


Abstract
Under certain culture conditions, exposure of the human colon adenocarcinoma cell line Caco-2 to 1,25-(OH)2-D3 induces expression of CYP3A4 to levels comparable to that in human small intestinal epithelium. To determine whether 1,25-(OH)2-D3 could be used to restore CYP3A expression in other culture models, we examined several cell lines derived from malignancies of human tissues known to express CYP3A enzymes: Hep G2 (liver), LS180 (colon), HPAC (pancreas), Hs746T (stomach). Primary cultures of human hepatocytes from two donors were also examined. 1,25-(OH)2-D3 increased CYP3A catalytic activity in LS180 (15-fold), HPAC (6-fold), and hepatocytes (2- to 3-fold); this was accompanied by induction of CYP3A4 mRNA and CYP3A immunoreactive protein. However, 1,25-(OH)2-D3had no effect on CYP3A expression in Hs746T or Hep G2. Known ligands for pregnane X receptor (PXR) (rifampin, dexamethasone, and dexamethasone t-butyl acetate) markedly induced CYP3A4 expression in human hepatocytes. In contrast, these ligands had little or no effect on CYP3A4 expression in Caco-2 cells, even at concentrations 1 to 2 orders of magnitude greater than effective concentrations of 1,25-(OH)2-D3 or two other vitamin D receptor (VDR) ligands (25-OH-D3 and 1-OH-D3). The retinoic acid receptor ligand all-trans-retinoic acid augmented the 1,25-(OH)2-D3-mediated induction of CYP3A4 catalytic activity up to 2-fold in Caco-2 cells, while having no demonstrable effect on levels of CYP3A4 mRNA or protein. The retinoid X receptor ligand 9-cis-retinoic acid appeared to slightly reduce CYP3A4 catalytic activity. We conclude that 1,25-(OH)2-D3 can be used to increase CYP3A4 expression in some, but not all, human cell lines derived from tissues known to express CYP3A enzymes. The mechanisms involved in this induction are unlikely to involve PXR and may involve VDR.
Cytochrome P450 (CYP2) 3A4 is the principal CYP isoform expressed in both human liver (Shimada and Guengerich, 1989) and small intestinal epithelial cells (enterocytes) (Watkins et al., 1987; Kolars et al., 1992). It is now well recognized that, along with hepatic CYP3A4, intestinal CYP3A4 can be a major factor in determining the extent of the first-pass metabolic extraction and, hence, oral bioavailability of some commonly prescribed drugs (Hall et al., 1999).
The human colon adenocarcinoma cell line Caco-2 has been widely used to study oral drug absorption although, under general culture conditions, it lacks expression of several drug-metabolizing enzymes, including CYP3A4. We have previously reported that, when Caco-2 cells are grown on extracellular matrix-coated permeable supports, treatment with the naturally occurring hormone 1α,25-dihydroxyvitamin D3[1,25-(OH)2-D3] induces expression of CYP3A4 mRNA, immunoreactive protein, and catalytic activity (midazolam 1′-hydroxylation) to levels comparable to those observed in human intestinal epithelial cells (Schmiedlin-Ren et al., 1997). This observation has led to a 1,25-(OH)2-D3-treated Caco-2 cell model that appears to more closely mimic the function of the human intestinal epithelium as a barrier to orally ingested CYP3A4 substrates, such as midazolam (Schmiedlin-Ren et al., 1997; Fisher et al., 1999a,b) and indinavir (Hochman et al., 2000). Using heterologous expression techniques, other investigators have developed CYP3A4-expressing Caco-2 cells (Crespi et al., 1996; Hu et al., 1999;Brimer et al., 2000). However, the use of 1,25-(OH)2-D3 treatment may result in a more physiologically relevant model since the endogenous CYP3A4 gene is up-regulated.
The human liver hepatoblastoma cell line Hep G2 is another commonly used experimental model that lacks CYP3A4 expression (Schuetz et al., 1993), limiting its usefulness in studying drug disposition. We reasoned that up-regulation of CYP3A4 gene expression in Hep G2 and other cell lines might lead to additional improved models for the study of drug disposition. We therefore used the same culture conditions found to maximize CYP3A4 expression and function in Caco-2 cells to examine the 1,25-(OH)2-D3responsiveness of Hep G2 cells, three other cell lines derived from malignancies of human tissues known to express CYP3A4 (Watkins et al., 1987; Kolars et al., 1994), and primary human hepatocytes.
We report that, although 1,25-(OH)2-D3 treatment results neither in CYP3A4 expression in Hep G2 cells nor in a human stomach carcinoma cell line (Hs746T), it does increase CYP3A4 expression in LS180 cells (human colon adenocarcinoma), HPAC cells (human pancreas adenocarcinoma), and in primary human hepatocytes. We also show that CYP3A4 is inducible in Caco-2 cells by the vitamin D receptor (VDR) ligand 1α-hydroxyvitamin D3(1-OH-D3), in addition to the VDR ligands 1,25-(OH)2-D3 and 25-hydroxyvitamin D3(25-OH-D3). However, CYP3A4 is not substantially induced in Caco-2 cells by known ligands for PXR (pregnane X receptor;Lehmann et al., 1998), also referred to as the pregnane-activated receptor (PHR; Bertilsson et al., 1998) or steroid and xenobiotic receptor (SXR; Blumberg et al., 1998). Collectively, our observations indicate that 1,25-(OH)2-D3 can be used to up-regulate CYP3A4 expression in some, but not all, human digestive tract cell lines and that this induction may involve VDR and not PXR.
Experimental Procedures
Materials.
Hep G2, LS180, HPAC, and Hs746T cells were obtained from American Type Culture Collection (Manassas, VA). The Caco-2 cell clone P27.7 was isolated from the parent cell line (ATCC HTB37), as previously described (Schmiedlin-Ren et al., 1997). Cryopreserved normal human hepatocytes were obtained from Clonetics Corporation (San Diego, CA).
Culture inserts with track-etched polyethylene terephthalate (PET) membranes (1-μm pore size) were purchased from Becton Dickinson Labware (Bedford, MA) and were either uncoated or coated with laminin (25 μg/cm2 as a dry film). Uncoated inserts were coated in-house with laminin (Becton Dickinson Labware) at 5 μg/cm2 according to the manufacturer's instructions. In a small trial, Caco-2 cells grown on inserts coated in-house with laminin at 1, 5, or 10 μg/cm2 and given a 4 μM apical dose of midazolam (MDZ) generated a concentration of 1′-hydroxymidazolam (1′-OH-MDZ), which was approximately 82% of the concentration generated by cells grown on commercially coated inserts. The CYP3A4 band on immunoblots was slightly more intense for cells grown on commercially coated inserts than for cells grown on the inserts coated in-house (not shown). There was essentially no difference in CYP3A4 expression or catalytic activity among cells grown on 1, 5, or 10 μg of laminin/cm2; 5 μg/cm2 was selected for routine use when commercially coated inserts became unavailable.
DMEM, medium supplements, and Hanks' balanced salt solution were obtained from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was obtained from Hyclone Laboratories (Logan, UT). Hepatocyte basal medium and supplements to prepare the hepatocyte growth medium were obtained from Clonetics Corporation. The hydrocortisone and gentamicin/amphotericin supplements were not added to the hepatocyte basal medium before use.
1,25-(OH)2-D3 (purity ≥99% by thin layer chromatography) and 25-OH-D3(purity ≥98% by thin layer chromatography) were obtained from Calbiochem (San Diego, CA). 1-OH-D3 (purity 99.7% by HPLC) was obtained from Tetrionics (Madison, WI). Pregnenolone 16α-carbonitrile (PCN) was a gift from Dr. Erin Schuetz (St. Jude Children's Research Hospital, Memphis, TN). Rifampin, dexamethasone, and pregnenolone were obtained from Sigma (St. Louis, MO). Dexamethasone t-butyl acetate (DtBA) was obtained from Research Plus (Bayonne, NJ).
Stock solutions of 1,25-(OH)2-D3, 25-OH-D3, and 1-OH-D3 were made 1000-fold concentrated in absolute ethanol. Stock solutions of rifampin, dexamethasone, DtBA, pregnenolone, and PCN were made 1000-fold concentrated in dimethyl sulfoxide. The midazolam stock solution was 4 mM in dimethyl sulfoxide.
N-methyl-N-(t-butyl-dimethylsilyl)trifluoroacetamide was purchased from Pierce (Rockford, IL). MDZ, [15N3]MDZ, 1′-OH-MDZ, and deuterated 1′-hydroxymidazolam (D2-1′-OH-MDZ) were gifts from Roche Laboratories (Nutley, NJ). The stable isotope-labeled internal standard ([15N3]1′-OH-MDZ) was generated from an incubation of [15N3]MDZ with human liver microsomes, as previously described (Schmiedlin-Ren et al., 1997). Other chemicals were obtained from Sigma and were of tissue culture or molecular biology grades where appropriate.
Cell Culture.
All cultures were maintained in a humidified 37°C incubator with a 5% carbon dioxide in air atmosphere.
Cell Line Studies.
Each cell line was grown and expanded in the recommended medium in plastic tissue culture dishes until sufficient cells were available for experimentation. The cells were then removed from plastic by trypsin digestion (Hep G2 at passage 80, HPAC at passage 122, Hs746T at passage 22) or by scraping (LS180 at passage 41) and were seeded onto laminin-coated culture inserts. The cells were allowed to grow in the inserts for 6 days (Hep G2 and HPAC) or 20 days (LS180 and Hs746T) in complete growth medium as used for Caco-2 cells (below). After the stated growth period, the medium was changed to complete differentiation medium as used for Caco-2 cells (below) for the remaining 3 weeks in culture. Because it was a component of the medium recommended by ATCC, 1 mM sodium pyruvate was added to the growth and differentiation media for Hep G2 cells. After 1 week, 0.25 μM 1,25-(OH)2-D3 or vehicle was added to the medium for the final 2 weeks in culture. At the end of the culture period, the medium was replaced, and 4 μM midazolam was applied apically. After a 4-h incubation at 37°C, the medium was recovered from both compartments, combined, and then assayed for 1′-OH-MDZ by gas chromatography/mass spectrometry (GC/MS) (Schmiedlin-Ren et al., 1997). Cells from a portion of the membrane were scraped into denaturing solution (Chomczynski and Sacchi, 1987), and isolated RNA was subjected to RT-PCR. The cells from the remainder of the membrane were scraped and homogenized in solution D [20% v/v glycerol, 100 mM Tris HCl, pH 7.4, 10 mM EDTA, 1 mM dithiothreitol (Bonkovsky et al., 1985)] containing 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, and 100 μg/ml aprotinin. These homogenates were subjected to SDS-PAGE.
Caco-2 Cell Studies.
Using a medium consisting of DMEM containing 25 mM glucose and 4 mMl-glutamine and supplemented with 0.1 mM nonessential amino acids, 45 nM dl-α-tocopherol, 100 U/ml sodium penicillin G, and 100 μg/ml streptomycin, complete growth medium was prepared by adding 20% heat inactivated FBS. Cells of the Caco-2 cell clone P27.7 (Schmiedlin-Ren et al., 1997) at passages 29 to 33 were seeded at 6 × 105 cells/cm2onto laminin-coated PET culture inserts using complete growth medium. Upon reaching confluence, the medium was changed to differentiation medium (95% DMEM containing 25 mM glucose and 4 mMl-glutamine and supplemented with 0.1 mM nonessential amino acids, 45 nM dl-α-tocopherol, 100 U/ml sodium penicillin G, 100 μg/ml streptomycin, 0.1 μM sodium selenite, 3 μM zinc sulfate, and 5 μM ferrous sulfate; 5% heat inactivated FBS) for an additional 2 weeks in culture. During this period, the cells were treated with varying concentrations of 1,25-(OH)2-D3, 25-OH-D3, 1-OH-D3, PCN, DtBA, pregnenolone, or rifampin. At the end of the culture period, the upper compartment (apical) medium was replaced with medium containing 4 μM midazolam, and the lower compartment (basolateral) medium was replaced with midazolam-free medium. After a 4-h incubation at 37°C, the medium from both compartments was recovered and assayed separately for 1′-OH-midazolam by GC/MS (Paine et al., 1996; Schmiedlin-Ren et al., 1997) or by liquid chromatography/mass spectrometry (LC/MS). In some cases, a portion of the membrane with the attached monolayer was cut out, fixed in formalin, embedded in paraffin, and sectioned. The sections were stained with H&E and examined by light microscopy. Cells from another portion of the membrane were lysed in denaturing solution (Chomczynski and Sacchi, 1987), and isolated RNA was subjected to RT-PCR. The cells from the remainder of the membrane were scraped and homogenized in solution D containing protease inhibitors as above, and the homogenates were subjected to SDS-PAGE.
Human Hepatocyte Studies.
Cryopreserved normal human hepatocytes from two different donors were obtained from Clonetics Corporation, thawed, and seeded at 1.4 × 105 cells/cm2 onto PET inserts commercially coated with laminin. FBS (5%) was present for only the first 24 h of culture. Treatments were begun on the third day after seeding and continued for 7 days. Cultures were done in duplicate. At the end of treatment, the medium was replaced (both apically and basolaterally) on one of each pair of inserts with medium containing 4 μM midazolam. After 4 h, the medium was recovered from both compartments, combined, and assayed for 1′-OH-midazolam by LC/MS. The cells were scraped into lysis buffer (0.5% Triton X-100, 5 mM EDTA, 150 mM NaCl, 8 mM TES, pH 7.5), containing 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, and 100 μg/ml aprotinin, and allowed to stand on ice for 1 h. The lysates were then centrifuged for 10 min at 10,000g at 4°C to pellet the nuclei. The cleared lysates were subjected to SDS-PAGE. The cells from the second set of cultures were lysed in denaturing solution (Chomczynski and Sacchi, 1987), and isolated RNA was subjected to RT-PCR.
1′-Hydroxymidazolam Assays.
Quantitation of 1′-hydroxymidazolam (1′-OH-MDZ) in culture medium was accomplished by GC/MS, as previously described (Paine et al., 1996;Schmiedlin-Ren et al., 1997) or by LC/MS; the method used for a given experiment was selected based on instrument availability. All culture samples were analyzed in duplicate, and the mean of the two measurements is reported.
In preparation for LC/MS, the volume of fresh culture medium needed to bring the total volume to 1 ml was added to a 150- to 600-μl portion of each sample. Standards were prepared in the same medium as the samples. Samples and standards were then spiked with 10 pmol of D2-1′-OH-MDZ as internal standard, extracted with ethyl acetate, and dried in a SpeedVac (Savant, Holbrook, NY). The final residue was redissolved and then injected into the HPLC system with a YMC J′sphere ODS-M80 column (150 × 2 mm; 4-μm particle size; C18; Waters, Milford, MA) fitted with a 30- × 10-mm YMC precolumn.
The LC/MS analyses of 1′-OH-MDZ were performed using a Hewlett Packard series 1100 HPLC system (Palo Alto, CA) coupled to a Thermo Finnigan (San Jose, CA) LCQ classic mass spectrometer operating in the positive ion electrospray ionization mode. Analytes with mass-to-charge ratios (m/z) of 342 (1′-OH-MDZ) or 346 (37Cl-isotope of D2-1′-OH-MDZ) were subjected to collision-induced dissociation using helium as the collision gas, and fragments were detected between m/z 220 to 360 in ms2 full scan mode. The mass spectrometric peak areas were determined for fragment (daughter) ions with m/z of 324 and 328, corresponding to 1′-OH-MDZ and the 37Cl-isotope of D2-1′-OH-MDZ, respectively. Concentrations of 1′-OH-MDZ were determined by peak area ratios. The interday coefficient of variation in the slope of the standard curve was 7.5%. Other than temporary time savings due to instrument availability, the LC/MS method did not offer any benefit over the GC/MS method and has since been abandoned.
Immunoblots.
The protein concentrations of the cell homogenates or lysates were measured by the method of Bradford (1976), using bovine serum albumin as the reference standard. The homogenates or lysates were electrophoresed in 10% polyacrylamide gels containing 0.1% SDS, and the separated proteins were electrophoretically transferred to a polyvinylidene difluoride membrane (0.45-μm pore size; Amersham Pharmacia Biotech, Piscataway, NJ). The immunoblots were developed as previously described (Lown et al., 1994). CYP3A proteins were detected using a mouse monoclonal antibody named 13-7-10 (Beaune et al., 1985), which was a gift from Dr. Pierre Kremers (Université de Liège, Liège, Belgium). This antibody detects all known forms of human CYP3A. CYP3A5 was detected using a rabbit antibody raised against a specific peptide of CYP3A5 (GENTEST, Woburn, MA). Villin and albumin were used as control proteins. Villin is a cytoskeletal protein associated with microvilli present on the apical membranes of enterocytes (West et al., 1988), the apical membranes of pancreatic acinar and ductular cells (Elsässer et al., 1991), and on the bile canalicular membrane of hepatocytes (Tsukada et al., 1995) and Hep G2 cells (Sormunen et al., 1993). Villin was detected using a mouse monoclonal antibody (Chemicon International, Temecula, CA). Albumin was detected using polyclonal rabbit anti-human albumin (ICN, Costa Mesa, CA). Rabbit anti-mouse IgG and goat anti-rabbit IgG/A/M conjugated with horseradish peroxidase were obtained from Zymed Laboratories (San Francisco, CA). Binding of secondary antibodies was detected using enhanced chemiluminescence reagents and film from Amersham Pharmacia Biotech. Immunoblots were repeated twice; bands from representative blots are shown.
mRNA Analyses.
Reverse transcriptase (from avian myeloblastosis virus), dNTPs, and Taq DNA polymerase were obtained from Roche (Indianapolis, IN) and oligo dT [12–18] from Amersham Pharmacia Biotech. cDNA was prepared from the total RNA, as previously described (Schmiedlin-Ren et al., 1993). The polymerase chain reaction was performed using a PTC-100 programmable thermal cycler (MJ Research, Watertown, MA). Primer sequences for amplification of CYP3A (CCTTACATATACACACCCTTTGGAAGT and AGCTCAATGCATGTACAGAATCCCCGGTTA; product size, 382 bp) and villin cDNA (product size, 303 bp) were as previously described (Kolars et al., 1994; Schmiedlin-Ren et al., 1997). The CYP3A primers were designed with the intention of amplifying CYP3A4 specifically. CYP3A5 cDNA cannot be amplified with these primers because the region complimentary to the antisense primer does not exist in the shorter CYP3A5 cDNA. However, it is possible that the primers could amplify CYP3A7 cDNA since the antisense primer contains only four mismatches, and there are no mismatches within the sense primer. Sequencing of the amplified fragments obtained with cDNA from 1,25-(OH)2-D3 treated Hep G2, HPAC, and LS180 cells was therefore done to confirm specificity. We have previously sequenced the PCR products obtained using these primers to amplify cDNAs from Caco-2 cells (Schmiedlin-Ren et al., 1997) and intestine; the sequences were consistent with the amplification of CYP3A4 in both of these instances.
PCR reaction mixtures were obtained as previously described (Schmiedlin-Ren et al., 1993). Each thermal cycle consisted of 95°C for 1 min, then 65°C for 1 min 15 s; after completing all cycles, a 10-min extension step at 65°C was performed. PCR products were electrophoresed on agarose gels and stained with ethidium bromide. A reagent control, included in each PCR run, was confirmed to be negative when run on the gel. PCR products were judged to be of the size appropriate for amplification of the specific cDNA by comparison with molecular weight standards included on each gel. PCR reactions were done in duplicate; bands from representative gels are shown.
Sequencing of PCR Products.
Sequencing was done by the University of North Carolina (Chapel Hill, NC) Automated DNA Sequencing Facility on a 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA). The sequencing reaction was done using the ABI PRISM BigDye terminator cycle sequencing ready reaction kit with AmpliTaq DNA Polymerase FS (Applied Biosystems).
Results
Under culture conditions that were previously shown to produce maximal induction of CYP3A4 in Caco-2 cells (Schmiedlin-Ren et al., 1997), a 2-week treatment with 1,25-(OH)2-D3 did not produce a detectable increase in expression of CYP3A immunoreactive protein, mRNA, or catalytic activity in Hep G2 or Hs746T cells (Fig.1). However, exposure to 1,25-(OH)2-D3 resulted in increases in CYP3A catalytic activity of 15-fold in LS180 cells and 6-fold in HPAC cells (Fig. 1). This increase in catalytic activity was accompanied in both cell types by an induction of CYP3A immunoreactive protein and an increase in CYP3A4 mRNA. Expression of the structural protein villin did not change with 1,25-(OH)2-D3 treatment of LS180 cells. Although villin immunoreactive protein was not detected in HPAC cells, villin mRNA was detectable by RT-PCR and increased in response to 1,25-(OH)2-D3treatment. CYP3A5 immunoreactive protein was detected only in the HPAC cell homogenates and did not appear to respond to 1,25-(OH)2-D3 treatment.

Effect of 1,25-(OH)2-D3 on expression of CYP3A4 in various cell lines derived from malignancies of human tissues known to express CYP3A enzymes.
Cells were seeded onto culture inserts commercially coated with laminin and grown for 13 days [Hep G2 (liver) and HPAC (pancreas)] or 21 days [LS180 (colon) and Hs746T (stomach)] before initiation of a 2-week treatment with 1,25-(OH)2-D3 (0.25 μM), applied apically and basolaterally. At the end of treatment, the medium was replaced, and 4 μM midazolam was applied apically. Following a 4-h incubation, the medium from the apical and basolateral compartments was recovered, combined, and assayed for 1′-hydroxymidazolam (1′-OH-MDZ). A portion of the cells was homogenized and subjected to SDS-PAGE. The remaining cells were lysed, and RNA was isolated and subjected to RT-PCR using primer pairs, described underExperimental Procedures. The CYP3A PCR products from the 1,25-(OH)2-D3-treated cells were sequenced. A, CYP3A catalytic activity as measured by 1′-hydroxylation of MDZ. The metabolite was assayed in culture medium by GC/MS. B, ethidium-stained gels of products from RT-PCR (34 cycles) using primers of the indicated specificities. The sequences of the amplified products for the HPAC and LS180 cells were consistent with amplification of CYP3A4 cDNA, whereas the sequence of the amplified product for Hep G2 cells was consistent with amplification of CYP3A7 cDNA. C, immunoblots probed with antibodies of the indicated specificities and developed using enhanced chemiluminescence (10 μg of homogenate protein/lane). BDL, below detectable limits.
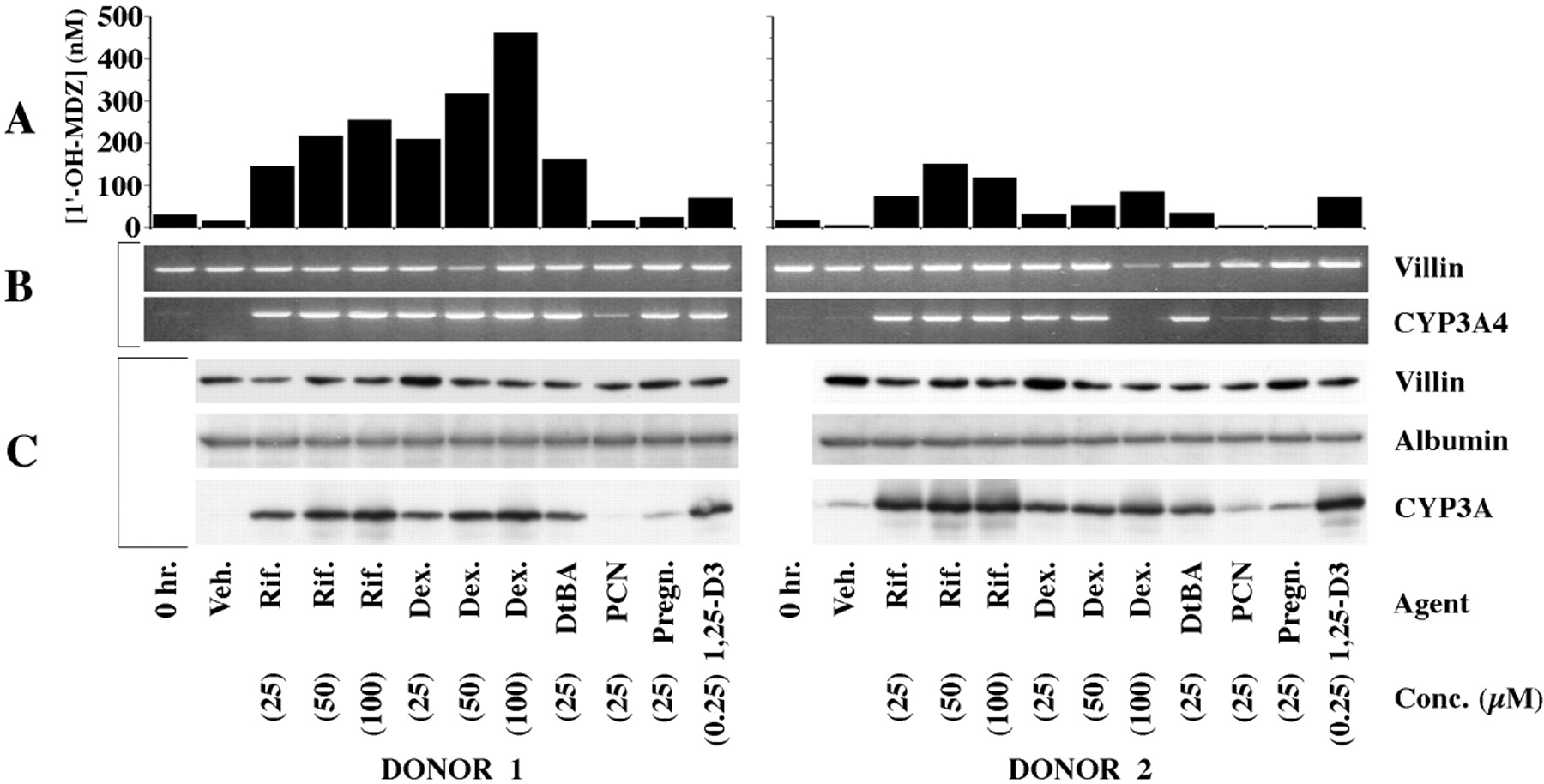
The lack of response of Hep G2 cells suggested that primary human hepatocytes might also not respond to 1,25-(OH)2-D3. To address this issue, cryopreserved human hepatocytes from two donors were seeded onto laminin-coated culture inserts and exposed to known CYP3A4 inducers in addition to 1,25-(OH)2-D3. Compared with vehicle-exposed cells, cells treated for 7 days with rifampin (25, 50, or 100 μM), dexamethasone (25, 50, or 100 μM), or DtBA (25 μM) had increased CYP3A4 mRNA and CYP3A immunoreactive protein and catalytic activity (Fig. 2). PCN had no detectable effect on CYP3A4 regulation. Pregnenolone appeared to increase CYP3A4 immunoreactive protein and mRNA in each donor but had minimal or no effect on catalytic activity. Treatment with 1,25-(OH)2-D3 produced an increase in CYP3A4 mRNA and CYP3A immunoreactive protein and catalytic activity in the hepatocytes of both donors. The magnitude of induction of CYP3A catalytic activity produced by 1,25-(OH)2-D3 was less than that observed with rifampin. CYP3A5 immunoreactive protein was not detected in the hepatocytes of either donor under any of the treatment conditions (not shown).

Effects of 1,25-(OH)2-D3 and PXR ligands on CYP3A4 expression in primary human hepatocytes prepared from two donors.
Cryopreserved normal human hepatocytes were thawed and seeded onto culture inserts commercially coated with laminin and grown for 3 days before initiation of a 7-day treatment with the indicated agents, applied apically and basolaterally. At the end of treatment, the apical and basolateral media were replaced with medium containing 4 μM midazolam. Following a 5-h incubation, the medium from the apical and basolateral compartments was recovered, combined, and assayed for 1′-OH-MDZ. The cells were lysed and subjected to SDS-PAGE. Cells from a parallel set of cultures were lysed, and RNA was isolated and subjected to RT-PCR. A, CYP3A catalytic activity as measured by 1′-hydroxylation of MDZ. The metabolite was assayed in culture medium by LC/MS. B, ethidium-stained gels of products from RT-PCR using primers of the indicated specificities (CYP3A4, 35 cycles; villin, 34 cycles). C, immunoblots probed with antibodies of the indicated specificities and developed using enhanced chemiluminescence (10 μg of lysate protein/lane). Veh, vehicle; Rif, rifampin; Dex, dexamethasone; Pregn, pregnenolone.
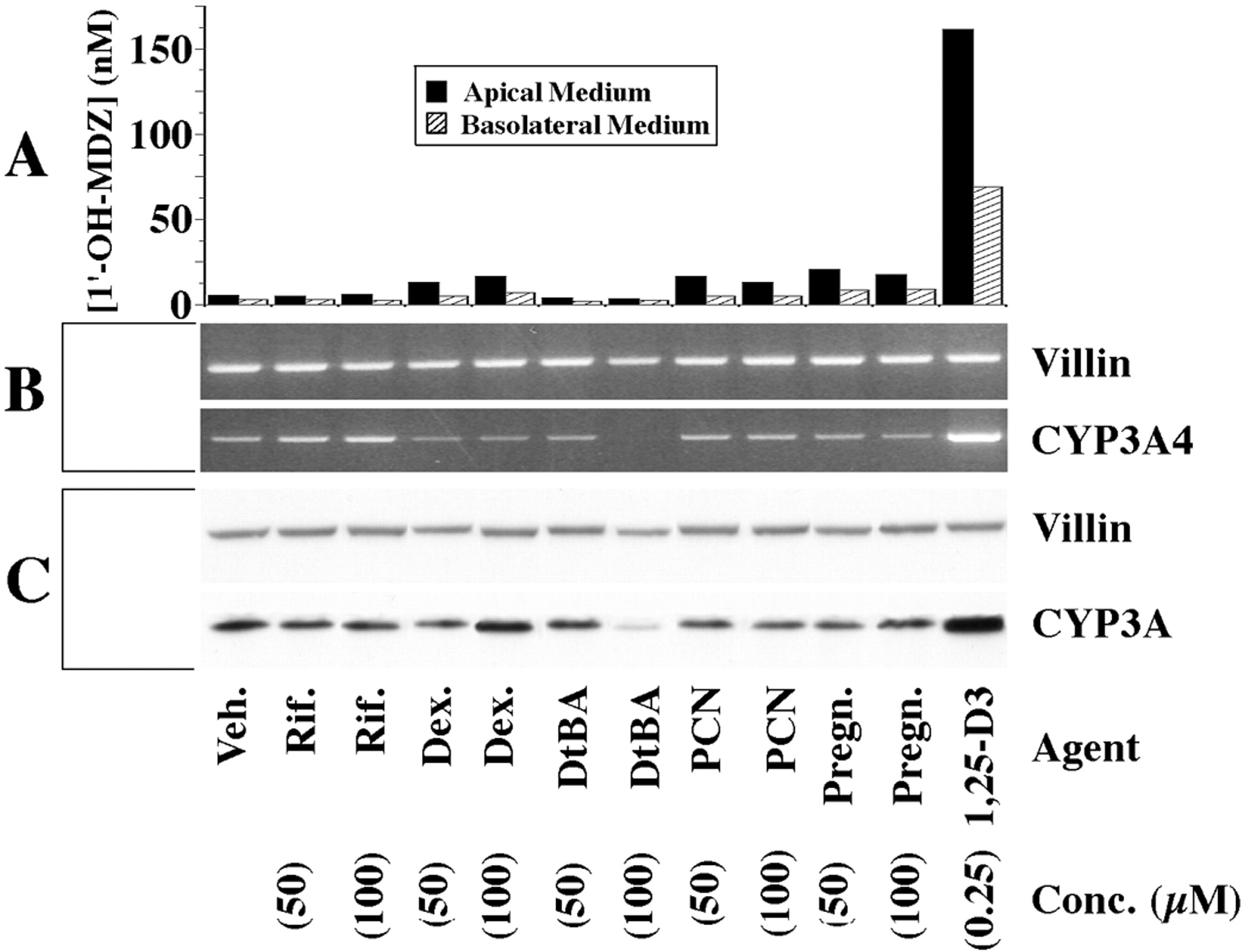
We next examined Caco-2 cells for their responsiveness to known PXR ligands. As shown in Fig. 3, treatment with rifampin or DtBA produced no detectable induction of CYP3A4 mRNA, protein, or catalytic activity. Treatment with PCN or pregnenolone appeared to increase CYP3A catalytic activity (relative to vehicle) but had minimal or no effect on the levels of CYP3A immunoreactive protein or CYP3A4 mRNA. Dexamethasone treatment also appeared to increase CYP3A catalytic activity and, at the highest dose tested, also appeared to increase CYP3A immunoreactive protein. The inductive effects of all of the examined PXR ligands were clearly small compared with those of 1,25-(OH)2-D3.

Effects of PXR ligands on expression of CYP3A4 in Caco-2 cells.
Clone P27.7 Caco-2 cells (passage 32) were seeded onto culture inserts commercially coated with laminin and grown for 9 days before initiation of a 2-week treatment with the indicated agents, applied apically and basolaterally. At the end of treatment, the medium was replaced, and 4 μM midazolam was applied apically. Following a 4-h incubation, the medium from the apical and basolateral compartments was recovered and assayed separately for 1′-OH-MDZ. A portion of the cells was homogenized and subjected to SDS-PAGE. The remaining cells were lysed, and RNA was isolated and subjected to RT-PCR. A, CYP3A catalytic activity as measured by 1′-hydroxylation of MDZ. The metabolite was assayed in culture medium by LC/MS. B, ethidium-stained gels of products from RT-PCR using primers of the indicated specificities (CYP3A4, 34 cycles; villin, 25 cycles). C, immunoblots probed with antibodies of the indicated specificities and developed using enhanced chemiluminescence (10 μg of homogenate protein/lane). The decrease in CYP3A immunoreactive protein and CYP3A4 mRNA in the cells treated with DtBA at 100 μM was probably due to toxicity secondary to incomplete solubility of DtBA (noted microscopically). Transepithelial electrical resistance across this monolayer progressively declined over the course of treatment, falling to a level below that considered to signify confluence (250 Ohm · cm2; Traber et al., 1987) by the end of 2 weeks. Veh, vehicle; Rif, rifampin; Dex, dexamethasone; Pregn, pregnenolone.
To further characterize the induction of CYP3A4 in Caco-2 cells by 1,25-(OH)2-D3, a dose-response experiment was done using 1-OH-D3, a high affinity vitamin D receptor analog not previously examined. Increases in CYP3A4 mRNA and CYP3A immunoreactive protein and catalytic activity were observed with the lowest dose used (0.01 μM), and each rose in a dose-dependent manner with maximal response achieved at a dose of approximately 2.5 μM (Fig. 4).

Effect of 1-OH-D3 on CYP3A4 expression in Caco-2 cells.
Clone P27.7 Caco-2 cells (passage 29) were seeded onto culture inserts commercially coated with laminin and grown for 5 days before initiation of a 2-week treatment with varying concentrations of 1-OH-D3, applied apically and basolaterally. At the end of treatment, the medium was replaced, and 4 μM midazolam was applied apically. Following a 4-h incubation, the medium from the apical and basolateral compartments was recovered and assayed separately for 1′-OH-MDZ. A portion of the cells was homogenized and subjected to SDS-PAGE. The remaining cells were lysed, and RNA was isolated and subjected to RT-PCR. A, CYP3A catalytic activity as measured by 1′-hydroxylation of MDZ. The metabolite was assayed in culture medium by GC/MS. B, ethidium-stained gels of products from RT-PCR using primers of the indicated specificities (CYP3A4, 35 cycles; villin, 25 cycles), C, immunoblots probed with antibodies of the indicated specificities and developed using enhanced chemiluminescence (10 μg of homogenate protein/lane). BDL, below detectable limits.
Treatments with 1,25-(OH)2-D3 and 25-OH-D3 were done in parallel cultures. In agreement with previous results (Schmiedlin-Ren et al., 1997), there were increases in CYP3A4 mRNA, immunoreactive protein, and catalytic activity in response to both of these analogs; midazolam 1′-hydroxylation results are shown (Fig.5). However, higher doses of 25-OH-D3 resulted in a decrease in CYP3A4 mRNA, protein, and catalytic activity, confirming the previous impression of toxicity of this analog at high doses (Schmiedlin-Ren et al., 1997). Microscopic examination of hematoxylin- and eosin-stained sections of the monolayers from these dose-response experiments showed columnar epithelium in the untreated culture and all of the cultures treated with 1,25-(OH)2-D3 or 1-OH-D3. Columnar epithelium was also seen in cultures treated with the lower doses of 25-OH-D3; but at doses of 10, 15, and 20 μM, the cells were cuboidal or flattened, and areas of the membrane lacking cells were seen (not shown).

Concentration-dependent effects of three VDR ligands on CYP3A4 catalytic activity in Caco-2 cells.
Clone P27.7 Caco-2 cells (passage 29) were seeded onto culture inserts commercially coated with laminin and grown for 5 days before initiation of a 2-week treatment with varying concentrations of 1,25-(OH)2-D3, 1-OH-D3 or 25-OH-D3, applied apically and basolaterally. At the end of treatment, the medium was replaced, and 4 μM midazolam was applied apically. Following a 4-h incubation, the medium from the apical and basolateral compartments was recovered and assayed separately for 1′-OH-MDZ by GC/MS. The means of the apical and basolateral concentrations are shown.
Vitamin D analogs are known to bind to VDR, which then forms a heterodimer with the retinoid X receptor (RXR). We therefore examined the effect of the RXR ligand 9-cis-retinoic acid (9-cis-RA) on the 1,25-(OH)2-D3-mediated induction of CYP3A4 in Caco-2 cells. For comparison, the effect of the retinoic acid receptor (RAR) ligand all-trans-retinoic acid (ATRA) was also examined. A 2-week treatment with either 9-cis-RA or ATRA at 0.25 μM did not induce CYP3A and appeared to cause a slight decrease in the levels of CYP3A4 mRNA (Fig.6). Concurrent treatment with 9-cis-RA and 1,25-(OH)2-D3 (both at 0.25 μM) appeared to slightly decrease CYP3A4 catalytic activity relative to cells treated with 1,25-(OH)2-D3 alone, with no clear effect on mRNA or protein levels. However, concurrent treatment of Caco-2 cells with 1,25-(OH)2-D3 and ATRA (both at 0.25 μM) was associated with a slight increase in CYP3A catalytic activity over that observed in Caco-2 cells treated with 1,25-(OH)2-D3 alone. The ATRA-mediated increase in CYP3A4 catalytic activity was not associated with detectable increases in the levels of CYP3A immunoreactive protein or CYP3A4 mRNA relative to treatment with 1,25-(OH)2-D3 alone (Fig.6).
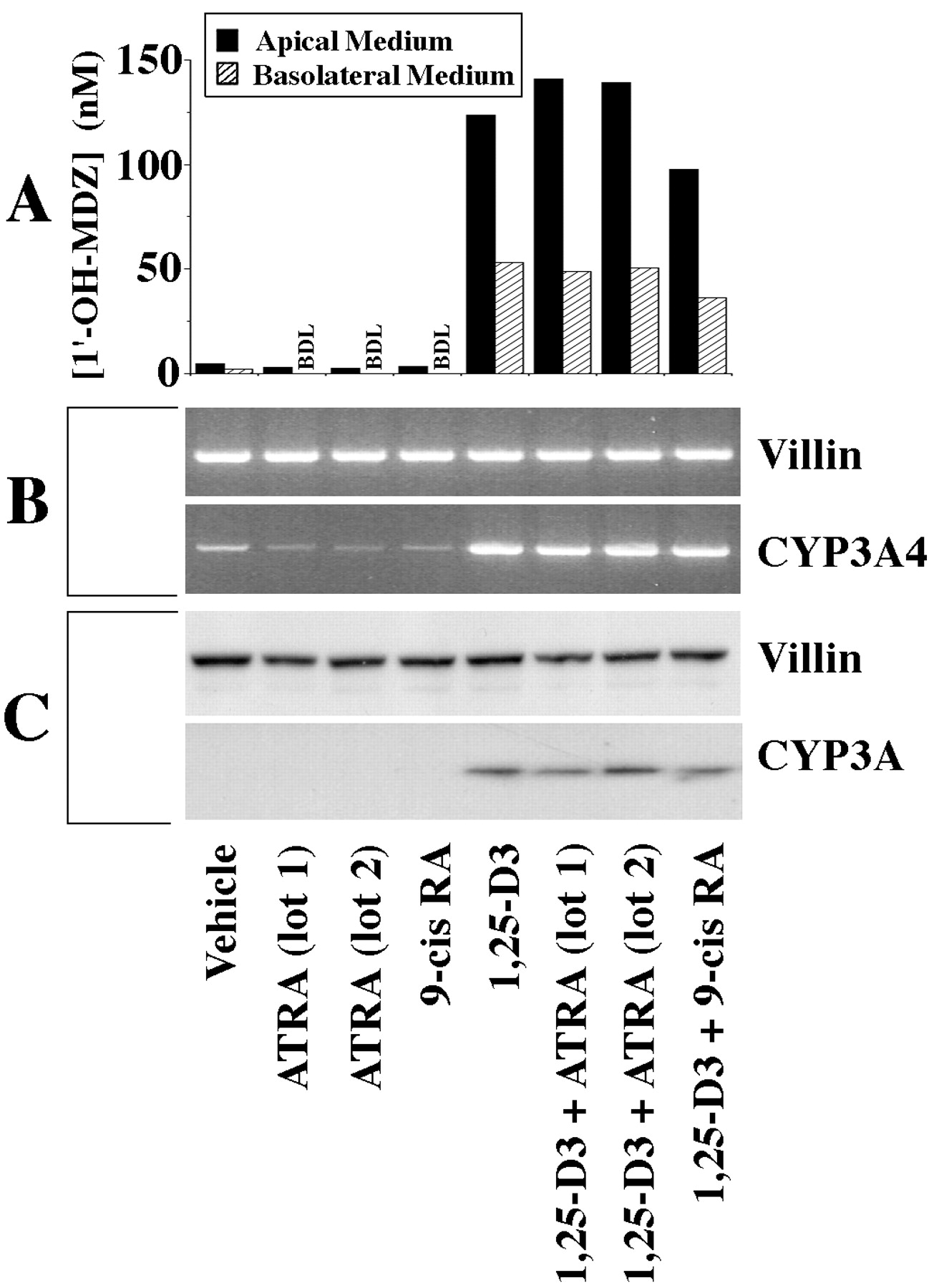
Effects of ATRA and 9-cis-RA on CYP3A4 expression in Caco-2 cells.
Clone P27.7 Caco-2 cells (passage 29) were seeded onto culture inserts commercially coated with laminin and grown for 2 days before initiation of a 2-week treatment with 0.25 μM 1,25-(OH)2-D3 and/or ATRA or 9-cis-RA, applied apically and basolaterally. At the end of treatment, the medium was replaced, and 4 μM midazolam was applied apically. Following a 4-h incubation, the medium from the apical and basolateral compartments was recovered and assayed separately for 1′-OH-MDZ. A portion of the cells was homogenized and subjected to SDS-PAGE. The remaining cells were lysed, and RNA was isolated and subjected to RT-PCR. A, CYP3A catalytic activity as measured by 1′-hydroxylation of MDZ. The metabolite was assayed in culture medium by GC/MS. B, ethidium-stained gels of products from RT-PCR using primers of the indicated specificities (CYP3A4, 34 cycles; villin, 25 cycles). C, immunoblots probed with antibodies of the indicated specificities and developed using enhanced chemiluminescence (10 μg of homogenate protein/lane).
To further examine whether ATRA could be used to augment CYP3A4 catalytic activity in our Caco-2 model, the cells were exposed for 2 weeks to 1,25-(OH)2-D3(0.25 μM) together with varying concentrations of ATRA (0.05, 0.1, 0.25, 0.5, or 1.0 μM). We found that 0.05 μM and 0.1 μM ATRA increased CYP3A4 catalytic activity roughly 2-fold over 1,25-(OH)2-D3 treatment alone (not shown). The effect of ATRA diminished at higher concentrations (not shown).
Discussion
We have previously shown that 1,25-(OH)2-D3 is a potent inducer of CYP3A4 in the human colon adenocarcinoma cell line Caco-2 when it is grown on extracellular matrix-coated permeable supports (Schmiedlin-Ren et al., 1997). We anticipated a similar effect on other commercially available human cells lines derived from organs known to express CYP3A enzymes. Inducibility of CYP3A4 by 1,25-(OH)2-D3 was found to be cell line-specific but not restricted to Caco-2 cells or to cell lines of intestinal origin.
In comparison to vehicle-treated cultures, treatment with 1,25-(OH)2-D3 induced CYP3A catalytic activity and immunoreactive protein and CYP3A4 mRNA in both LS180 and HPAC cells (Fig. 1). LS180 cells are known to express CYP3A4 (Schuetz et al., 1996) and, like Caco-2 cells, are derived from a human colon adenocarcinoma. That both cell lines respond similarly to 1,25-(OH)2-D3 may suggest that this is a characteristic of intestinal epithelial cells. It has not yet been possible to culture human intestinal epithelial cells to directly test this hypothesis. The pancreatic adenocarcinoma cell line HPAC appeared to contain both CYP3A4 and CYP3A5 (Fig. 1); but, the inductive effect of 1,25-(OH)2-D3 appeared to be restricted to CYP3A4 expression. This observation is consistent with our previous studies in Caco-2 cells (Schmiedlin-Ren et al., 1997) in which CYP3A5 immunoreactive protein was not induced by 1,25-(OH)2-D3 treatment. This suggests that the mechanisms involved in CYP3A4 induction by 1,25-(OH)2-D3 are not shared with CYP3A5. Although villin immunoreactive protein was not detected in either treated or untreated HPAC cells, the apparent increase in villin mRNA in these cells may reflect a general differentiation-promoting effect of 1,25-(OH)2-D3.
In the human stomach carcinoma cell line Hs746T, there was no detectable midazolam 1′-hydroxylase activity or any detectable CYP3A protein or CYP3A4 mRNA, with or without exposure to 1,25-(OH)2-D3 (Fig. 1). Normal stomach has been shown to express primarily CYP3A5 (Kolars et al., 1994). The Hs746T cells appear to have lost the ability to express CYP3A enzymes (Fig. 1).
1,25-(OH)2-D3 also produced no detectable change in CYP3A mRNA, immunoreactive protein, or catalytic activity in Hep G2 cells (Fig. 1). We had hoped for a positive effect because expression of catalytically active CYP3A4 in these cells could expand the application of this widely used cell model in drug metabolism research. However, the only CYP3A protein that has been demonstrated in Hep G2 cells to date is CYP3A7 (Schuetz et al., 1993). The CYP3A immunoreactive protein we detected in our Hep G2 cells appeared to migrate slightly slower than CYP3A4 during electrophoresis (Fig. 1), consistent with expression of only CYP3A7 in these cells. Additionally, despite a previous report of the presence of CYP3A4 mRNA in Hep G2 cells (Sumida et al., 2000), the sequence of our PCR product was consistent with the amplification of CYP3A7 rather than the intended CYP3A4.
To address the question of responsiveness of normal human hepatocytes to 1,25-(OH)2-D3, we conducted studies in hepatocytes prepared from two donors (Fig. 2), comparing the effects of 1,25-(OH)2-D3with the effects of CYP3A4 inducers known to act through the recently described receptor PXR [Lehmann et al., 1998; also referred to as the pregnane-activated receptor (PHR; Bertilsson et al., 1998) or steroid and xenobiotic receptor (SXR; Blumberg et al., 1998)]. As expected, CYP3A4 induction was most marked in response to treatment with the known human PXR ligands rifampin and dexamethasone and not evident after treatment with PCN, a ligand for the rodent but not human PXR (Kliewer et al., 1999). Treatment with 1,25-(OH)2-D3 at concentrations 1 to 2 orders of magnitude less than those used for the PXR ligands also induced CYP3A4 mRNA, protein, and catalytic activity in cells from both donors. In donor 2, the magnitude of the effect of 1,25-(OH)2-D3 was comparable to that observed with rifampin and dexamethasone.
To explore whether the induction of CYP3A4 by 1,25-(OH)2-D3 involves PXR, we examined the ability of the PXR ligands to induce CYP3A4 in Caco-2 cells under culture conditions optimal for the inductive effects of 1,25-(OH)2-D3. Unlike in the primary hepatocytes, the effects of the PXR ligands in Caco-2 cells was minimal compared with the inductive effects of 1,25-(OH)2-D3 (Fig. 3). As in the hepatocyte studies, the concentration of 1,25-(OH)2-D3 effective in inducing CYP3A4 was 1 to 2 orders of magnitude lower than the doses of the PXR ligands used. The relative lack of responsiveness to PXR ligands suggests that PXR is not functional in the Caco-2 cells. It therefore seems unlikely that PXR is essential for CYP3A4 induction by 1,25-(OH)2-D3.
We have previously shown that CYP3A4 is induced in Caco-2 cells by 25-OH-D3 in addition to 1,25-(OH)2-D3(Schmiedlin-Ren et al., 1997). Since both of these vitamin D analogs are known ligands for VDR, we sought to determine whether another high affinity VDR ligand, 1-OH-D3, could also induce CYP3A4 in these cells. Treating confluent Caco-2 cells for 2 weeks with 1-OH-D3 resulted in a CYP3A4-inductive response very similar to that elicited by 1,25-(OH)2-D3 (Fig. 4), with dose-dependent increases in CYP3A4 mRNA and CYP3A immunoreactive protein and catalytic activity. The fact that three ligands for VDR at submicromolar concentrations have now been shown to mediate induction of CYP3A4 supports a role for VDR in CYP3A4 regulation. Although VDR has been demonstrated to be present in Caco-2 cells (Giuliano et al., 1991), a search of the literature revealed no data concerning the expression of VDR or PXR in the other cell lines examined. However, it has been reported that VDR is present in intestinal epithelial cells, pancreas, and hepatocytes (Berger et al., 1988; Johnson et al., 1994), the origins of the cells we found to be responsive to 1,25-(OH)2-D3. Additionally, PXR is not present in pancreas (Bertilsson et al., 1998;Blumberg et al., 1998). These observations are consistent with the concept that the induction of CYP3A4 by 1,25-(OH)2-D3 probably involves VDR.
The consensus VDRE sequence is considered by many to be a DR3 (direct repeat of six nucleotides with a 3-nucleotide spacer between the half-sites) (Haussler et al., 1997; Toell et al., 2000). The sequence in the proximal promoter of CYP3A4, which responds to PXR with bound ligand, is not a DR3 but an imperfect ER6 [everted repeat with a 6-nucleotide spacer; TGAACTcaaaggAGGTCA; −168 to −151 (Barwick et al., 1996)]. The CYP3A4 gene contains imperfect DR3 sequences in the distal enhancer (TGAACTtgcTGACCC; −7733 to −7719; Goodwin et al., 1999) and at a site between the distal enhancer and the proximal promoter (GGGTCAgggAGCTCA; −1289 to −1275). It is possible that VDR activates CYP3A4 transcription through binding at one or both of these DR3 sites.
It is generally believed that VDR with bound ligand (1,25-(OH)2-D3) preferentially forms heterodimers with the RXR; the heterodimers then bind to vitamin D responsive elements (reviewed in Haussler et al., 1997) to influence transcription. Caco-2 cells have been reported to express RXRα and RXRγ mRNAs (Kane et al., 1996), indicating that heterodimer formation with RXR is possible in these cells. Because RXR, which has bound ligand, does not form heterodimers with VDR, we expected that concurrent exposure of Caco-2 cells to 1,25-(OH)2-D3 and 9-cis-retinoic acid (the preferred ligand of RXR) might result in a reduction in CYP3A4 expression relative to that observed in cells treated with 1,25-(OH)2-D3 alone. This occurred to a mild degree (Fig. 6).
ATRA is a ligand for the RAR; RAR with bound ligand preferentially forms heterodimers with RXR (Haussler et al., 1997). Caco-2 cells have been shown to express RARβ (McCormack et al., 1996). RAR has not been reported to ordinarily form heterodimers with VDR and has not been implicated in CYP3A4 gene expression. It was therefore expected that concurrent treatment with ATRA and 1,25-(OH)2-D3 would not increase levels of CYP3A4 mRNA or CYP3A immunoreactive protein. However, an up to 2-fold increase in CYP3A4 catalytic activity was observed. The mechanism whereby ATRA increased CYP3A4 activity in the absence of an effect on gene regulation is unknown, although it may involve transcriptional regulation of a protein influencing CYP3A activity (e.g., cytochrome P450 reductase). Although the response to the retinoids examined does not help to clarify the potential role of VDR in CYP3A4 gene regulation, ATRA treatment does provide a rather inexpensive method to augment CYP3A4 catalytic activity in 1,25-(OH)2-D3-treated Caco-2 cells.
In conclusion, 1,25-(OH)2-D3 can be used to increase CYP3A4 expression in some but not all human cell lines derived from tissues known to express CYP3A enzymes. Although the mechanisms involved in this induction are not known, our observations suggest that it is unlikely that PXR is involved and support a role for VDR. The finding that CYP3A4 is induced in multiple gastrointestinal cell lines and in primary human hepatocytes raises the possibility that dietary and/or other exogenous vitamin D analogs may play a role in CYP3A4 regulation in vivo.
Acknowledgments
We thank Dr. Michael E. Fitzsimmons for development of the LC/MS method for 1′-hydroxymidazolam quantitation and for sample analyses using that method. We also thank Justina C. Calamia for the assay of 1′-hydroxymidazolam in the samples for which results are shown in Fig.6.
Footnotes
-
↵1 J.M.F. is presently affiliated with Pfizer, Groton, CT.
-
This research was supported by National Institutes of Health Grants GM 38149 (to P.B.W.) and GM 48349 (to K.E.T.).
- Abbreviations used::
- CYP
- cytochrome P450
- 1,25-(OH)2-D3
- 1α,25-dihydroxyvitamin D3
- VDR
- vitamin D receptor (intracellular)
- 1-OH-D3
- 1α-hydroxyvitamin D3
- 25-OH-D3
- 25-hydroxyvitamin D3
- PXR
- pregnane X receptor
- PET
- polyethylene terephthalate
- MDZ
- midazolam
- 1′-OH-MDZ
- 1′-hydroxymidazolam
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- HPLC
- high-pressure liquid chromatography
- PCN
- pregnenolone 16α-carbonitrile
- DtBA
- dexamethasonet-butyl acetate
- D2-1′-OH-MDZ
- deuterated 1′-hydroxymidazolam
- GC/MS
- gas chromatography/mass spectrometry
- RT-PCR
- reverse transcription-polymerase chain reaction
- PAGE
- polyacrylamide gel electrophoresis
- LC/MS
- liquid chromatography/mass spectrometry
- RXR
- retinoid X receptor
- 9-cis-RA
- 9-cis-retinoic acid
- RAR
- retinoic acid receptor
- ATRA
- all-trans-retinoic acid
- TES
- N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid
- Received April 16, 2001.
- Accepted August 6, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}