


Abstract
The expression of three cytochromes P450 (CYP3A4, CYP2C9, and CYP2B6) was investigated in primary human hepatocyte cultures following treatment with four calcium channel modulators (CCM) of the dihydropyridine family, three antagonists (nifedipine, nicardipine, and isradipine), and one agonist (BK8644). Induction of CYP3A4 was studied by Northern blot, Western blot, and enzymatic activity. Induction began between 1 and 10 μM CCM and was dependent on the presence of dexamethasone (100 nM) in the medium. CYP3A4 mRNA accumulation started only after 16 h of treatment because pregnane X receptor (hPXR) synthesis was needed. Cotransfection experiments showed that the proximal and the distal PXR response elements of the CYP3A4 promoter and hPXR (HepG2 cells) or dexamethasone-induced hPXR (primary hepatocytes) were necessary to obtain full induction. Furthermore, glutathione S-transferase pull-down assays demonstrated that the CCM tested can act as hPXR ligands. In addition, cotransfection experiments in CV1 cells showed that these compounds failed to reverse CAR (constitutively activated receptor) inactivation by androstenol. Finally, 10 μM CCM induced both CYP2C9 and CYP2B6, strengthening the evidence that hPXR is involved in the regulation of these genes. All together, these results widen the field of hPXR activators to a new class of ligand, namely the CCM of the dihydropyridine family.
Calcium channel modulators (CCM1) belonging to the dihydropyrine family have been used widely for the treatment of hypertension, angina pectoris, and other cardiovascular diseases since first introduced in the 1960s. With such widespread use, there has been a number of reports on significant pharmacokinetic drug interactions associated with CCM (Pedersen et al., 1981; Rameis et al., 1984; Katoh et al., 2000). Induction of cytochrome P450 (P450) expression by CCM has been suggested as one possible explanation for these interactions.
Induction of cytochromes P450 by xenobiotic chemicals is a common cellular defense mechanism, usually leading to increased detoxification of xenobiotics but sometimes paradoxically to formation of metabolites that are more toxic and carcinogenic (Conney, 1982). An inductive effect of calcium channel antagonists on the P450 system would be clinically significant because these drugs are used for long-term treatment. The major P450 form that has been reported to metabolize nicardipine and nifedipine is CYP3A4 (Guengerich et al., 1986;Guengerich, 1991), which can be induced by some of its own substrates.
Two nuclear receptors, pregnane X receptor (PXR) (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998) and the constitutively activated receptor (CAR) (Honkakoski et al., 1998), have recently been shown to mediate P450 gene induction in response to xenobiotics (Sueyoshi et al., 1999; Moore et al., 2000), and we have shown that the glucocorticoid receptor mediates PXR and CAR expression in human hepatocytes (Pascussi et al., 2000a,b). PXR is activated by a wide spectrum of xenobiotics, including rifampicin (RIF), nifedipine (Bertilsson et al., 1998), and clotrimazole, and steroids, including some glucocorticoids. The CAR receptor is involved in the phenobarbital induction of CYP2B genes. This receptor is cytoplasmic; but following activation by xenobiotics, it translocates to the nucleus where it undergoes a second activation step, leading to the final activated form. In addition, the mCAR can be deactivated by androstane (Forman et al., 1998) in contrast to human CAR (Moore et al., 2000). Both the CAR and the PXR form heterodimers with the retinoid X receptor, and these complexes bind to and transactivate several response elements located in both the proximal and distal P450 gene promoters (Lehmann et al., 1998; Goodwin et al., 1999; Sueyoshi et al., 1999; Moore et al., 2000).
In this study, we examined the induction profiles of several CCMs [nifedipine, nicardipine, isradipine (ISRA), and BK8644 (BK)[ on the expression of three major P450 isoenzymes (CYP3A, CYP2B, and CYP2C) in human hepatocytes. Our results suggest that these CCM activate the hPXR in human hepatocytes to induce these cytochromes.
Experimental Procedures
Materials.
Ham's F-12 and Williams' E culture media, vitamins and hormones, collagenase (type IV), dimethyl sulfoxide (DMSO), and dexamethasone were purchased from Sigma (St. Quentin Fallavier, France). Collagen-coated culture dishes were obtained from Corning (Iwaki, Japan). α-[32P]dCTP, α-[32P]UTP, and enhanced chemiluminescence-developing reagents were purchased from Amersham Pharmacia Biotech (Cardiff, Wales).
Isradipine [4-(4-benzofurazanyl)-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylic acid methyl 1-methylethyl ester] was kindly provided by Dr. Pierre Charnet (IFR24, Montpellier, France). Nifedipine [1,4-dihydro-2,6-dimethyl-4-(2-nitrophenyl)-3,5-pyridinedicarboxylic acid dimethyl ester], nicardipine [1,4-dihydro-2,6-dimethyl-4-(3-nitrophenyl)-3,5-pyridinedicarboxylic acid methyl 2-[methyl-(phenylmethyl)amino]ethyl ester], and BK8644 [1,4-dihydro-2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]pyridine-3-carboxylic acid methyl ester] were purchased from Sigma-Aldrich (St. Louis, MO). TCPOBOP [1,4-bis[2-(3,5-dichloropyridyloxy)]-benzene] was a gift from P. Lesca (INRA, Toulouse, France).
Plasmid Constructs.
The CYP3A4 5′-flanking fragments (−263/+11) and (−163/+11) were generated by PCR from a previously isolated genomic clone (Jounaidi et al., 1994) acting as a template using oligonucleotides, which create artificial cloning sites at the 5′ (KpnI) and 3′ (SmaI) positions. These fragments were cloned into pGL3 basic reporter genes (Promega, Madison, WI). The XREM/263/11- and XREM/163/11-CYP3A4 constructs were generated by inserting the PCR-amplified XREM region (nucleotides −7836 to −7208) of the CYP3A4 promoter (Goodwin et al., 1999) from human genomic DNA into theKpnI cut (−263/+11)- and (−163/+11)-CYP3A4 constructs, respectively.
The ΔATG-hPXR expression plasmid was generated by PCR amplification of cDNA-encoding amino acids 1 to 434 of hPXR (kindly provided by Dr. S. Kliewer, Glaxo Wellcome, Research Triangle Park, NC) using oligonucleotides 5′-GGGTGTGGGGAATTCACCACCATGGAGGTGAGACCCAAAGAAAGC and 5′-GGGTGTGGGGGATCCTCAGCTACCTGTGATGCCG and insertion into pSG5 digested with EcoRI/BamHI. The mCAR expression vector (pCR3-mCAR) was kindly provided by M. Negishi.
Cell Culture and Transfections.
HepG2 cells (human hepatoma) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (Invitrogen, Carlsbad, CA). Transfection of plasmid DNA was performed in single batches with Fugene-6 (Roche Diagnostics Corporation, Indianapolis, IN), as instructed by the manufacturer. Transfections were performed using 100,000 cells, 250 ng of reporter plasmid, and 80 ng of expression vector. Plasmid pSV-β-galactosidase (Progema; 50 ng) was added as an internal control of transfection. pSG5 empty vector (Stratagene, La Jolla, CA) was added to equalize the total concentration of transfected plasmid DNA. After 12 to 16 h, the medium was changed, and fresh medium containing 0.1% DMSO or inducers was added. Cells were harvested in reporter lysis buffer (Promega) 24 h after changing the medium, and cell extracts were analyzed for luciferase and β-galactosidase activities, as described elsewhere (Pascussi et al., 2000a).
Tissue Source and Hepatocyte Cultures.
Hepatocytes were prepared from lobectomy segments resected from adult patients for medical purposes unrelated to our research program. The use of these human hepatic specimens for scientific purposes has been approved by the French National Ethics Committee. Hepatocytes were prepared and cultured according to a previously published procedure (Pichard et al., 1990). The cells were plated into 100-mm plastic dishes, precoated with collagen at 10 × 106 cells/plate in a total volume of 6 ml of a hormonally and chemically defined medium consisting of a mixture of Williams' E and Ham's F-12 (1:1 in volume). Forty-eight hours after plating, dexamethasone was withdrawn from the culture medium for 16 h (or 24 h for CYP2C study). Cells were then cultured in the presence or absence of the indicated inducers for 6 to 48 h.
Ribonuclease Protection Assays and Northern Blot.
Total RNA and protein were isolated using Trizol reagent (Invitrogen) from 107 cultured hepatocytes, according to the manufacturer's instructions. Plasmids for CYP3A4, CYP2C9, and CYP2B6 RNase protection assays have been previously described. Total RNA (30 μg) was analyzed by the RNase protection assay using a specific riboprobe, as previously described (Greuet et al., 1997) with minor modifications. Total RNA was hybridized with radiolabeled antisense RNA probe (100,000–150,000 cpm) overnight at 37°C, after a 10-min incubation at 95°C. For Northern blot experiments, 30 μg of total RNA was analyzed using α-[32P]dCTP-CYP3A4 cDNA probe and autoradiography was carried out by exposing the dried gel to Kodak X-AR film (Eastman Kodak, Rochester, NY). The signals were quantified by analyzing the radioactivity with a PhosphorImager apparatus and ImageQuant software (Amersham Pharmacia Biotech). For quality control, 30 μg of total RNA was analyzed by Northern blot using a rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA probe (J. M. Blanchard, Institut de Genetique Moleculaire de Montpellier, Montpellier France).
Coactivator Receptor Ligand Assay (CARLA).
For this CARLA, human PXR [35S]methionine-labeled protein was prepared by in vitro translation using the coupled transcriptional and translation system, as described by the manufacturer (Promega, Madison, WI). GST-steroid receptor coactivator (SRC)-1 fusion protein was expressed in the Escherichia coli BL21 strain and purified using glutathione-Sepharose-4B bead affinity chromatography (Amersham Pharmacia Biotech). The beads were subsequently washed and suspended in NETN (20 mM tris, pH 8.0, 100 mM NaCl, and 0.1% NP-40) buffer. GST proteins bound to glutathione-Sepharose were incubated with 5 μl of [35S]methionine-labeled proteins in the presence of NETN buffer and the compounds to be tested or 1% DMSO. After overnight incubation at 4°C with gentle agitation, agarose beads were extensively washed with NETN buffer, and bound proteins were eluted in sample buffer and analyzed by SDS-PAGE. Gels were then stained with Coomassie blue, incubated in an autoradiography enhancer (PerkinElmer Life Sciences, Boston, MA), dried, and subjected to autoradiography at −70°C.
Immunoblot Analysis.
Thirty micrograms of Trizol-isolated protein from 107 hepatocytes were separated by SDS-PAGE (10%) and electroblotted onto a nitrocellulose membrane (Sartorius, Gottingen, Germany). Membranes were incubated with specific antibodies against CYP3A4 or hPXR (Santa Cruz Biotechnology, Santa Cruz, CA) and developed with the enhanced chemiluminescence detection system (Amersham Pharmacia Biotech). Quantification was performed using densitometric analysis software (NIH Image; National Institutes of Health, Bethesda, MD).
Measurement of CYP3A4-Dependent Cyclosporin A (CSA) Oxidase Activity.
Human hepatocytes were treated for 96 h with CCM and then incubated in the absence of inducer for 4 h, with or without 5 μM CSA and 5 μCi [3H]CSA. Metabolites of CSA were quantified by high-pressure liquid chromatography, as previously described (Pichard et al., 1990).
Results
Induction of CYP3A4 mRNA and Protein by Various CCM in Cultured Primary Human Hepatocytes: Kinetics and Dose-Response.
We first treated for 48 h human hepatocytes prepared from different patients in primary cultures with 50 μM four different CCM (nifedipine, BK8644, isradipine, and nicardipine) or the prototype CYP3A4 inducer rifampicin (Fig. 1A). Induction of the CYP3A4 was then analyzed at the mRNA, protein, and enzymatic activity levels.
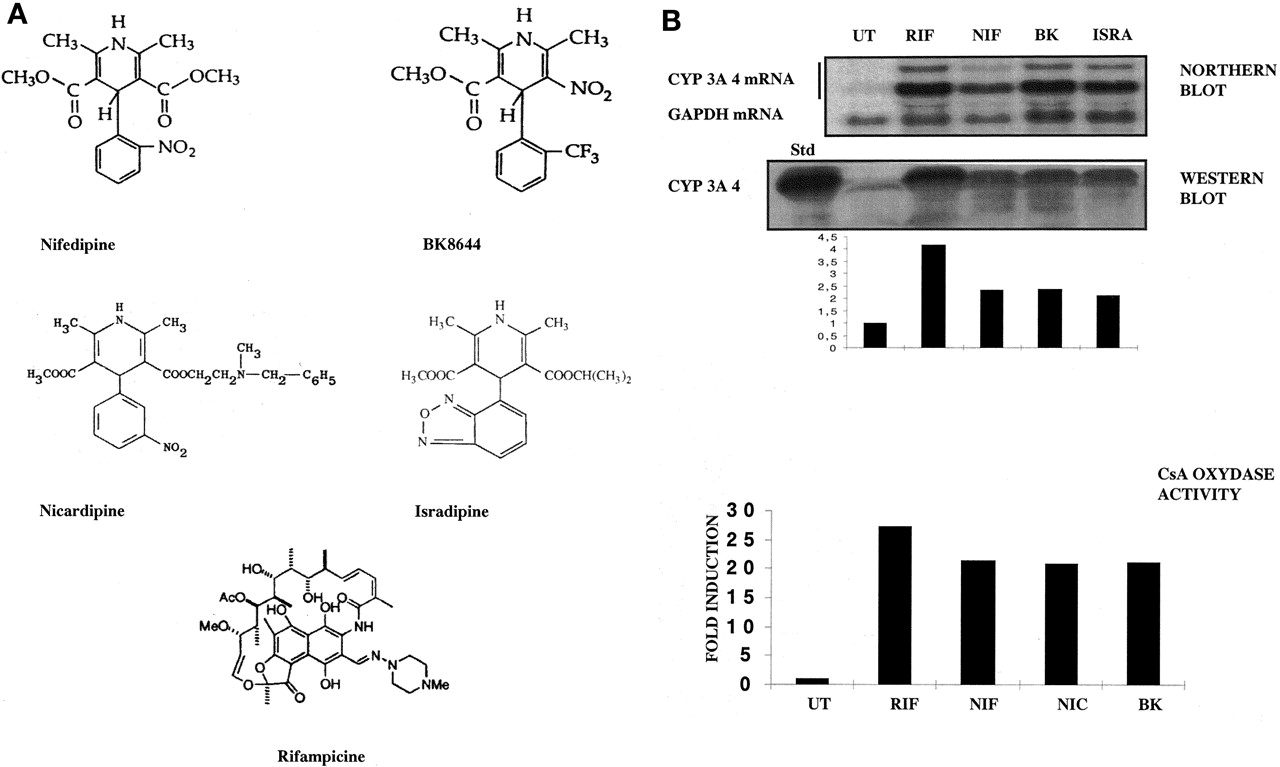
A, chemical structure of the CCM examined in this study; B, CYP3A4 induction by different CCM in primary cultures of human hepatocytes (FT159; similar results were obtained with liver FT160).
Forty-eight hours after plating, 107 human hepatocytes were treated by 0.1% DMSO, 50 μM NIF, BK, ISRA, or RIF in DEX-containing medium for another 48 h. CYP3A4 induction was studied at three levels. At the RNA level, total RNA (30 μg) was analyzed by Northern blot using both GAPDH and CYP3A4 [32P]dCTP-radiolabeled cDNA probes. Radioactivity in the bands corresponding to CYP3A4 (two higher bands) and GAPDH (lower band) mRNA was measured by PhosphorImager and ImageQuant software. At the protein level, Trizol-isolated proteins (30 μg) were analyzed by Western blot using CYP3A4-specific antibody. Quantification of the higher band was obtained using NIH Image software. Values are expressed in -fold induction compared with DMSO-treated cells. Protein levels were evaluated by bicinchoninic acid and assayed by amino black staining. At the enzyme activity level (FT169; similar results were obtained with livers FT155, FT156, and FT159), induction of the cyclosporin A oxidase activity was assessed after treatment with 10 μM inducers using high-pressure liquid chromatography, as detailed underExperimental Procedures. Values are expressed in -fold induction compared with DMSO-treated cells.
As shown on Fig. 1B, nifedipine (NIF), BK8644 (BK), and isradipine (ISRA) were potent inducers at the mRNA level [7-, 11-, and 10-fold, respectively, compared with 12-fold for rifampicin (RIF)] and at the protein level (2.3-, 2.4-, and 2.1-fold, respectively, for the CCM and 4.2-fold for rifampicin). In addition, cyclosporine A oxidase activity, a reference assay to measure CYP3A4 activity, was found to be increased in the hepatocytes after treatment with NIF, nicardipine (NIC), and BK8644.
Next, we investigated how CYP3A4 responds to the concentration of CCM and rifampicin. Human hepatocytes were cultured for 16 h and then exposed for 48 h to increasing concentrations (0.1 to 10 μM) of CCM. As shown in Fig. 2, A–B, we found that induction of both CYP3A4 mRNA and protein began at concentrations between 1 and 10 μM of the tested CCM, depending on the liver.

Concentration-dependent induction of CYP3A4 by CCM in primary cultures of human hepatocytes (FT176).
Forty-eight hours after plating, 107 human hepatocytes were treated by 0.1% DMSO, and increasing concentrations (0.1–10 μM) of NIF, BK, NIC, or RIF in DEX-containing medium. A, CYP3A4 induction was studied at the RNA level; total RNA (30 μg) was analyzed by Northern blot using both GAPDH and CYP3A4 [32P]dCTP-radiolabeled cDNA probes. Radioactivity in the bands corresponding to CYP3A4 (two higher bands) and GAPDH (lower bands) mRNA were measured by PhosphorImager and ImageQuant software. B, at the protein level, Trizol-isolated proteins (30 μg) were analyzed by Western blot using CYP3A4-specific antibody. Quantification of the higher band was obtained using NIH Image software. Values are expressed in -fold induction compared with DMSO-treated cells. Protein levels were evaluated by bicinchoninic acid and assayed by amino black staining.
We observed recently that both hPXR and human CAR (Pascussi et al., 2000a,b) are up-regulated by dexamethasone in human hepatocytes, most likely through the glucocorticoid receptor pathway. As a consequence, the levels of both PXR and CAR decrease in cells cultured in the absence of dexamethasone. This accounts for the previous observations that induction of CYP3A4 in response to compounds known to activate PXR (i.e., rifampicin) is potentiated by treatment of hepatocytes with dexamethasone. We report a similar observation here with respect to CCM-mediated CYP3A4 induction. Indeed, Fig.3A shows that induction of CYP3A4 mRNA in response to CCM was significantly enhanced in cells cultured in the presence of dexamethasone (high-level hPXR) compared with cells cultured in the absence of dexamethasone (low-level hPXR). It should be noted that the potentiation factor in response to dexamethasone varies slightly from one CCM to another and from one culture to another due to interindividual variability. Control experiments clearly confirmed that in the presence of dexamethasone, glucocorticoid receptor, PXR, and CAR mRNA (as assessed by RNase protection assay) were expressed in our cultures at a level similar to that observed in tissue [data not shown and as previously reported (Pascussi et al., 2000a,b)].
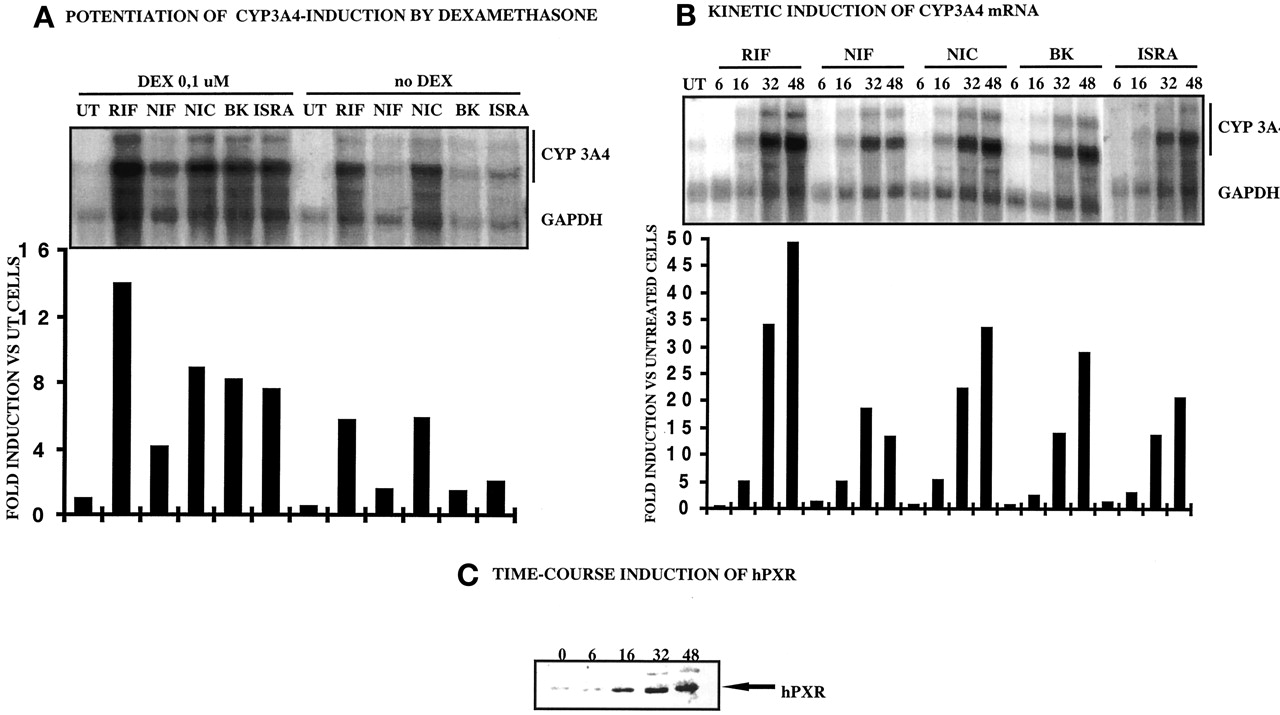
Potentiation of CYP3A4 induction by dexamethasone (A), kinetic induction of CYP3A4 mRNA (B), time course induction of hPXR (C).
A, CYP3A4 induction potentiation by dexamethasone. B, time course induction of CYP3A4 mRNA in primary cultures of human hepatocytes (FT178; similar results were obtained with liver FT168). Forty-eight hours after plating, 107 human hepatocytes were cultured in dexamethasone-free medium for 48 h, then passed in DEX-containing medium (or DEX-free medium for A), and then treated by 0.1% DMSO and 10 μM NIF, NIC, BK, ISRA, and RIF. Cells were harvested after 6, 16, 32, and 48 h of treatment, and total RNA (30 μg) was analyzed by Northern blot using both GAPDH and CYP3A4 [32P]dCTP-radiolabeled cDNA probes. Radioactivity in the bands corresponding to CYP3A4 (two higher bands) and GAPDH mRNA were measured by PhosphorImager and ImageQuant software. Values are expressed in -fold induction compared with DMSO-treated cells. C, time course induction of hPXR protein in dexamethasone-treated primary cultures of human hepatocytes (FT178; similar results were obtained with liver FT176) Forty-eight hours after plating, 107 human hepatocytes were cultured in dexamethasone-free medium for 48 h, then passed in DEX-containing medium. Cells were harvested after 6, 16, 32, and 48 h of treatment and 30 μg of Trizol-isolated protein were analyzed by Western blot using anti-hPXR antibody.
The time dependence of CCM-mediated CYP3A4 mRNA induction was similar to RIF induction (Fig. 3B). In fact, induction began only after 16 h of 10 μM CCM treatment. Interestingly, these kinetics fit well with the kinetics of synthesis of the hPXR protein, detected only between 6 and 16 h and increasing continuously between 32 and 48 h (Fig. 3C).
In summary, these observations suggest that calcium channel modulators of the dihydropyridine family are CYP3A4-inducers and exhibit similar behavior to RIF in terms of kinetics and potentiation by dexamethasone, and this response is likely to be mediated by PXR.
Nifedipine and Analogs Transactivate a CYP3A4 Promoter Construct via Activation of the Human PXR.
To study the mechanism of this induction, HepG2 cells and human hepatocytes were transfected with different constructs of the CYP3A4 promoter-driven luciferase reporter (Fig.4), with or without the hPXR expression vector. The different constructs were: the (−263/+11) CYP3A4 promoter region containing the proximal hPXR-responsive element (ER6) alone; the above fragment in association with the XREM distal enhancer (−7836/−7208); and the (−163/+11) fragment lacking the proximal ER6 in association with the distal XREM.

Human hepatocytes (A), HepG2 cells (B), CV1 cells (C).
A, upper panel, nifedipine-mediated transactivation of CYP3A4 promoter construct needs dexamethasone-induced hPXR synthesis in primary cultures of human hepatocytes (FT167, FT168, FT175). Hepatocytes were cotransfected with the XREM/263/11- or 263/11-CYP3A4 pGL3 plasmids and the pSV-β-galactosidase control vector. Twenty-four hours later, cells were treated with 10 μM RIF or NIF. Forty-eight hours later, luciferase activity was determined and normalized by β-galactosidase activity. Values are given as mean of three independent experiments in duplicate in -fold induction versus untreated cells. Lower panel, schematic representations of the constructs use in the different experiments. B, CCM-mediated transactivation of CYP3A4 promoter construct via hPXR activation in HepG2 cells. Upper panel, HepG2 cells were cotransfected with the XREM/263/11-, XREM/163/11-, or 263/11-CYP3A4 pGL3 plasmids, the pSG5-hPXR expression vector, and the pSV-β-galactosidase control vector. Twenty-four hours later, cells were treated with 10 μM RIF or NIF. Twenty-four hours later, luciferase activity was determined and normalized by β-galactosidase activity. Values are given as mean of three independent experiments in duplicate in -fold induction versus untreated cells. Lower panel, cotransfection of the XREM/263/11 construct, hPXR expression vector or empty pSG5 expression vector, and treatment with 10 μM RIF and CCM. C, nifedipine or nicardipine do not reverse the inhibition of mCAR by androstenol. CV1 cells were cotransfected by a (NR1)3-tk-Luc plasmid, a mCAR expression vector, and the pSV-β-galactosidase control vector. Twenty-four hours later, cells were treated with 10 μM RIF, NIF, NIC, BK8466, and 250 nM TCPOBOP in the presence or absence of 4 μM 3α-androstenol (AN). Forty-eight hours later, luciferase activity was determined and normalized by β-galactosidase activity.
We first transfected these constructs in human hepatocytes cultured with or without 100 nM dexamethasone during 48 h. Cotreatment by RIF or NIF and nanomolar concentrations of dexamethasone produced a synergistic effect only for the XREM/263/11 construct containing both the proximal and the distal ER6, whereas no induction was found for the construct (−263/+11) containing only the proximal ER6 (Fig. 4A). This synergistic effect on both CYP3A4 promoter activity and endogenous CYP3A4 mRNA accumulation suggests that this construct is able to mimic both glucocorticoid receptor-mediated and PXR- or CAR-mediated components of the CYP3A4 induction.
Figure 4B (upper) shows results obtained in the human hepatoma cell line HepG2 after cotransfection of the described constructs and the hPXR expression vector following treatment with 10 μM rifampicin or nifedipine versus DMSO-treated cells. Induction was greater (5.6-fold for rifampicin and 6.4-fold for nifedipine) for the XREM/263/11 construct but was reduced by half when construct-lacking proximal ER6 was tested. Weak induction was found for rifampicin and the CCM in the absence of hPXR, perhaps due to some PXR induced by dexamethasone analogs present in the fetal calf serum used for cell cultures. However, full induction was obtained only with cotransfection of the receptor and the XREM/263/11 construct, showing the need for both the hPXR and its responsive elements to obtain full activation (Fig. 4B, lower).
The nuclear orphan receptor CAR was originally characterized as constitutively activated and repressed by 3α-androstenol and androstenol, and its implication in P450 gene regulation was recently reported (Honkakoski et al., 1998; Moore et al., 2000). We therefore cotransfected a CAR response element (NR1)-driven luciferase reporter gene with the mCAR expression vector into CV1 cells treated with 3α-androstenol, and the activity was restored by treatment with TCPOBOP, as reported by Sueyoshi et al. (1999). In contrast, all compounds tested including RIF failed to restore the constitutive activity of the mCAR receptor on the NR1-containing reporter (Fig. 4C). These results strongly suggest that these compounds do not act as mCAR ligands (Xie et al., 2000).
CCM Act as hPXR Ligands In Vitro.
Using CARLA based on ligand-induced interaction between nuclear receptor and coactivator, it has previously been reported that pregenolone-16 α-carbonitrile and rifampicin induced interaction of GST-PXR with a SRC-1 fragment, indicating that these two molecules can act as PXR ligands (Kliewer et al., 1998; Lehmann et al., 1998). SRC-1 is a positive AF-2 coactivator member of the P160/SRC-1 family and binds to the ligand binding domain/AF-2 part of the ligand-activated nuclear receptor (Feng et al., 1998). To prove that the CCM can act as a human PXR ligand, we examined whether high concentrations of these compounds were able to produce ligand-induced SRC-1/PXR interaction. We observed that CCM and rifampicin at 20 μM all induced the binding of radiolabeled in vitro translated ΔATG-hPXR to purified GST-SRC-1 (amino acids 580–750) fusion protein, whereas solvent alone failed to do so (Fig.5). Note that the two PXR bands present in the electrophoresis gel reflect the use of two ATG codons in the hPXR gene (Bertilsson et al., 1998). Taken together, these data demonstrate that at high concentrations (20 μM) CCM may act as human PXR ligands.

Nifedipine and its analog act as an hPXR ligand.
GST-SRC-1 proteins bound to glutathione-Sepharose were incubated with 5 μl of [35S]methionine hPXR in the presence of NETN buffer and 20 μM NIF, RIF, BK, NIC, ISRA, and 1% DMSO. After overnight incubation at 4°C with gentle agitation, agarose beads were extensively washed with NETN buffer, and bound proteins were eluted in sample buffer and analyzed by SDS-PAGE. Gels were then stained with Coomassie blue, incubated in an autoradiography enhancer, dried, and subjected to autoradiography at −70°C.
CCM Induces CYP2C9 and CYP2B6 in Primary Cultures of Human Hepatocytes with a Time Dependence Similar to that of Rifampicin.
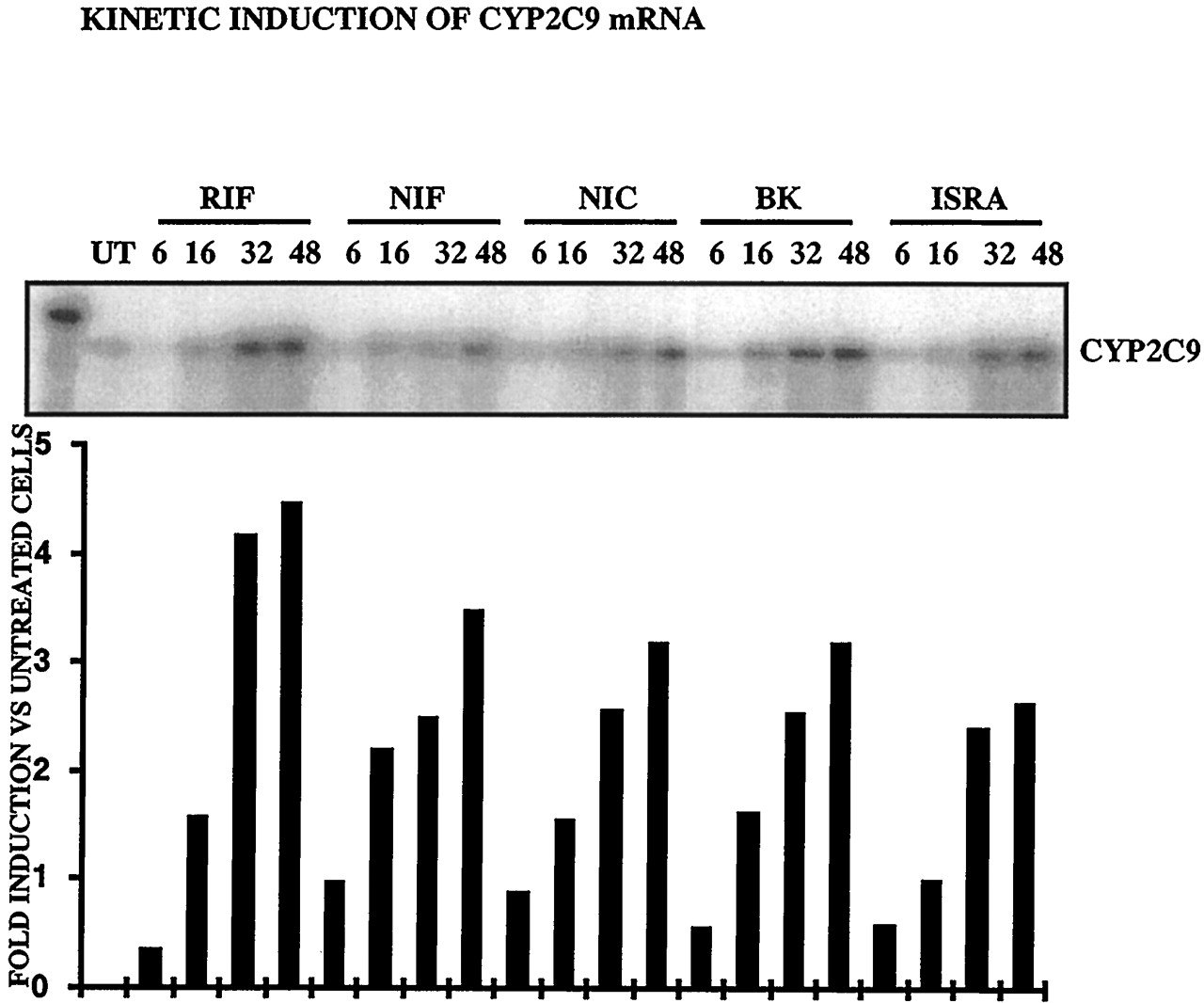
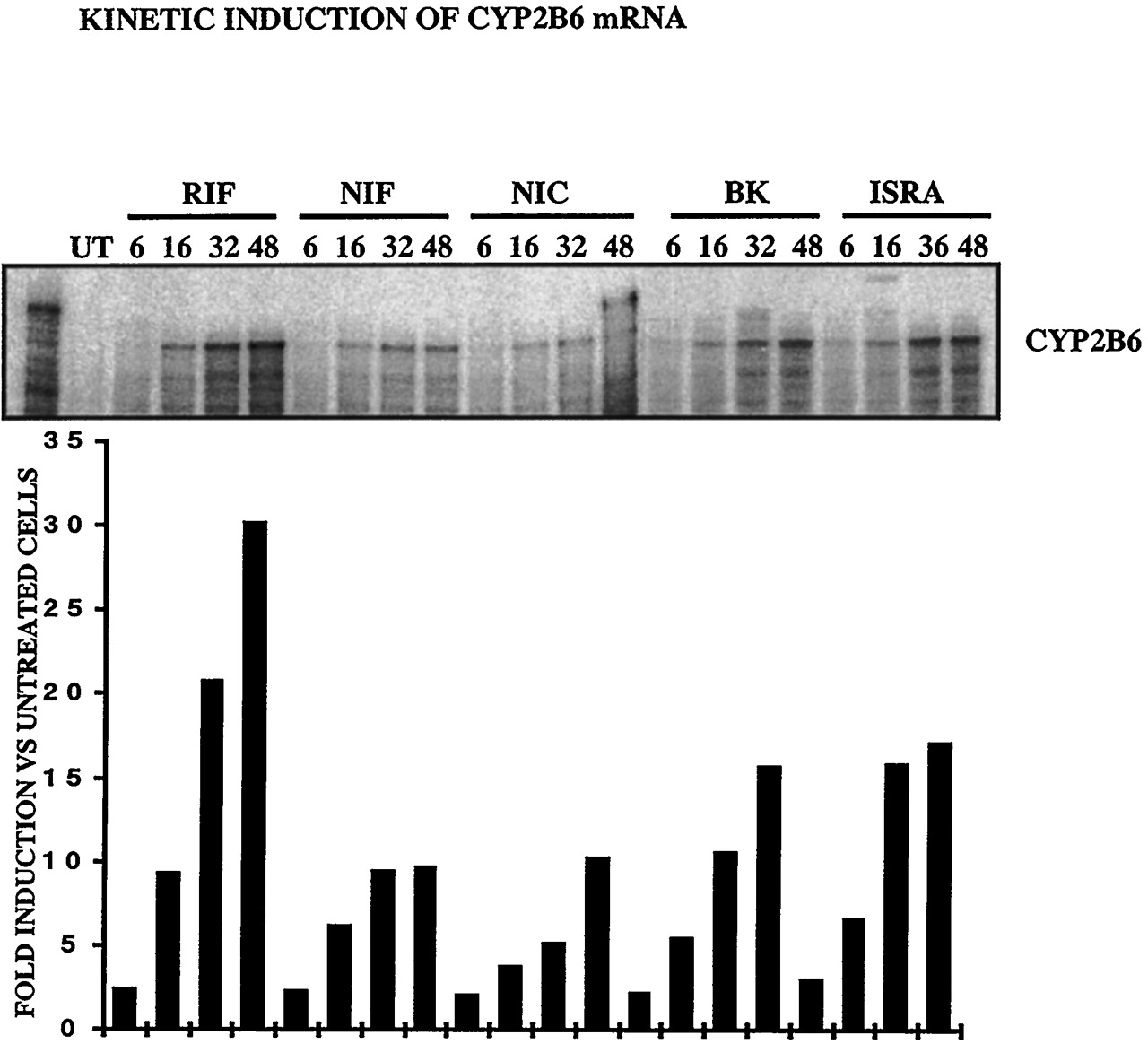
These results show that CCM bind and activate hPXR. Other compounds, such as rifampicin, known to activate the pregnane X receptor, have been shown to induce CYP2B6 and to a lesser extent, CYP2C9 mRNAs and proteins (Gerbal-Chaloin et al., 2001). We then investigated whether the CCM were inducers of these cytochromes. Figure6 shows that nifedipine, nicardipine, BK8644, and isradipine were inducers of CYP2C9 at the RNA level (3.5-, 3.2-, 3.18-, and 2.64-fold, respectively, with respect to untreated cells). Similarly, all the CCM tested induced an accumulation of the CYP2B6 mRNA (9.6-, 10.2-, 15.6-, and 17-fold for nifedipine, nicardipine, BK8644, and isradipine, respectively) (Fig.7). The finding that nifedipine, nicardipine, isradipine, and BK8644 are inducers of CYP2B6 and 2C9, as is rifampicin, is in favor of a possible implication of the PXR in the regulation of these genes.

CCM induce CYP2C9 in primary cultures of human hepatocytes (FT178; similar results were obtained with livers FT166, and FT167).
Twenty-four hours after plating, 107 hepatocytes were treated by 10 μM BK, RIF, NIF, NIC, ISRA, or 0.1% DMSO for 6, 16, 32, or 48 h. Total RNA (20 μg) was analyzed by ribonuclease protection assay with antisense CYP2C9. Radioactivity in the band was measured by PhosphorImager and ImageQuant software. Values are expressed in -fold induction compared with DMSO-treated cells.

CCM induce CYP2B6 in primary cultures of human hepatocytes (FT178; similar results were obtained with livers FT166, and FT167).
Twenty four hours after plating, 107 hepatocytes were treated by 10 μM BK, RIF, NIF, NIC, ISRA, or 0.1% DMSO for 6, 16, 32, or 48 h. Total RNA (20 μg) was analyzed by ribonuclease protection assay with antisense CYP2B6. Radioactivity in the higher band was measured by PhosphorImager and ImageQuant software. Values are expressed in -fold induction compared with DMSO-treated cells.
Discussion
In this work we have shown that nifedipine and three other calcium channel modulators of the dihydropyridine family induce CYP3A4 at the mRNA, protein, and activity levels in a dose-dependent manner. We also found that this induction is delayed due to dexamethasone-mediated intermediate synthesis, following a pathway similar to rifampicin. Transfection studies showed that full induction was obtained only for the CYP3A4 construct containing both the proximal and the distal hPXR response elements and that cotransfection of hPXR was also required, suggesting CCM as hPXR ligands. This was confirmed 1) in GST pull-down experiments where CCM produced a ligand-induced interaction between hPXR and SRC-1; and 2) by the transactivation of the (XREM/263/11) CYP3A4 construct only in the primary culture of human hepatocytes containing the dexamethasone-induced hPXR. Finally, we found that the CCM tested also induced the CYP2C9 and 2B6 in cultured human hepatocytes.
Koleva reported that administration of nifedipine to rats shortened hexobarbital sleeping times and increased the metabolism of several drug substrates (Koleva and Stoytchev, 1993, 1995). On the basis of these results, these authors suggested that CYP2B, 2C, and 3A may be induced by nifedipine. Indeed, they are induced by the same compounds, including PB, RIF, and DEX (Gerbal-Chaloin et al., 2001) in humans. This is the first report showing that they are also inducible by nifedipine and its analogs in primary cultures of human hepatocytes, a model that represents the most reliable in vitro system for evaluating the inducibility of P450 genes in response to xenobiotics in man.
The CYP2B are models of the PB induction, which has been shown to be mediated by the CAR via its activation and translocation in the nucleus. For CYP2C genes, the receptors implicated in induction have not yet been characterized. For CYP3A4, induction is mediated by hPXR, which has been shown to be activated by PB, RIF (Kliewer et al., 1998), and by CCM of the dihydropyridine family (this work). The question remains whether several receptors could be responsible for the induction of CYP2C and 2B as, to date, RIF and nifedipine have been found to activate hPXR. We have shown that, in contrast to DEX, which increases the expression of CAR and its nuclear translocation, PB increases only its nuclear translocation, and RIF has no effect on either expression or translocation of CAR (not shown). These results suggest that hPXR possibly has a role in the transactivation of CYP2B and 2C.
During this study, we found that nicardipine strongly induced CYP3A4 in contrast to the action of nifedipine. Indeed, nicardipine differs from nifedipine in that it contains anN-benzyl-N-methylaminoethyl side chain, and its nitro group is located at position C3 rather than at C2. Previous studies demonstrated that release of theN-benzyl-N-methylaminoethyl side chain of nicardipine occurs rapidly in the rat, and the hydrolysis product represents a major metabolite in the rat and other species (Higuchi et al., 1977; Higuchi and Shiobara, 1980). Because of the importance of this chain and of the position of the nitro group in nicardipine, further examination is required to determine how these functional groups affect CYP3A induction. In addition, nicardipine induces both human CYP3A4 (this work) and rat CYP3A23 (Zangar et al., 1999) in contrast to nifedipine, which seems restricted to the human isoform. Since our results suggest that compounds of the dihydropyridine family can act as activators of hPXR, structure-activity studies should be performed to find compounds keeping their CCM activity but lacking the inducing capability.
Acknowledgments
We are grateful to Drs. Colin Young and Paul Bello for careful reading of the manuscript.
Footnotes
-
This work was supported in part by GlaxoWellcome (L.D.) and la Ligue Nationale contre le Cancer (J.-M.P.).
- Abbreviations used are::
- CCM
- calcium channel modulators
- P450
- cytochrome P450
- PXR
- pregnane X receptor
- hPXR
- human PXR
- CAR
- constitutively activated receptor
- mCAR
- mouse CAR
- DMSO
- dimethyl sulfoxide
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)]-benzene
- PCR
- polymerase chain reaction
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GST
- glutathione S-transferase
- PAGE
- polyacrylamide gel electrophoresis
- CSA
- cyclosporin A
- NIF
- nifedipine
- BK
- BK8644
- ISRA
- isradipine, RIF, rifampicin
- NIC
- nicardipine
- SRC
- steroid receptor coactivator
- PB
- phenobarbital
- DEX
- dexamethasone
- CARLA
- coactivator receptor ligand assay
- ER6
- everted repeat 6
- Received March 26, 2001.
- Accepted July 9, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}