


Abstract
A novel and convenient in vitro method for predicting in vivo metabolic clearance in the liver (CLH) was developed. The CLH of a drug is usually predicted by using both the unbound fraction in serum and the intrinsic hepatic clearance of the unbound fraction, but this procedure is labor-intensive. We simplified the method by directly measuring intrinsic hepatic clearance using isolated rat hepatocytes suspended in rat serum and called this “the serum incubation method”. Sixteen commercially available compounds reported to be mainly excreted by liver metabolism were evaluated using our method. The remaining ratio of the unchanged drug after incubation was measured to calculate the rate of metabolism, and then CLH was predicted based on the dispersion model. The predicted CLH values of the drugs estimated by the serum incubation method were in good agreement with their in vivo plasma clearance values. In addition, the intrinsic hepatic clearance values obtained by the serum incubation method were comparable with those obtained by conventional methods. Furthermore, oral bioavailability values were equal to or lower than hepatic availability values predicted from the serum incubation method. These results indicate that compounds showing poor oral bioavailability can be excluded before in vivo pharmacokinetic study by using this method. In conclusion, the serum incubation method is a convenient and useful tool at the early stage of drug discovery.
The importance of pharmacokinetic studies at the early stage of drug discovery is increasing, and the progress of chemical synthetic techniques such as combinatorial chemistry and other parallel syntheses is increasing the number of candidates to be evaluated. As in vivo pharmacokinetic studies are time-consuming and labor-intensive, an in vitro approach for the quantitative prediction of in vivo parameters is desirable as a primary screen. Since avoidance of first-pass metabolism is one of the important issues in developing an orally available drug, a screening method that could predict metabolic hepatic clearance and/or availability would be useful in the drug discovery stage. Theoretical backgrounds for in vitro to in vivo extrapolation of hepatic clearance of a drug have been developed (Pang and Rowland, 1977; Rane et al., 1977; Wilkinson, 1987), and successful prediction of in vivo hepatic metabolic clearance has been reported (Houston, 1994; Houston and Carlile, 1997; Iwatsubo et al., 1997). Parameters such as unbound fraction in serum (fub1) and intrinsic hepatic clearance of unbound fraction (CLH, u int), which can be obtained under experimental conditions that properly reflect the situation in vivo, are necessary for this type of prediction, making the procedure labor-intensive for screening many compounds at the early stage of drug discovery. Therefore, we developed a more convenient method, called “the serum incubation method”, that can quantitatively predict in vivo metabolic clearance in the liver. The method involves direct measurement of intrinsic hepatic clearance (CLH, int) of a drug by using serum as an incubation medium for hepatocytes, which allows a prediction of hepatic clearance (CLH) and hepatic availability (FH) without considering serum protein binding.
In this method, we used isolated rat hepatocytes to reflect CLH, u int and rat serum to reflect fub. Hepatocytes possess not only phase I metabolic activity but also phase II metabolic activity in the liver and cell membranes in which drug transporters are functioning. In these respects, isolated hepatocytes are regarded as a more appropriate in vitro source for reflecting metabolic clearance in the liver than are liver microsomes, which mainly represent the microsomal phase I metabolic activity (Houston, 1994; Houston and Carlile, 1997; Iwatsubo et al., 1997). In addition, serum contains major drug-binding proteins. From these considerations, a method using hepatocytes and serum would be ideal for prediction. In this report, we describe the usefulness of our method for the prediction of in vivo CLH and FH of drug candidates.
Experimental Procedures
Chemicals.
Ibuprofen and 7-ethoxy coumarin were purchased from Aldrich (Milwaukee, WI). Tolbutamide and (S)-warfarin were obtained from Salford Ultrafine Chemical and Research Ltd. (Manchester, England). Alprenolol, quinidine, propranolol and trypsin inhibitor (Type II-S: Soybean) were obtained from Sigma Chemical Co. (St. Louis, MO). Lidocaine, theophylline and hexobarbital were purchased from Tokyo Kasei Kogyo Co. (Tokyo, Japan). Antipyrine, caffeine, chlorpromazine, diazepam, phenytoin, and collagenase were obtained from Wako Pure Chemical Industries (Osaka, Japan). Compounds X and Y were synthesized in our laboratories. William's E medium was from Life Technologies (Grand Island, NY). All other chemicals were obtained from either Wako Pure Chemical Industries (Osaka, Japan) or Junsei Chemical Co. (Tokyo, Japan).
Serum Preparation.
Male Sprague-Dawley rats, 7 to 10 weeks of age, purchased from Charles River Japan, Inc. (Kanagawa, Japan), were used in all of the experiments. Rat blood was collected via the abdominal aorta under ether anesthesia. After stabilization for 3 h at room temperature to coagulate the blood, samples were centrifuged (10 min, 1800g). Serum was collected as the clear supernatant and stored at −80°C until use.
Hepatocyte Preparation.
Hepatocytes were freshly isolated from rats by a procedure similar to that described by Baur et al. (1975). After isolation, the hepatocytes were suspended in William's E medium (WE), pH 7.4, and kept on ice until use. Cell viability was routinely checked by the 0.4% Trypan Blue exclusion test, and only hepatocytes with viability greater than 90% were used in this study.
Effect of Serum on Antipyrine Metabolism.
Incubation media containing 0, 25, 50, and 100% serum were prepared by mixing rat serum and William's E medium (v/v). Hepatocytes were resuspended at a density of 2 × 106cells/ml in each incubation medium at ice-cold temperature. An aliquot of DMSO solution of 12.5 mM antipyrine was pipetted at a volume of 1 μl per well into a 48-well plate. Each hepatocyte suspension was added at a volume of 250 μl per well at ice-cold temperature (the final concentration of antipyrine was 50 μM, DMSO was 0.4%). The final antipyrine concentration was set lower than its Michaelis-Menten constant, Km = 2.2 mM (Buters and Reichen, 1990). The samples were incubated at 37°C with shaking at 150 rpm under an atmosphere of 95% O2/5% CO2. After 0.5-, 1-, and 2-h incubation times, the reaction was terminated by adding 500 μl of ice-cold CH3CN/MeOH solution (2:1, v/v). The samples were centrifuged (10,000g × 10 min), and the amount of antipyrine remaining in the supernatant was measured by HPLC-UV (254 nm) (SPD10A; Shimadzu, Tokyo, Japan).
Serum Incubation Method.
The metabolism studies were typically performed as follows. Hepatocytes were resuspended at a density of 1 × 106cells/ml in rat serum at ice-cold temperature. An aliquot of DMSO solution of a compound was pipetted at a volume of 1 μl per well into a 48-well plate with the exception of (S)-warfarin, which was added as an aliquot of water solution. The hepatocyte suspension was added in a volume of 250 μl per well at ice-cold temperature (final concentration of DMSO: 0.4%). The samples were incubated at 37°C with shaking at 150 rpm under an atmosphere of 95% O2/5% CO2. After a 1-h incubation, the reaction was terminated by adding 500 μl of ice-cold CH3CN/MeOH solution (2:1, v/v). The samples were centrifuged (10,000g × 10 min), and the amount of the compound remaining in the supernatant was measured by HPLC-UV or liquid chromatography-tandem mass spectrometry. For low-clearance compounds such as antipyrine, caffeine, ibuprofen, theophylline, tolbutamide, and (S)-warfarin, cell density and incubation time were modified to 3 × 106 cells/ml and 2 h, respectively. All substrate concentrations were set lower than their Michaelis-Menten constants (Kmvalues). Compounds with unknown Km values, such as chlorpromazine, propranolol, verapamil, compound X, compound Y, and phenobarbital, were incubated at 1 μM, while theophylline was incubated at 5 μM.
Conventional Hepatocyte Incubation Method.
Hepatocytes were resuspended at a density of 0.2 × 106 cells/ml in William's E medium, pH 7.4, at ice-cold temperature. Incubation and subsequent steps were performed as described under Serum Incubation Method. For low-clearance compounds such as antipyrine, caffeine, ibuprofen, theophylline, tolbutamide, and (S)-warfarin, cell density and incubation time were changed to 1 × 106 cells/ml and 2 h, respectively. Compounds with unknown Kmvalues, such as chlorpromazine, verapamil, and phenobarbital, were incubated at 1 μM, while propranolol was incubated at 0.2 μM, compounds X and Y at 0.1 μM, and theophylline at 5 μM.
Theory and Calculation
The in vitro clearance of a drug is commonly expressed as follows:
When the CE of a drug is much lower than itsKm value, the CL is calculated by the following eq. 2, using cell density (D), incubation time (T), and the ratio of unchanged compound remaining after incubation (R).
When the compound is metabolized by hepatocytes suspended in William's E medium (the conventional incubation method), the intrinsic clearance of unbound fraction in William's E medium (CLu int, WE) is calculated by eq. 2. Also, when an incubation is performed with hepatocytes suspended in serum (the serum incubation method), the intrinsic clearance in serum (CLint, serum) is also calculated by eq. 2. To scale these in vitro clearance values up to in vivo liver values, the hepatocyte number per kilogram of body weight calculated from a report by Houston (1994) is used as a scaling factor (SF). The calculation is as follows:
SF = liver weight × hepatocyte number per gram of liver = 45 (g/kg of body weight) × 1.35 × 108 (cells/g of liver) = 6075 × 106 (cells/kg).
Then, CLH, int is calculated as follows:
In the case of the conventional method:
Results
Effects of Serum on Antipyrine Metabolic Activity in Isolated Rat Hepatocytes.
The effects of serum on the intrinsic metabolic potency of isolated rat hepatocytes were investigated using antipyrine as a test compound because antipyrine was reported not to bind to serum proteins (fub = 1; Singh et al., 1991). Aliquots of serum were added to the incubation medium (William's E medium) containing hepatocytes to give 0, 25, 50, and 100% serum. The metabolism of antipyrine in these incubation media was studied as described underExperimental Procedures.
No marked difference was observed in the metabolic rate of antipyrine in the different incubation media (Table1). Moreover, a linear metabolic reaction was observed over the 2 h following the start of the reaction. These observations demonstrate that serum has no effect on the intrinsic metabolic potency of antipyrine and can be used as an incubation medium for suspended isolated rat hepatocytes, at least during short-term metabolism studies.
Effects of serum on antipyrine metabolic activity in isolated rat hepatocytes
Prediction of Metabolic Clearance and Availability in Liver by the Serum Incubation Method.
Eighteen compounds eliminated mainly by liver metabolism, including 16 commercial and 2 novel compounds, were evaluated by the serum incubation method in terms of prediction of CLH and FH in the liver. The following parameters of the compounds are listed in Table2: Michaelis-Menten constant (Km), fub, in vivo plasma clearance (CLp), and oral bioavailability (Fpo). In vivo CLH, int was calculated by the dispersion model with the assumption that CLp was the same as the metabolic clearance. Incubation was performed as described under Experimental Procedures. The following parameters were calculated by the equations described under Theory and Calculation: CLint, serum, CLH, int, serum, CLH, and FH. As shown in Fig.1, a good correlation (r2 = 0.94) was observed between the predicted CLH and in vivo CLp. Low-clearance drugs (CLp < 15 ml/min/kg), such as antipyrine, caffeine, ibuprofen, phenytoin, tolbutamide, theophylline, and (S)-warfarin, were predicted to be low-clearance compounds. Moderately cleared drugs (15 < CLp < 40 ml/min/kg), such as compounds X and Y, were predicted to be moderate-clearance compounds. Moreover, drugs showing high clearance (CLp > 40 ml/min/kg), such as alprenolol, chlorpromazine, diazepam, 7-ethoxy coumarin, hexobarbital, lidocaine, propranolol, quinidine, and verapamil, were predicted to be high-clearance compounds. These results indicate that the serum incubation method is able to predict in vivo hepatic clearance with appropriate rank order.
Pharmacokinetic parameters and Km values for the metabolism of the compounds tested

Prediction of metabolic clearance in the liver by the serum incubation method.
CLH was determined by the serum incubation method as described under Experimental Procedures. Each value represents the mean of three incubations. In vivo CLp in rats was quoted from references. r2 = 0.94 [1, alprenolol; 2, chlorpromazine; 3, diazepam; 4, 7-ethoxy coumarin; 5, hexobarbital; 6, lidocaine; 7, phenytoin; 8, propranolol; 9, quinidine; 10, verapamil; 11, compound X; 12, compound Y; 13, antipyrine; 14, caffeine; 15, ibuprofen; 16, tolbutamide; 17, theophylline; 18, (S)-warfarin].
Figure 2 shows the correlation between the predicted FH and in vivo Fpo values. Compounds such as alprenolol, hexobarbital, lidocaine, propranolol, and verapamil, defined as having low oral bioavailability in vivo (Fpo < 0.2), were predicted to have poor hepatic availability because of extensive hepatic metabolism (predicted FH < 0.2). This finding indicates that compounds extensively metabolized by first-pass metabolism in the liver can be predicted by the serum incubation method. Also, compounds reported to have good oral bioavailability (Fpo > 0.7), such as antipyrine, caffeine, ibuprofen, tolbutamide, and theophylline, were predicted to have good hepatic availability (predicted FH > 0.7). However, phenytoin was also predicted to have FH> 0.7, while the in vivo Fpo is reported as 0.26 (Scriba et al., 1995). Phenytoin was reported to show poor absorption in the intestine because of low solubility (Scriba et al., 1995). For such compounds with insufficient availability in the intestine, Fpo cannot be predicted only by FH because Fpo is calculated as the product of FH and intestinal availability. Therefore, for compounds with excellent availability in the intestine, the serum incubation method predicts in vivo Fpo values in rank order.
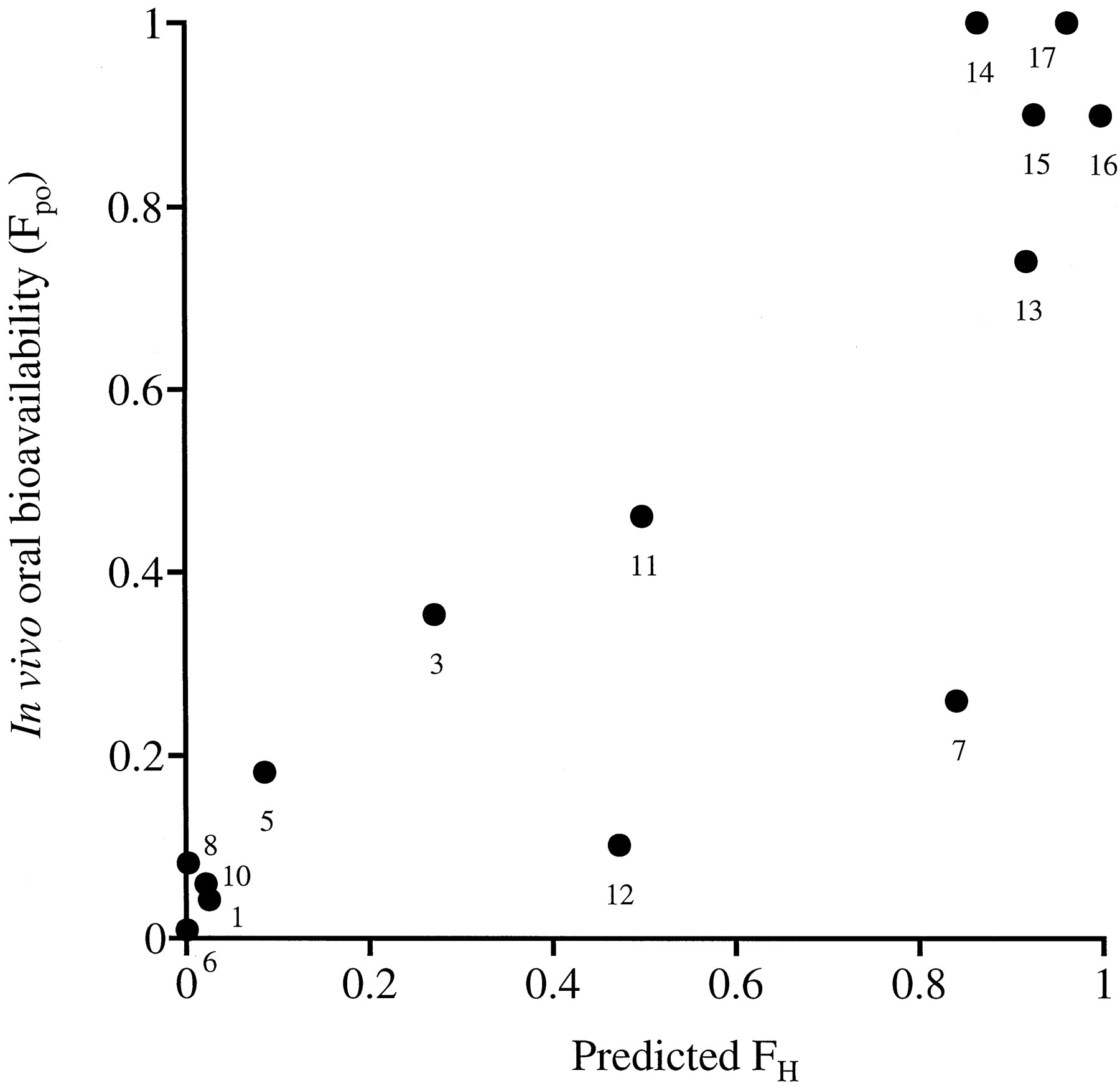
Comparison of FH and in vivo oral bioavailability Fpo.
FH was determined by the serum incubation method as described under Experimental Procedures. Each value represents the mean of three incubations. In vivo Fpo in rats was quoted from references. [1, alprenolol; 3, diazepam; 5, hexobarbital; 6, lidocaine; 7, phenytoin; 8, propranolol; 10, verapamil; 11, compound X; 12, compound Y; 13, antipyrine; 14, caffeine; 15, ibuprofen; 16, tolbutamide; 17, theophylline].
In conclusion, these results indicate that the serum incubation method can be used to predict in vivo CLH and maximum Fpo values without the fubvalue.
Comparison with the Conventional Method.
Conventional hepatocyte incubation was performed as described underExperimental Procedures. The following parameters were calculated by the equations described under Theory and Calculation: CLint, WE, CLH, u int, WE, and CLH, int, WE. The correlation coefficient value in this section is calculated based on low- to moderate-clearance compounds with in vivo CLp values under 40 ml/min/kg [phenytoin, compounds X and Y, antipyrine, caffeine, ibuprofen, tolbutamide, theophylline, and (S)-warfarin] to focus on the relationship of intrinsic clearance and free fraction in serum.
Figure 3A shows the correlation between in vivo CLH, int and in vitro CLH, u int, WE (r2 = 0.55). Because fub was not reflected, the values of CLH, u int, WE were greater than those of in vivo CLH, int, especially in the case of compounds with high serum protein binding (fub < 0.02), such as ibuprofen, tolbutamide, and (S)-warfarin, while the values of compounds with low serum protein binding, such as antipyrine, caffeine, theophylline, and hexobarbital, were in good agreement. On the other hand, when CLH, u int, WE was converted to CLH, int, WE by multiplying the fub, the values were well correlated with in vivo CLH, int (Fig. 3B,r2 = 0.86). These observations suggest that the fub value is necessary for precise prediction of in vivo hepatic clearance if the conventional incubation method is adopted.

Comparison of in vitro and in vivo intrinsic clearance values obtained by the conventional and the serum incubation methods.
A, CLH, u int, WE versus in vivo CLH, int. B, CLH, int, WE versus in vivo CLH, int. C, CLH, int, serum versus in vivo CLH, int. In vivo CLH, int values were calculated by the dispersion model assuming that hepatic clearance was the same as in vivo CLp. [1, alprenolol; 2, chlorpromazine; 3, diazepam; 4, 7-ethoxy coumarin; 5, hexobarbital; 6, lidocaine; 7, phenytoin; 9, quinidine; 10, verapamil; 11, compound X; 12, compound Y; 13, antipyrine; 14, caffeine; 15, ibuprofen; 16, tolbutamide; 17, theophylline; 18, (S)-warfarin]. Phenytoin, compounds X and Y, antipyrine, caffeine, ibuprofen, tolbutamide, theophylline, and (S)-warfarin are classified as low- to moderate-clearance compounds. A, CLH, u int, WE was compared with in vivo CLH, int. In vitro CLH, u int, WE was determined using the conventional hepatocyte incubation method under serum-free conditions as described underExperimental Procedures.r2 = 0.55 in low- to moderate-clearance compounds; r2 = 0.89 in all compounds. B, CLH, int, WE was compared with in vivo CLH, int. In vitro CLH, int, WE was calculated by multiplying CLH, u int, WE and fub as quoted from references. r2 = 0.86 in low- to moderate-clearance compounds; r2 = 0.90 in all compounds. C, CLH, int, serum was compared with in vivo CLH, int. In vitro CLH, int, serum was determined by the serum incubation method as described underExperimental Procedures.r2 = 0.98 in low- to moderate-clearance compounds; r2 = 0.85 in all compounds.
Figure 3C shows the correlation between in vivo CLH, int and in vitro CLH, int, serum, which was directly predicted by the serum incubation method. As in Fig.3B, a good correlation was observed between the in vitro and in vivo values (r2 = 0.98). This finding shows that the serum incubation method gives comparable prediction results without considering the fub.
Discussion
Many methods for predicting the in vivo metabolic hepatic clearance of drugs from in vitro values have been reported. These methods usually require two independent in vitro parameters, fub and CLH, u int(Houston, 1994; Houston and Carlile, 1997; Iwatsubo et al., 1997). Because we used isolated rat hepatocytes to reflect CLH, u int and rat serum to reflect fub, parameters of in vitro CLH, u int obtained from incubation with isolated hepatocytes and fub are essential to predict in vivo metabolism.
However, the conventional methodology is inadequate as a screening technique in the early stage of drug discovery because independent measurements of these two parameters are time-consuming. Moreover, it is difficult to measure fub and/or CLH, u int accurately. Although fub is usually obtained by ultrafiltration or equilibrium dialysis methods, some compounds adsorb to the membrane and/or apparatus, which causes an inaccurate estimation of the results (Bertilsson et al., 1979; Desoye, 1988). The same problem would also affect the calculation of CLH, u int because some drugs may not only adsorb to the apparatus but may also bind to microsomes or hepatocytes in the incubates (Obach, 1997, 1999). Therefore, for accurate calculation of CLH, u int, the concentration of a drug in the incubate should be corrected as the free fraction of the drug (Obach, 1997, 1999). Thus, additional studies would be required for this purpose.
However, with the serum incubation method, direct measurement of CLH, int at the expected concentration can be performed without considering the unbound fraction in serum as demonstrated in Fig. 3C, and quantitative prediction of in vivo CLH and FH can be performed as shown in Figs. 1 and 2. Because the number of samples in the analysis step is thereby reduced at least by half compared with the conventional method, we can obtain the prediction result much faster. In addition, serum can reduce the adsorption of compounds to the apparatus or to hepatocytes because serum proteins such as albumin are sometimes used to prevent adsorption of compounds to the apparatus. There are several reports of in vitro metabolism studies with hepatocytes or microsomes performed with media containing purified albumin or other serum proteins (Gariepy et al., 1992; Obach, 1997). But as in vivo serum includes many drug-binding proteins, it would be difficult to reflect in vivo fub correctly using only purified serum proteins. Lavé et al. (1997) ranked the compounds according to the ratio of hepatic extraction into three groups (low, middle, and high) after calculation of CLH, u int based on human hepatocyte suspensions, and their observations correspond fundamentally to those shown in Fig. 3A. Thus, the serum incubation method seems to be ideal for predicting in vivo metabolic CLH and FH, and the simplicity of the method may save considerable time and labor in the drug discovery stage. As far as we know, this is the first report to describe a method that can quantitatively predict in vivo metabolic CLH and FH using isolated hepatocytes directly suspended in serum.
It is true that serum protein binding improves FHand CLH, but the unbound drug concentration in serum also has significant effects on many aspects of pharmacodynamics and should also receive attention at the drug screening stage. Therefore, the fub should be confirmed by an appropriate method such as ultrafiltration or equilibrium dialysis after screening by the serum incubation method. The fub can be roughly estimated according to the difference in metabolic rate of CLH, int, serum versus CLH, u int, WE by a procedure similar to that reported by Gariepy et al. (1992). However, the relevance of this approach remains unclear because the activation of metabolism by serum protein was reported (Ludden et al., 1997), and adsorption of a drug to the apparatus and/or hepatocytes, especially in serum-free condition, should be considered. A high unbound fraction is desirable for accessibility to the target, but a low unbound fraction may not always be unfavorable if the drug has good pharmacokinetic properties and good pharmacodynamic effects, for the in vivo drug effect is also dependent on its intrinsic potency to the target.
When orally available compounds are screened in the drug discovery stage, the prediction of in vivo CLH and/or FH by the serum incubation method can facilitate the exclusion of undesirable candidates that have high clearance and low oral bioavailability due to extensive hepatic metabolism, without the performance of labor-intensive and time-consuming in vivo pharmacokinetic studies. In addition, when an in vivo pharmacokinetic result has already been obtained, predicted CLHand FH values obtained from in vitro experiments may help to estimate the contribution of hepatic metabolism to in vivo oral bioavailability and systemic clearance.
Conclusion
The serum incubation method appears to be more convenient than conventional methodology for predicting in vivo metabolic clearance and availability in the liver because these values are achieved without assessing fub. This method may be a useful tool for screening orally available compounds in the early stage of drug discovery.
Acknowledgment
We thank Dr. Allen Nielsen Jones, Merck Research Laboratories, for kindly reviewing the manuscript.
Footnotes
-
Send reprint requests to: Yoshihiro Shibata, Drug Metabolism, Tsukuba Research Institute, Banyu Pharmaceutical Co., Ltd., Okubo 3, Techno-park Oho, Tsukuba, Ibaraki 300-2611, Japan.
- Abbreviations used are::
- fub
- unbound fraction in serum
- CL
- clearance
- CLu int, WE
- intrinsic clearance of unbound fraction obtained from incubation in WE
- CLint, serum
- intrinsic clearance obtained from incubation in serum
- CLH
- hepatic clearance
- CLH, u int
- hepatic intrinsic clearance of unbound fraction
- CLH, u int, WE
- hepatic intrinsic clearance of unbound fraction obtained from incubation in WE
- CLH, int
- hepatic intrinsic clearance
- CLH, int, WE
- hepatic intrinsic clearance obtained from incubation in WE
- CLH, int, serum
- hepatic intrinsic clearance obtained from incubation in serum
- CLp
- plasma clearance
- DMSO
- dimethyl sulfoxide
- DN
- dispersion number
- EH
- hepatic extraction ratio
- FH
- hepatic availability
- Fpo
- oral bioavailability
- SF
- scaling factor
- QH
- hepatic blood flow rate
- R
- ratio of intact drug remaining after incubation
- RB
- blood to plasma concentration ratio
- WE
- William's E medium (pH 7.4)
- Received April 29, 2000.
- Accepted September 14, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}