


Abstract
Between 45 and 60% of all drugs currently used are metabolized by the CYP3A4 protein. CYP3A4 expression in liver varies up to 60-fold in the general population, which can lead to ineffective drug therapy (high CYP3A4) or, on the other hand, to harmful drug reactions (low CYP3A4). Most of this variability has been attributed to genetic factors, but to date their identity remains unknown. Recently, it was shown that CYP3A expression is largely controlled by the pregnane X receptor (PXR). We, therefore, hypothesized that polymorphisms in PXR may contribute to CYP3A4 variability. The presence of PXR variants was investigated in two ethnic groups, Caucasians and Africans. Six missense mutations leading to variant PXR proteins were identified, and their consequences on CYP3A4 expression were analyzed. Expressed in LS174T cells, three protein variants, V140M, D163G, and A370T, exhibited altered basal and/or induced transactivation of CYP3A promoter reporter genes. Thus, these natural PXR protein variants may play a role in the observed interindividual variability of CYP3A4 expression and may be involved in rare, atypical responses to drugs or altered sensitivities to carcinogens.
Among the human cytochrome P450 (CYP1) proteins, the members of the CYP3A subfamily occupy a unique position due to their abundance in liver and gut and to their collective large substrate spectrum. Indeed, CYP3A proteins account for up to 50% of total cytochrome P450 activity in the liver, and they metabolize up to 60% of all drugs currently in use. The predominant CYP3A protein expressed in adult human livers is the CYP3A4 isoform (Thummel and Wilkinson, 1998). The expression levels of CYP3A4 in liver vary up to 60-fold in the general population (Lown et al., 1994; Shimada et al., 1994; Thummel et al., 1994; Özdemir et al., 2000).
CYP3A4 is transcriptionally regulated by a variety of hormones, including glucocorticoids and xenobiotics, such as phenobarbital, clotrimazole, and rifampicin. Induction by rifampicin and other xenobiotics as well as inhibition of CYP3A4 activity by drugs such as ketoconazole underlies many reported drug interactions and is therefore of considerable clinical importance (Michalets, 1998). In addition, the interindividual variability in CYP3A4 activity could also influence individual predisposition to tumors caused by environmental carcinogens, including liver and lung cancer (Forrester et al., 1990;Paolini et al., 1999). The reasons for the variable CYP3A4 expression are still unknown. Clinical studies with CYP3A4 substrates indicate that approximately 90% of the interindividual variability in the hepatic CYP3A4 activity is genetically determined (Özdemir et al., 2000). Several variants have been described in the CYP3A4 gene. However, their allelic frequencies and/or the available functional data indicate a limited role of these variants in the interindividual variability of CYP3A4 expression and activity (Rebbeck et al., 1998; Ball et al., 1999;Westlind et al., 1999; Sata et al., 2000; Wandel et al., 2000; Eiselt et al., 2001).
Recently, it was shown that the induction of rat CYP3A23 and human CYP3A4 expression is regulated by the nuclear receptor NR1I2 [pregnane X receptor (PXR)]. Heterodimers consisting of PXR and a retinoid X receptor bind to nuclear receptor response elements [AG(G/T)TCA repeats] in the 5′-flanking regions ofCYP3A genes and, upon treatment with CYP3A inducers, stimulate their transcription (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998). Through the use of a scintillation proximity binding assay, Jones et al. (2000)demonstrated that many of the compounds that induce CYP3Aexpression bind directly to human PXR. In addition, an analysis of mice transgenic for human PXR indicates that established inducers ofCYP3A genes act via PXR (Xie et al., 2000).
Genetic variants of PXR, which change the transactivation potential of its protein product, could have a dramatic impact onCYP3A4 expression (Eichelbaum and Burk, 2001). In this article, we describe the identification of six PXR protein variants in Caucasians and Africans. We demonstrate an altered basal and/or induced transactivation of CYP3A4 by three PXR variants with single amino acid substitutions. This is a first report of genetic variants in a transcriptional regulator of CYP3A4, which may contribute to the variable activity of the gene's product in vivo.
Materials and Methods
DNA Samples.
Blood or liver samples were obtained from Middle- and West-European Caucasians and Africans from the Ivory Coast. Samples were collected by the Institute of Clinical Pharmacology (University Medical Center Charite, Humboldt University in Berlin, Germany), by the Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology (Stuttgart, Germany), and by the Division of Pharmacology/Neurobiology (Biozentrum, University of Basel, Switzerland) under consideration of all necessary ethical and legal requirements. Genomic DNA was isolated using blood and tissue DNA isolation kits (QIAGEN, Hilden, Germany). The number of individuals analyzed for mutations in the different exons were as follows. Caucasians: exons 1a, 1b, 2, 3, and 5 (150); exon 4 (209); exons 6 and 7 (185); and exons 8 and 9 (156); Africans: exons 1a, 6, and 7 (28); exon 1b (30); exons 2 and 4 (37); exon 3 (31); exon 5 (27); and exon 8, 9 (32).
Oligonucleotides for PXR Genotyping.
Exon 1a (F: 5′-TCA AGT GCT GGA CTT GGG AC-3′; R: 5′-CCC ACT ATG ATG CTG ACC TC-3′; 460 bp), Exon 1b (F: 5′-CAC ATA CAA CCA GCT CCC TG-3′; R: 5′-CCA CAT GCA GGC AAG ACT C; 345 bp), Exon 2 (F: 5′-CTG AGG CCT CTA CAC ATC-3′; R: 5′-AGG CCC TGA GAT GTT ACC-3′; 345 bp), Exon 3 (F: 5′-CTG GGA CGC AAA GGC TAG TG-3′; R: CCT GTT GCA CAC GGA CAC-3′; 417 bp), Exon 4 (F: 5′-TAA CGG CTT CTG CTG CCT TG-3′; R: 5′-AGC TCT CCA AAT CTA CCC TC-3′; 423 bp), Exon 5 (F: 5′-CTG AGT TGG GAC CTG TCT-3′; R: 5′-CCA GGC CCT TTG AAC CTC-3′; 415 bp), Exon 6 and 7 (F: 5′-CTG CTG GTG CCG GCC TGT-3′; R: 5′-GAC TGG GAC CTT CCC TGG-3′; 598 bp), Exon 8 (F: 5′-GAG CAA TGC CCT GAC TCT-3′; R: 5′-CCC TCT GGC CAT GAA GTC-3′; 271 bp), Exon 9 (F: 5′-TGC TTG TGC AGC CTC AGA-3′; R: 5′-GCT CTT GGC AGT GTC CAT-3′; 324 bp).
PCR Amplification, Sequencing, and Sequence Analysis.
Genomic DNA (20–60 ng) was added to a reaction mix containing 1 to 2.25 mM MgCl2, 0.25 μM each oligonucleotide, 200 μM dNTPs, and 1 U of Taq polymerase (QIAGEN). PCR conditions were one cycle at 94°C for 2 min, 34 cycles at 94°C for 40 sec, 57°C to 60°C for 45 sec, and 72°C for 60 sec, and a final cycle of 72°C for 5 min. PCR products were purified using a QIAquick PCR purification kit (QIAGEN) and directly sequenced by cycle sequencing using the ABI BigDye terminator cycle sequencing kit. Sequencing reactions were determined using PE Biosystems' capillary 3700 DNA Analyzers (Foster City, CA). The sequences were analyzed for the presence of polymorphisms using the PHRED/PHRAP/POLYPHRED/CONSED software package (University of Washington, Seattle). All genotypes resulting in amino acid exchanges were confirmed by independent PCR followed by sequencing.
Construction of PXR Expression Plasmids.
Missense mutations were introduced into the human PXRexpression plasmid (Geick et al., 2001) using a QuickChange site-directed mutagenesis kit (Stratagene, Heidelberg, Germany) and were confirmed by sequencing. The PXR-2 variant in which amino acids 174 to 210 are deleted (Dotzlaw et al., 1999) was generated by sequential PCR steps and sequenced.
Reporter Gene Plasmids.
DR3 construct:
The DR3 motif of CYP3A23 (Kliewer et al., 1998) was multimerized by annealing the single-stranded oligonucleotides 5′-GAT CCT AGA TGA ACT TCA TGA ACT GTC TA-3′ and 5′-GAT CTA GAC AGT TCA TGA AGT TCA TCT AG-3′, which contain BamHI and BglII sites, to generate a double-stranded oligonucleotide, followed by a ligation. BamHI/BglII restriction digest of the ligated DNA was cloned into the luciferase reporter gene vector pGL3-Basic (Promega, Madison, WI), upstream of a minimal thymidine kinase promoter (−105 to +31). The construct used [pGL3(DR3)3TK further referred to as DR3] contains three copies of the DR3 motif.
ER6 construct:
A luciferase reporter gene (pGL3(ER6)2TK(−105)) containing two copies of the ER6 motif of the proximal promoter region of CYP3A4 (Lehmann et al., 1998) was constructed as described above by using the oligonucleotides 5′-GAT CCA ATA TGA ACT CAA AGG AGG TCA GTG A-3′ and 5′-GAT CTC ACT GAC CTC CTT TGA GTT CAT ATT G-3′.
CYP3A4 construct:
The CYP3A4 promoter region was amplified by PCR from human genomic DNA using the oligonucleotides 5′-ATG GTA CCT GCA GTG ACC ACT GCC CCA T-3′ and 5′-ACA AGC TTG CTC TTT GCT GGG CTA TGT GCA-3′. TheKpnI- and HindIII-digested PCR fragment was cloned into the corresponding sites of pGL3-Basic to generate pGL3-CYP3A4(−1105). The CYP3A4 distal enhancer module was amplified by PCR out of a bacterial artificial chromosome (BAC) clone GS23577 (Genome Systems, St. Louis, MO) using the primers 5′-GCG GTA CCG AGA TGG TTC ATT CCT TTC AT-3′ and 5′-CGA GAT CTC GTC AAC AGG TTA AAG GAG AA-3′, which introduce KpnI and BglII sites, respectively. The fragment was then cloned into theKpnI and BglII sites of pGL3-CYP3A4(−1105) to generate the pGL3-CYP3A4(−7830Δ7208−364) plasmid (further referred to as CYP3A4), which encompasses the nucleotides −7830 to −7209 and −363 to +51 of the CYP3A4 promoter.
CYP3A4 (mut prox. ER6):
Mutation of the ER6 element in the proximal CYP3A4 promoter region was introduced into pGL3-CYP3A(−374), a nested deletion mutant of pGL3-CYP3A4(−1105), by using a sequential PCR strategy, resulting in the sequence 5′-TGTTCT CAA AGG AGAACA-3′ for the mutated ER6 element (mutated bases are underlined). Then the BglII/HindIII fragment of this plasmid (pGL3-CYP3A4(−374/mER6), containing the proximal CYP3A4 promoter with the mutated ER6, and theKpnI/BglII fragment of pGL3-CYP3A4(−7830Δ7208−364), containing the distal enhancer module, were ligated into KpnI/HindIII-digested pGL3-Basic. The resulting plasmid pGL3-CYP3A4(−7830Δ7208−364)m1 is here further referred to as CYP3A4(mut prox. ER6). The plasmid p3A4-362(7836/7208ins) mut dNR1+NR2 (Goodwin et al., 1999) [here referred to as CYP3A4(mut dNR1+NR2)] was kindly provided by C. Liddle (Department of Clinical Pharmacology, University of Sydney, Sydney, Australia).
Western Blot Analysis.
LS174T cells were cotransfected with expression plasmids for wild-type or variant PXR and the β-galactosidase reference plasmid pCMVβ (CLONTECH, Palo Alto, CA) using a calcium phosphate coprecipitation method. Twenty-four hours after transfection cells were harvested, and cell pellets were boiled in a protein sample buffer. Total cellular proteins were resolved on a 10% SDS-polyacrylamide gel. Protein amounts were adjusted according to transfection efficiency as measured by β-galactosidase activity. Western blotting was performed as described (Klempnauer et al., 1986). PXR protein was detected with a polyclonal goat antiserum (anti-PXR N-16; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and PXR protein-antibody complexes were visualized using the ECL detection system (Amersham Pharmacia Biotech, Freiburg, Germany) and a digital CCD-camera (LAS-1000, Fuji Photo Film, Kanagawa, Japan). Signal quantification was performed with AIDA software (Raytest, Straubenhardt, Germany).
Cell Culture.
The human colon adenocarcinoma cell line LS174T (Tom et al., 1976) was obtained from American Type Culture Collection (Manassas, VA) and grown in Dulbecco's modified Eagle's medium buffered with 25 mM HEPES and supplemented with 10% fetal calf serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, 1% MEM nonessential amino acids, 2 mMl-glutamine, and 1 mM sodium pyruvate. During treatment with inducers, cells were cultured in phenol red-free medium supplemented with fetal calf serum that was pretreated with dextran-coated charcoal (Walker and Enrietto, 1995).
Transient Transfections, Luciferase, and β-Galactosidase Assays.
LS174T cells were plated in 24-well plates at a density of 1.5 × 105 cells/well 16 to 18 h before transfection. The plasmid DNA was transfected by using the Effectene Transfection Reagent (QIAGEN) according to the manufacturer's recommendations. Transfections were done two to five times in triplicate with at least two different plasmid DNA preparations. Usually, 0.15 μg of either reporter gene construct, 0.02 μg of β-galactosidase reference plasmid pCMVβ, and 0.01 μg ofPXR expression plasmid were used per well. Six to 7 h after transfection, the cells were treated with inducers or solvent control (0.1% DMSO). Cells were harvested 40 to 42 h after the start of the treatment and lysed with passive lysis buffer (Promega). After centrifugation, cleared lysates were used for reporter gene assays done in duplicate. For luciferase measurements, 300 μl of an assay solution (25 mM glycylglycine, pH 7.8/50 μM luciferin/2 mM ATP/10 mM MgCl2/27 μM coenzyme A/30 mM dithiothreitol) were automatically injected into 20 μl of cell lysate, and luminescence was measured immediately for 4 sec with an AutoLumat Plus (Berthold, Bad Wildbad, Germany). β-Galactosidase assays were done according to Jain and Magrath (1991) and measured as described above. Luciferase activity was normalized with respect to transfection efficiency using the corresponding β-galactosidase activity. To analyze statistically significant differences, one-way analysis of variance with Student-Newman-Keuls posttest was performed using GraphPad InStat version 3.05 for Windows 95 (GraphPad Software, San Diego, CA).
Results and Discussion
A BAC (GS21908) containing the entire human PXR gene was identified by PCR in the Genome Systems' human genomic library. The genomic structure of PXR was determined by sequencing PCR fragments generated with oligonucleotides located in two neighboring exons and by direct sequencing of the BAC DNA. Oligonucleotides used for PCR and sequencing were derived from a humanPXR cDNA sequence (GI: 3769538) upon the assumption of similarities between PXR and human VDR (vitamin D receptor) gene structures (Miyamoto et al., 1997; Bertilsson et al., 1998). The gene consists of 10 exons and 9 introns, and it spans approximately 20 kilobases of genomic DNA (Fig.1A). Exon-intron boundaries and sizes are shown in Fig. 1B. The first two exons (1a and 1b) are used as alternative 5′ ends of PXR transcripts both in liver and small intestine (data not shown). Transcripts using exon 1b (Fig. 1C) would be expected to be translated in a protein with an extended N terminus (Bertilsson et al., 1998).

Genomic organization of the human PXR gene and structure of human PXR transcripts.
A, protein-coding regions are depicted as filled boxes and the 5′ and 3′ untranslated regions as white boxes. Introns are shown by horizontal lines. Arrowheads represent the positions of oligonucleotides used in the screen for genetic variants. The 5′ boundaries of exons 1a and 1b are based on published cDNA sequences (Bertilsson et al., 1998). B, exon-intron organization and sizes of the human PXR gene were determined by sequencing PCR fragments generated with primers derived from two neighboring exons and sequencing BAC GS21908. Results were compared with the genomic sequence AC069444.7 (GenBank). Exon sequences are shown in capital letters and intron sequences in small letters. C, structure of human PXR transcripts. Transcript 1 originates from exon 1a and corresponds to the protein described here as PXR wild-type. Transcript 2 originates from exon 1b and corresponds to the cDNA published as PAR2 (Bertilsson et al., 1998). Transcript 3 represents an in-frame deletion of 111 bp of the 5′ part of exon 5 (depicted as a white box) by using a cryptic splice acceptor site within this exon. This transcript encodes the PXR splice variant PXR-2. DBD, DNA-binding domain.
The protein-coding regions of PXR, including some flanking intron sequences and parts of the 5′- and 3′-UTR, were amplified by PCR from genomic DNA of 150 to 209 Caucasians and 27 to 37 Africans. The PCR products were subsequently sequenced. A total of 28 variants were found in the samples screened, including six which result in missense mutations of the PXR protein (Table 1). Three variants (V140M, D163G, and A370T) are located within or close to the putative ligand-binding domain (LBD) of PXR and three (E18K, P27S, and G36R) in its N-terminal part. With the exception of G36R and V140M, all missense mutations affect amino acids that are conserved in the PXR proteins of human, rabbit, rat, and mouse (Moore et al., 2000). The most frequent protein polymorphism (P27S) occurs in 14.9% African chromosomes. All other protein variants have an allelic frequency of ≤3% and are specific for either Caucasians or Africans (Table 1).
Allelic frequencies of PXR protein variants in Caucasians and Africans
We investigated the effect of the missense PXR mutations on the expression and activity of the PXR protein. We also investigated the effect of the PXR-2 splice variant that has been found in normal and neoplastic human breast tissue (Dotzlaw et al., 1999) and that results from a cryptic splice acceptor site within exon 5 (Fig. 1C), leading to a deletion of 37 amino acids from the LBD of PXR. PXR-2 transcripts were also expressed in human liver and small intestine (data not shown). To this end, we constructed eukaryotic expression plasmids for all six PXR protein variants and for the PXR-2 splice variant. Western blot analysis of LS174T cells transiently transfected with these plasmids showed that the plasmids encoding the variants P27S, D163G, A370T, and PXR-2 directed the expression of similar amounts of protein compared with wild-type, whereas E18K, G36R, and V140M were expressed slightly weaker (Fig.2). This effect was strongest for V140M, which showed an expression level of only 50% of wild-type. The apparent sizes of the variants were in agreement with the calculated molecular weight of PXR (49.7 kDa) and PXR-2 (45.7 kDa) with the exception of the E18K variant, which showed an apparent molecular weight of 47 to 48 kDa (Fig. 2).
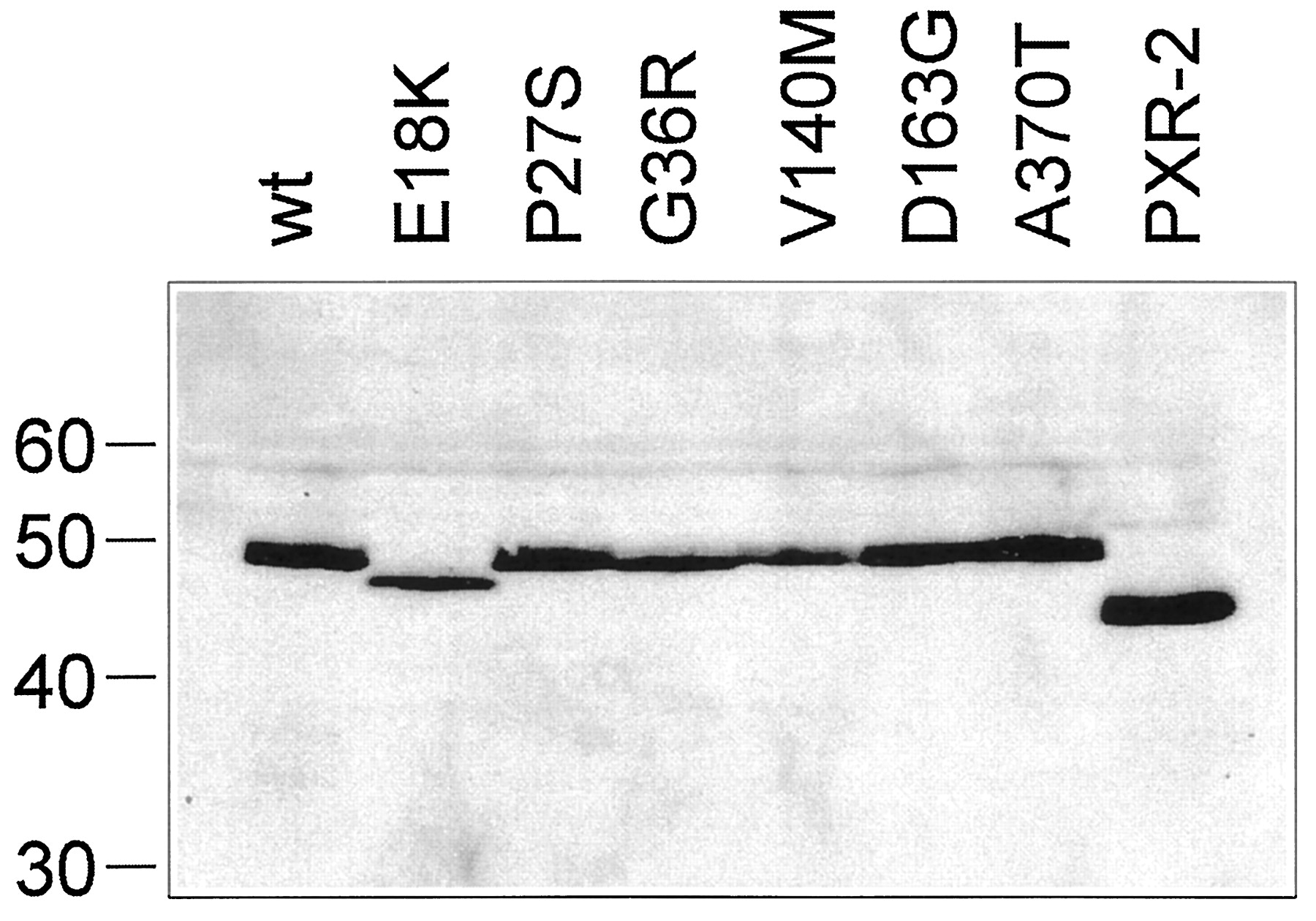
Recombinant PXR protein expression.
Western blot analysis of total cellular protein of LS174T cells transiently transfected with expression plasmids for wild-type (wt) or variant PXR proteins. Protein amounts were adjusted according to transfection efficiency as estimated by the activity of the β-galactosidase cotransfected into the cells. Blots were probed with a PXR-specific antibody. Molecular weight markers (in kilodaltons) are shown on the left.
The functional consequences of the variant PXR proteins were investigated in LS174T cells. These cells were chosen because endogenous CYP3A4 mRNA expression can be induced by a variety of CYP3A4 inducers in a manner virtually identical to that observed in primary human hepatocytes. LS174T cells expressPXR mRNA; albeit endogenous PXR protein expression is not detectable with the antibody used to detect transfected PXR. Transfection experiments with increasing amounts of PXRexpression plasmid suggest that the heterodimerization partner retinoid X receptor is not a limiting factor and expressed in sufficient amounts. A more detailed analysis of CYP3A4 induction in LS174T cells will be presented elsewhere. In one experimental series, LS174T cells were cotransfected with a vector expressing wild-type or variant human PXR and with plasmid pGL3(DR3)3TK(−105) (in the following referred to as DR3 construct), which contains three copies of the PXR-binding DR3 motif of the rat CYP3A23 gene (Kliewer et al., 1998), cloned upstream of a minimal thymidine kinase promoter and a luciferase gene. In another experimental series, PXRexpression plasmids were cotransfected with a luciferase reporter gene under the control of the proximal CYP3A4 promoter and of the distal xenobiotic-responsive enhancer module [pGL3-CYP3A4(−7830Δ7208−364), in the following referred to as CYP3A4 construct]. The construct contains one DR3-type and two ER6-type PXR response elements identified in the CYP3A4promoter (Lehmann et al., 1998; Goodwin et al., 1999) and was used to examine PXR activity in a more physiological context.
Transfection of wild-type PXR expression plasmid activated the DR3 and CYP3A4 constructs 5- and 16-fold, respectively, even in the absence of endogenous inducers, demonstrating a basal transcriptional activity of PXR (Fig. 3A, B). We first asked whether the PXR protein variants showed an altered basal activity. All variants with the exception of D163G and PXR-2 showed a statistically significant basal activity compared with control levels. Irrespective of the reporter construct used, variants located N terminal of the DNA-binding domain of PXR (E18K, P27S, and G36R) had no statistically significant impact on the basal activity in comparison to wild-type PXR (Fig. 3A, B). On the other hand, variants located within the LBD of PXR (D163G and A370T), close to the LBD (V140M) and the deletion variant PXR-2, showed a moderate though dramatic effect. Independent of the reporter gene construct used the A370T variant showed a 2- (P < 0.001) or 1.5-fold (P < 0.05) enhanced basal activity, respectively. The V140M variant showed a 1.6-fold enhanced basal activity toward the CYP3A4 construct (P < 0.05) and a 1.3-fold enhanced basal activity toward the DR3 construct, which was, however, statistically not significant. In contrast, the D163G and PXR-2 variants did not show any basal activity; they even appeared to inhibit the DR3 and CYP3A4 constructs, but this inhibition was statistically not significant (Fig.3, A and B).
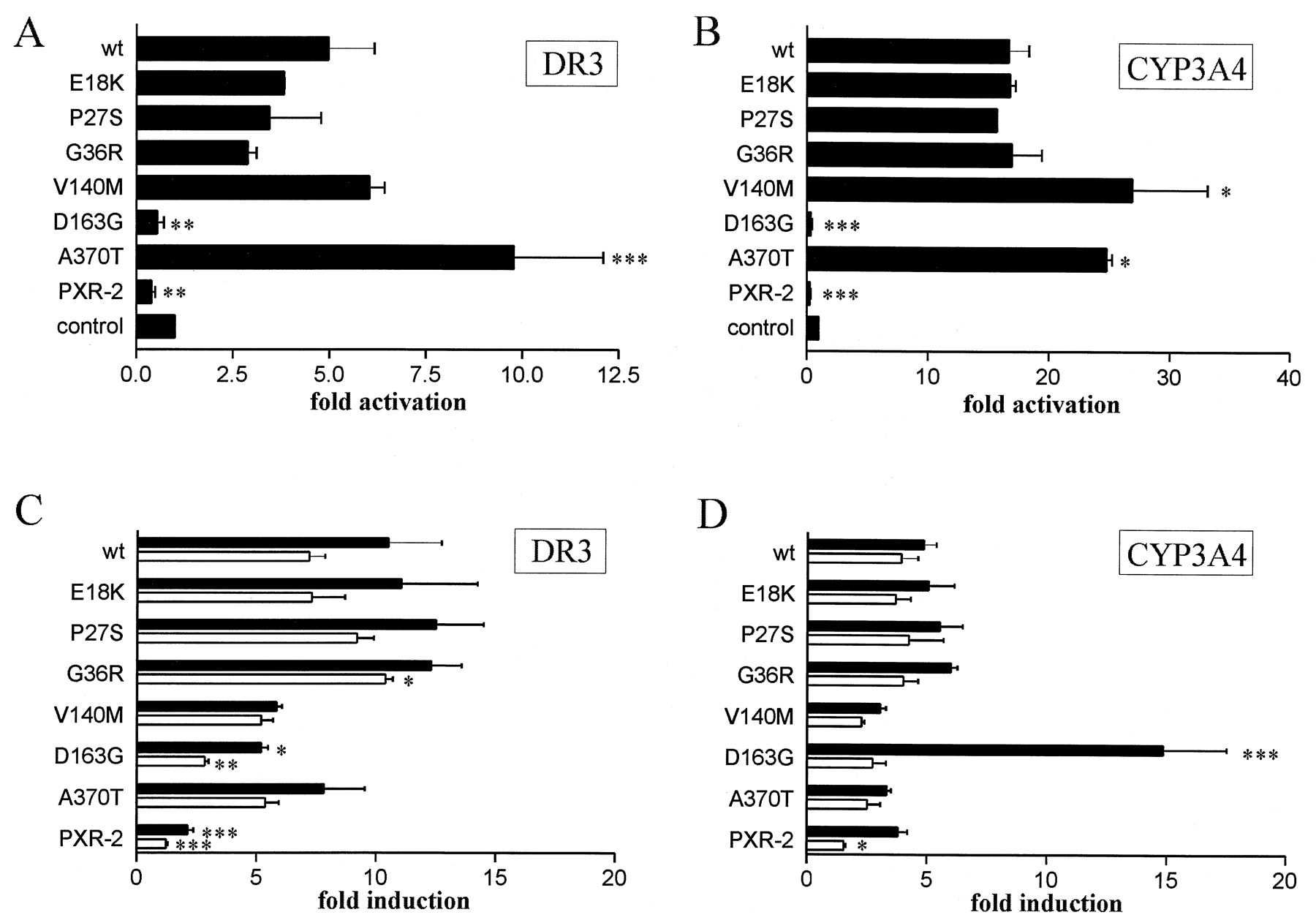
The effect of PXR protein variants on basal (A and B) or induced (C and D) transcriptional activity.
LS174T cells were cotransfected either with the DR3 or CYP3A4 reporter gene constructs and with PXR wild-type (wt) or variant expression plasmids as indicated. A and B, the effect of PXR variants in the absence of exogenous inducers. The activity of the respective reporter gene in the presence of empty expression vector (control) was designated as 1. C and D, the effect of PXR variants following 42-h treatment with 10 μM rifampicin (black bars) or 10 μM corticosterone (open bars). The activity of each reporter/expression plasmid combination in the presence of DMSO only was designated as 1. Data are shown as mean value ± S.D. The numbers of independent transfection experiments done with the different PXR expression plasmids in combination with either reporter gene were as follows: E18K, P27S, G36R (2); V140M, D163G, PXR-2 (3), A370T (4), wt (5). Statistically significant differences as compared with wild-type are indicated by asterisks. ∗, p < 0.05; ∗∗,p < 0.01; ∗∗∗, p < 0.001.
In the following, we investigated the effect of the variants on the response to rifampicin or corticosterone, two known activators of PXR-mediated CYP3A4 transcription (Blumberg et al., 1998;Lehmann et al., 1998). The variants E18K and P27S stimulated transcription of both reporter genes nearly as efficiently as wild-type PXR following the rifampicin or corticosterone treatment (Fig. 3, C and D). The activity of the G36R toward the DR3 construct in response to corticosterone was increased by 40% (P < 0.05). In contrast, transcriptional activation of the DR3 construct in response to either inducer and toward the CYP3A4 construct in response to corticosterone was significantly reduced by the PXR-2 variant (Fig. 3C, D). Interestingly, the D163G variant resulted in a 50% reduced induction of the DR3 construct expression following treatment with either inducer (P < 0.05 and P < 0.01, respectively; Fig. 3C), whereas treatment with rifampicin but not corticosterone revealed a 3-fold stronger activating effect of this variant on the expression of the CYP3A4 construct (P < 0.001; Fig. 3D) than wild-type PXR. V140M and A370T showed a reduced induction of both reporter gene constructs after treatment with either inducer. However, these effects were not statistically significant.
We hypothesized that the opposite effects of the D163G variant on the transactivation of the two different reporter gene constructs in response to rifampicin could be caused by the presence of different types of PXR response elements in the reporter gene constructs used. ER6-type PXR response elements were present only in the CYP3A4 construct. Therefore, we investigated the induction by rifampicin of wild-type PXR and D163G variant in more detail, using further reporter genes (Fig. 4). In accordance with our hypothesis, D163G showed an 8-fold higher induction by rifampicin than wild-type PXR of a reporter gene containing two ER6 elements cloned in front of the TK promoter (ER6). Figure 4 also shows that the induction properties of wild-type PXR and D163G variant were not significantly different if a CYP3A4 construct with a mutated proximal ER6 element was used. In contrast, mutation of the two PXR response elements dNR1 (DR3-type) and dNR2 (ER6-type) in the distal xenobiotic-responsive enhancer module increased the difference in the induction properties of wild-type PXR and D163G compared with wild-type CYP3A4 construct. With this latter construct, D163G showed a 15-fold higher induction by rifampicin than wild-type PXR. Altogether, these data indicate that the enhanced induction by rifampicin of D163G is dependent on the ER6 element in the proximal promoter of CYP3A4.
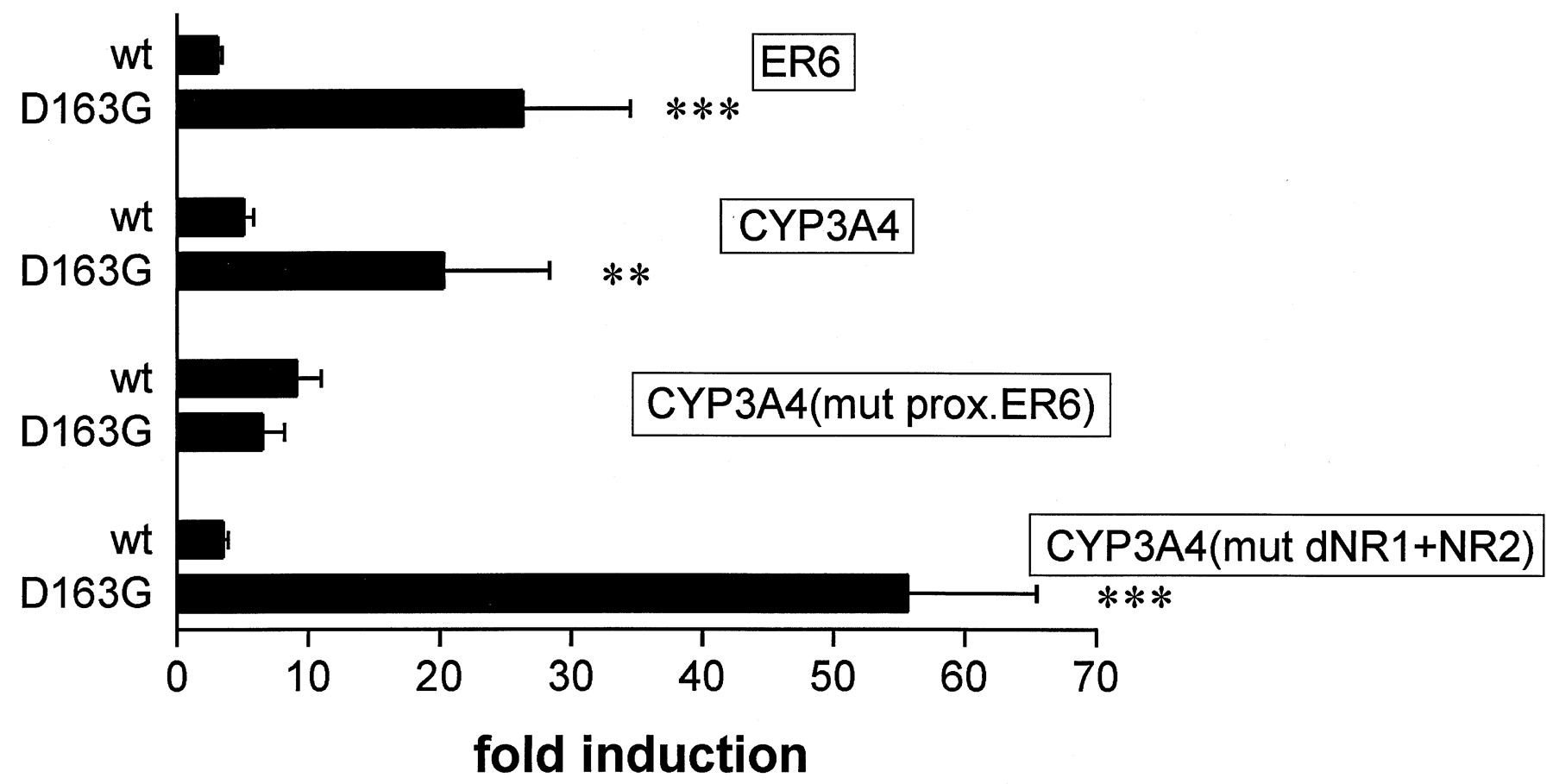
Enhanced induction by D163G depends on the ER6 element in the proximal CYP3A4 promoter.
LS174T cells were cotransfected with the indicated reporter gene constructs and with PXR wild-type (wt) or D163G expression plasmids. Bars show the induction following a 42-h treatment with 10 μM rifampicin. The activity of each reporter/expression plasmid combination in the presence of DMSO only was designated as 1. Data are shown as mean value ± S.D. The numbers of independent transfection experiments done with the different reporter genes were as follows: ER6, CYP3A4 (5); CYP3A4(mut prox. ER6), CYP3A4(mut dNR1+NR2) (3). Statistically significant differences as compared with wild-type are indicated by asterisks. ∗∗, p < 0.01 and ∗∗∗, p < 0.001.
The three variants caused by single amino acid substitutions, which affect the basal and/or induced expression of the reporter gene constructs, are V140M, D163G, and A370T. The PXR-2 splice variant, which lacks a contiguous stretch of 37 amino acids (Dotzlaw et al., 1999), has a similar effect. A similar deletion of 41 amino acids in the same region of the LBD of mouse PXR, termed mPXR.2, has also been shown to strongly reduce the responsiveness toward synthetic steroid and naturally occurring pregnane ligands of PXR (Kliewer et al., 1998). A common feature of these variants is their localization within (D163G, A370T, and PXR-2) or at the boundary (V140M) of the LBD of PXR. The effects of D163G and PXR-2 protein variants on the response to rifampicin and corticosterone are consistent with an impaired or altered ligand binding to the LBD. Based upon the recently published crystal structure of the human PXR LBD (Watkins et al., 2001), amino acid D163 is not expected to be directly involved in ligand binding because it is not lining the ligand binding cavity. In contrast, some of the amino acids, which are deleted in PXR-2, are lining the ligand binding cavity. Therefore, direct effects on ligand binding have to be expected and could explain the altered induction properties of PXR-2. The enhanced induction by rifampicin but not corticosterone of D163G is not a unique phenomenon. Rifampicin but not the high-affinity ligand SR12813 appears to be a more potent activator of experimental mutant D205A than of wild-type PXR (Watkins et al., 2001). To our knowledge, the data obtained with D163G variant for the first time demonstrated that the type of a PXR response element influences induction. The PXR LBD-variants V140M, D163G, A370T, and PXR-2 show an altered basal transcriptional activity (in the absence of exogenous inducers). Similar altered basal activity has been reported for mutants generated in the LBD of the PXR protein (Watkins et al., 2001). Basal activity could be explained by the existence of an endogenous PXR ligand in LS174T cells. As reported previously, pregnanes (Kliewer et al., 1998) or bile acids (Staudinger et al., 2001) are likely candidates for endogenous ligands. Alternatively, it could reflect a ligand-independent, intrinsic capability of PXR to transactivate its targets. Altered basal and ligand-dependent activity often seem to be coupled. This favors the hypothesis that endogenous ligands are responsible for the basal activity of PXR and not a true ligand-independent mechanism as shown to be present in the related nuclear receptor CAR (Tzameli et al., 2000).
The allelic frequencies of PXR protein variants found in this study predict that individuals homozygous or double heterozygous for a PXR protein variant constitute a minuscule fraction of the Caucasian and the West-African populations. A number of single nucleotide polymorphisms have been identified, some of which affect either constitutive or inducible activity, but further studies are needed to elucidate whether variants with altered transcriptional activity towardCYP3A4 may represent reliable predictors for CYP3A4 activity useful in drug development or treatment. On the other hand, it has to be shown whether these variants play a role in atypical responses to drugs or altered sensitivity to carcinogens that are CYP3A4 substrates. The verification of this hypothesis will be provided by genotyping of patients from clinical or epidemiological studies.
Acknowledgments
We thank J. Brockmoller, U. A. Meyer, and U. Zanger for DNA samples used in this study. C. Liddle kindly provided us with the plasmid p3A4-362(7836/7208ins) mut dNR1+NR2.
Footnotes
-
This work was supported in parts by the Deutsche Forschungsgemeinschaft (Germany), Grant Bu 1249/1–1 (to O.B.), by the Robert Bosch Foundation (Germany), and by Grant 01GG9848–15 from the German Ministry for Education, Science, Research, and Technology.
- Abbreviations used are::
- CYP
- cytochrome P450
- PXR
- pregnane X receptor
- bp
- base pair
- PCR
- polymerase chain reaction
- DMSO
- dimethyl sulfoxide
- BAC
- bacterial artificial chromosome
- LBD
- ligand-binding domain
- TK
- thymidine kinase
- Received May 25, 2001.
- Accepted August 6, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}