.4 (98% activity remaining), formation of this metabolite was inhibited by n-octylamine at pH 8.3 (30% activity remaining).
Effect of CYP inhibitors on the formation of xanomeline N-oxide by human liver microsomes

Effect of heat (55°C, 2 min), with or without 1 mM NADPH present, incubation at pH 8.3, and 2.5 mM methimazole on xanomeline N-oxide formation by human liver or kidney or rat liver, kidney, or lung microsomes after incubation with xanomeline.
Incubation conditions known to be optimal for FMO-mediated activity were utilized to examine the formation of xanomeline N-oxide by all microsomal samples examined (Fig. 3). The formation of xanomeline N-oxide was enhanced at pH 8.3 as compared with pH 7.4 in all tissues examined except rat lung microsomes. In all microsomal samples examined, the formation of xanomelineN-oxide was sensitive to heating at 55°C for 1 min without NADPH present at pH 7.4. However, all microsomal samples but those prepared from rat lung were fairly insensitive to heating at 55°C for 1 min if NADPH was present. Rat lung microsomes exhibited only 22% of control activity when heated with NADPH present at pH 7.4. Results similar to those obtained at pH 7.4 were observed with respect to heating the samples with and without NADPH when incubated at pH 8.3 (data not shown).
Discussion
The studies reported herein confirm a primary role of FMO in the formation of xanomeline N-oxide in liver and kidney from both humans and rats. This conclusion was reached through the examination of the formation of xanomeline N-oxide under a variety of incubation conditions. Results from correlation studies, which used a characterized human liver microsomal bank, determined that the formation of xanomeline N-oxide correlated with FMO activity. In addition, expressed FMO1 and FMO3 formed this metabolite at a significantly greater rate than that observed for expressed CYP. As additional evidence of the role of FMO in this biotransformation by liver and kidney microsomes from these two species, formation of xanomeline N-oxide exhibited characteristics of FMO-mediated reactions including sensitivity to heat treatment (Uehleke, 1973;McManus et al., 1987) and enhanced activity at basic pH (Atta-Asafo-Adjei et al., 1993; Itagaki et al., 1996; Kawaji et al., 1997). The FMO substrate methimazole (Overby et al., 1995; Itagaki et al., 1996) inhibited formation of N-oxide xanomeline, but most inhibitors of CYP-mediated reactions only exhibited slight, if any, inhibition of this reaction by liver and kidney microsomes. The one exception observed was the inhibition of xanomelineN-oxide formation after incubations with a high concentration of xanomeline (234 μM) and a substrate of CYP2A6, coumarin. This may mean that CYP2A6 is a low-affinity enzyme capable of catalyzing this metabolic conversion. However, the expressed enzyme data would suggest that this route of metabolism would play a minor role in the formation of xanomeline N-oxide. Finally, as reported by Ziegler (1988), n-octylamine, an effector of some FMO-mediated reactions, was found not to be a reliable probe in identifying FMO involvement in the N-oxidation of xanomeline.
Previous examination of human liver has determined that the dominant FMO present in human liver is FMO3 (Phillips et al., 1995; Overby et al., 1997), and that in human kidney is FMO1 (Phillips et al., 1995). Therefore, enzyme kinetics were performed with human liver or kidney microsomes to examine the affinity of what was presumed to be FMO1 and FMO3 for xanomeline. A single enzyme was found to form xanomeline N-oxide in both human liver and human kidney microsomes. The enzyme forming xanomeline N-oxide in the human kidney, assumed to be FMO1, exhibited a 20-fold greater affinity (mean KM (apparent) = 5.5 μM) for xanomeline than the enzyme forming this metabolite in human liver (presumably FMO3) (mean KM (apparent) = 107 μM).
The enzyme(s) responsible for the formation of xanomelineN-oxide by rat liver or rat kidney microsomes exhibited a behavior in forming xanomeline N-oxide similar to that exhibited by human kidney microsomes under conditions of incubation at pH 8.3 or in the presence of methimazole. This suggests the involvement of a form of FMO in the formation of xanomeline N-oxide in rat liver and rat kidney that is similar to that responsible for this biotransformation in human kidney (FMO1). These results would agree with previously reported high levels of expressed FMO1 in both rat liver and rat kidney (Larsen-Su and Williams, 1996).
Pulmonary microsomes from a human and rats were also examined for their ability to form xanomeline N-oxide under various incubation conditions. Little formation of xanomeline N-oxide was seen by human lung microsomes. This agrees with previous reported data which showed that, although the FMO2 gene is expressed in adult human lung (Phillips et al., 1995), the enzyme produced has lost its catalytic activity due to the loss of 64 amino acids from the carboxy-terminal end (Dolphin et al., 1998). The enzyme present in rat lung that formed xanomeline N-oxide exhibited a different behavior after heating and at pH 8.3 from that observed in the other tissues examined. Although rat lung is known to contain FMO5 (Atta-Asafo-Adjei et al., 1993), these data do not unequivocally confirm or disprove the involvement of an FMO present in the rat lung as the enzyme responsible for the formation of xanomelineN-oxide in this tissue.
The higher affinity of FMO1 for this biotransformation present in this extrahepatic tissue may explain why there is a greater prevalence ofN-oxidized metabolites of xanomeline after s.c. dosing versus the profile seen after oral dosing. Under this scenario, xanomeline exposure via the s.c. route would be presented initially to the high-affinity FMO present in the kidney leading toN-oxidation. The N-oxidized metabolites may then be substrates for other xenobiotic metabolizing enzymes, leading to the prevalence of N-oxidized metabolites observed (Shipley et al., 1995). However, if xanomeline were dosed orally, other xenobiotic metabolizing enzymes present in the liver would have an opportunity to compete for the metabolism of xanomeline. The resultant metabolites may not be substrates of the hepatic or extrahepatic FMOs; therefore, there would be proportionately fewer N-oxidized metabolites of xanomeline formed after oral dosing as compared with s.c. dosing.
An alternative explanation for these metabolic differences after different dosing regimes is suggested by Hui et al. (1994), who showed that the extent of the FMO mediated S-oxidation of cimetidine (Cashman et al., 1993; Piyapolrungroj and Fleisher, 1997) was related to substrate concentration and time of exposure to drug. In the Hui et al. (1994) study it was observed that the percentage of cimetidine metabolically converted to the sulfoxide metabolite in the intestine was greater in the presence of lower cimetidine concentrations in combination with an extended time of exposure to the drug. If this is a characteristic of FMO-mediated reactions, where drug concentration and exposure time affects the extent of metabolism, an alternative explanation for the metabolic differences observed after different routes of administration of xanomeline can be proposed. Under this scenario, oral dosing of xanomeline would lead to a bolus dose of drug entering the systemic circulation after first-pass metabolism, which would then be followed by an elimination phase. However, a distinctly different systemic exposure to xanomeline is observed after dosing from a s.c. depot. In this case, xanomeline is released over a sustained period of time at low concentrations. This leads to a lower circulating concentration of xanomeline being presented to the FMOs mediating the N-oxidation of xanomeline; and, with the slower, sustained release of drug, the time of exposure of these enzymes to xanomeline would be increased. If FMOs are more efficient with exposures to lower concentrations and sustained exposure to a drug, as suggested by the studies with cimetidine, greaterN-oxidation of xanomeline should occur.
These data and those in the literature suggest that the increased prevalence of N-oxidized metabolites of xanomeline after s.c. dosing as compared with oral dosing may be due to differences in the affinity of various FMO family members for xanomeline. Alternatively, the different levels of exposure to xanomeline that FMOs may receive under different dosing regimens may lead to an altered metabolic profile for xanomeline.
Footnotes
-
Send reprint requests to: Barbara Ring, Drop 0825, Lilly Research Laboratories, Eli Lilly and Co., Indianapolis, IN 46285. E-mail: ring_barbara_j{at}lilly.com
- Abbreviations used are::
- FMO
- flavin-containing monooxygenase
- CYP
- cytochrome P-450
- Received January 22, 1999.
- Accepted June 11, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}