


Abstract
Testosterone, 7-benzyloxyquinoline, and 7-benzyloxy-4-trifluoromethyl-coumarin, marker substrates for cytochrome P450 3A4 are commonly used within the pharmaceutical industry to screen new chemical entities as inhibitors of CYP3A4 in a high-throughput manner to predict the potential for drug-drug interactions. However, it has been observed that inhibition data obtained with a given CYP3A4 probe substrate may not correlate well with results from a different probe. As a consequence, the choice of the probe compound becomes an important consideration in such screens. In the present study, kinetic interactions between either two of the above three substrates were evaluated, and three-dimensional nonlinear regression analysis was performed to understand the kinetic mechanisms of drug interaction. Our results demonstrate that the kinetic interaction between each pair of substrates does not appear to be competitive and that the interactions are characterized by an unchanged or a decrease in both apparent Km (a = 0.21−0.72, a change of Km in the absence of the effector) and Vmax (α andβ = 0.09−0.75, changes ofVmax in the absence of the effector). These data suggest that 1) the three substrates bind to different domains; 2) at least two substrates can coexist in the active site of CYP3A4; and 3) the two bound substrates interact kinetically with each other (e.g., through steric hindrance), thereby leading to a change in both apparent kinetic parameters and partial inhibition. Selection of multiple substrates, which are shown not to be competitive, is necessary to accurately predict CYP3A4 inhibition and the potential for drug-drug interaction.
In the early stages of the drug discovery process, compounds are subjected to a battery of screens to maximize the success rate in clinical trials (Lin and Lu, 1997; Bajpai and Adkison, 2000; White, 2000). Cytochromes P450 (P450s1), a superfamily of drug-metabolizing enzymes, are recognized to play an important role in clinically relevant drug-drug interactions, as a result of which new chemical entities (NCEs) typically are subjected to high-throughput P450 inhibition screening (Lin and Lu, 1997; Crespi, 1999; Moody et al., 1999; Rodrigues et al., 2001). In vitro studies that use human liver microsomes containing multiple P450s or recombinant enzymes are widely used for this purpose, and fully automated high-throughput assays, such as those that use LC-MS/MS with short run times (Ayrton et al., 1998; Yin et al., 1999; Chu et al., 2000) or 96-well microtiter plates with fluorescent probes (Chauret et al., 1999; Crespi et al., 1999), have been developed. Because of the broad substrate specificity of CYP3A4, which catalyzes the metabolism of a wide spectrum of therapeutic agents, this isoform has been the focus of many screening efforts. However, recent studies have shown a poor correlation in inhibition data obtained with different CYP3A4 substrates. Moreover, many substrates are not mutually competitive, thereby making this simple approach unreliable (Kenworthy et al., 1999;Wang et al., 2000a,b). Therefore, in vitro CYP3A4 inhibition screening should be performed with caution.
CYP3A4 metabolizes structurally diverse substrates with a wide range of molecular size, shape, enzyme affinity, and turnover number. Unusual kinetic interactions involving CYP3A4 have been found to include sigmoidicity or substrate activation (Ueng et al., 1997; Shou et al., 1999), partial inhibition (Wang et al., 1997, 2000a; Kenworthy et al., 1999), substrate inhibition (Tang et al., 2000; Lin et al., 2001;Schrag and Wienkers, 2001), activation (Shou et al., 1994; Domanski et al., 1998; Ngui et al., 2000), and differential kinetics (Shou et al., 2001). These phenomena are thought to be due to the binding of multiple substrates within the active site of the enzyme (Shou et al., 1994,2001; Koley et al., 1995; He et al., 1997; Harlow and Halpert, 1998;Korzekwa et al., 1998; Hosea et al., 2000). As a result, the interaction of an inhibitor with one CYP3A4 probe may differ from that observed with other CYP3A4 substrates. The atypical interactions observed in vitro can be attributed to differential binding and steric restriction for interacting agents (substrates and inhibitors), resulting in both substrate- and inhibitor-dependent inhibition. This may lead to the erroneous evaluation of drug interaction potential. In the present study, we used testosterone, 7-benzyloxyquinoline (BQ), and 7-benzyloxy-4-trifluoromethyl-coumarin (BFC), commonly used probes for CYP3A4 inhibition screening, to elucidate the mechanism of kinetic interaction of any two substrates using three-dimensional fitting of a two-binding site model. Our results demonstrate that the three CYP3A4 substrates can bind to different domains at the active site of CYP3A4 and that any pair can interact with each other in a manner that is distinct from simple competitive inhibition. These results show that the use of more than one substrate may be required to accurately predict the potential for drug-drug interactions that occur at the level of CYP3A4 inhibition.
Materials and Methods
Chemicals.
Testosterone, 6β-hydroxytestosterone, cortisone, and NADPH were purchased from Sigma (St. Louis, MO). BQ, BFC, and their metabolites were obtained from Ultrafine Chemicals (Manchester, England, UK).
CYP3A4 Expression in Baculovirus.
Plasmids pGem-7/CYP3A4 and pGem-3Z/oxidoreductase (OR) containing the full-length cDNAs for CYP3A4 and OR (15) were provided by Dr. Frank J. Gonzalez (National Cancer Institute, Bethesda, MD). The entire coding region of each cDNA was excised from the vectors and inserted into baculovirus shuttle vector, pBlueBac (CYP3A4,XbaI/KpnI and OR, EcoRI). Recombinant virus was constructed according to the manufacturer's procedure (Invitrogen, Carlsbad, CA). CYP3A4 and OR proteins were coexpressed in Sf21 insect cells (Invitrogen). The multiplicity of infection ratio of viruses encoding CYP3A4 and OR was 1:0.1. Total P450 content was measured by the CO-difference spectrum and the molar ratio of CYP3A4 to OR in microsomal preparation was 1:2.8. The specific activity of CYP3A4 in testosterone 6β-hydroxylation was 42 nmol/min, nmol.
Metabolism of CYP3A4 Substrates.
Incubations of each substrate pair (testosterone-BFC, BQ-testosterone, and BQ-BFC) were performed using 96-well microtiter plates. The incubation mixture (usually a 0.2-ml final volume) consisted of 5 pmol of microsomal CYP3A4 protein, 0.1 M potassium phosphate buffer (KPi, pH = 7.4), and substrate in the presence or absence of the second substrate/effector. The concentrations of the two substrates varied from 3.1 to 100 μM in a total of 64 incubations. The incubation mixture was preincubated for 5 min at 37°C in a shaking water bath, and the reaction was initiated by the addition of 1 mM NADPH and incubated further for 15 min. The reactions were terminated by the addition of 2 volumes of acetonitrile containing the appropriate internal standards (cortisone for testosterone metabolism and 7-OH-4-methylcoumarin for BQ and BFC metabolism). Samples were centrifuged at 4000 rpm for 10 min, and supernatants were divided into two aliquots for metabolite quantitation from each substrate. Standard curves for the metabolites of interest were prepared using the corresponding authentic reference materials. The consumption of each substrate at all concentrations studied was limited to less than 10%, and the rates of product formation were expressed as nanomoles of product formed per minute per nanomoles of P450.
HPLC Analysis.
HPLC was performed using an HP1100 liquid chromatograph equipped with an HP model 1100 autosampler, a ternary solvent delivery system, and a fluorescence detector controlled by the Hewlett-Packard HPLC2D ChemStation software. Metabolites of BQ, BFC, or both were separated on a Zorbax SB-C18column (5 μm, 4.6 mm × 15 cm; MAC-MOD Analytical, Chadds Ford, PA), eluted using a mobile phase with a linear aqueous acetonitrile gradient (6 min from 15–40% and then 4 min from 40–90% acetonitrile in water containing 20 mM NH4Ac) at a flow rate of 1 ml/min. Samples were detected at emission and excitation wavelengths of 420 nm and 503 nm, respectively. Retention times of BFC and BQ metabolites and of internal standard were 10, 5.3, and 7.2 min, respectively. The metabolites were quantified by normalization against the internal standard.
LC-MS/MS.
Quantitative analysis of testosterone and its metabolite, 6β-hydroxytestosterone, was achieved using a PerkinElmer HPLC system, comprised of a Series 200 LC pump and LEAP autosampler coupled to a Sciex API 2000 mass spectrometer operated in the positive ion mode. Aliquots (20 μl) of incubation mixtures were chromatographed on a BDS hypersil C8 column (5-μm particle; 2 × 50 mm; Keystone Scientific, Inc., Bellefonte, PA) with a mobile phase consisting of solvent A (90% water/10% MeOH/0.1% trifluoroacetic acid) and solvent B (10% water/90% acetonitrile/0.1% trifluoroacetic acid). Samples were eluted using a 1.9-min linear gradient from 100% solvent A to 50% solvent B at a flow rate of 1.5 ml/min. Metabolite and internal standard were detected under atmospheric pressure chemical ionization conditions and identified using the transitionsm/z 305.5 → 269 (6β-OH-testosterone) andm/z 361 → 163.2 (internal standard). The rate of 6β-hydroxytestosterone formation was determined according to a standard curve generated concurrently.
Kinetic Model for Drug-Drug Interaction.
According to the observed changes in apparentKm and Vmax in the presence of the two substrates (Tables1-3), we proposed the two-site model (Scheme FS1) that states that the two different substrates bind specifically to their respective domains at the active site to form S2E, ES1, and S2ES1 complexes, respectively, which break down to product (P1 or P2). Since the two substrates within the active site interact kinetically, the dissociation (KS) and rate constants (kp) for the doubly bound enzyme complex (S2ES1) may no longer resemble those of the singly bound forms (S2E and ES1), such that the greatest changes in apparent kinetic constants are reflected by factors a, α, and β, as designated in the model (Scheme FS1) when [E]total = [S2ES1]. The kinetic scheme for an enzyme with two binding sites where the two substrates interact is depicted in Scheme FS1, whereas the corresponding velocity equations (Eqs. 1 and 2) express the kinetic relationship during the course of metabolism. The definition of kinetic parameters in the equations is described in Scheme FS1. We tested the hypothesis that the derived equations provide a reasonable fit to the experimental data and that the resulting kinetic parameters can be used to interpret the nature of the observed interactions of the particular CYP3A4 substrates within the active site. A more precise description of molecular events incorporating the binding of multiple substrate molecules can be achieved with a steady state and rapid equilibrium approach to the analysis of the interactions. The two domains in the model, which are bound singly and simultaneously by the substrates, can be distinguished from one another in such a way that a change in the binding affinity of the substrate and rate of product formation can be observed.
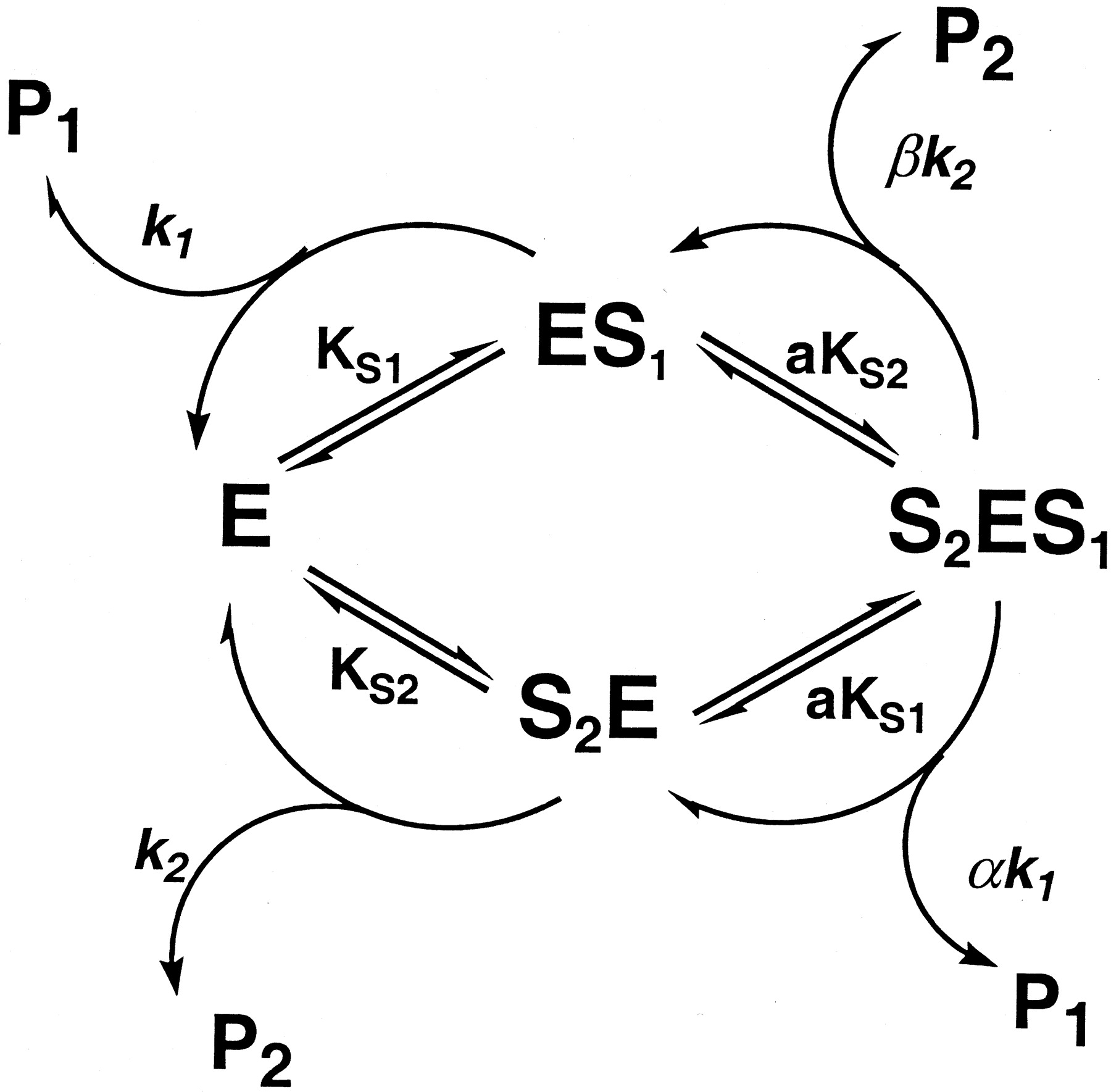
Proposed kinetic model for the two substrate interaction involving CYP3A4-mediated metabolism.
The model contains two binding domains in the active site of the enzyme, which can be occupied by two different substrates (S1 and S2) to form ES1 and S2E, respectively. The two singly bound enzyme complexes are further bound with the second substrate molecule to give S2ES1. KS1,KS2, k1, andk2 are dissociation and rate constants for ES1 and S2E, respectively. Factors a, α, and β represent changes of dissociation and rate constants when the second domains binds with an additional substrate molecule (e.g., S2ES1).Vmaxp1 orVmaxp2 =k1[E]total ork2[E]total. Velocity equations for P1 and P2 are shown in the text.
Kinetic Derivation and Analysis.
The values of all kinetic parameters in the equations were determined by nonlinear regression analysis of the experimental data using Mathematica 4.0 (Wolfram Research, Inc., Champain, IL). Each surface plot consists of both observed experimental data (scatter points) and predicted results (meshed plot) using either Eq. 1 or 2. An evaluation of goodness of the data fit was performed with statistical analyses (RSS and R2).
Results
Studies on the time dependence of product formation from testosterone, BQ, and BFC at their respectiveKm concentrations were performed to conform linearity up to at least 20 min (data not shown). Less than 10% of the substrate was consumed, and secondary metabolism was minimal during the course of the incubation. The linearity of product formation as a function of incubation time suggests that CYP3A4-catalyzed reactions were at steady state. Thus, all enzyme-substrate complexes were formed at the fixed rate at which they decomposed (e.g.,d[S2E]/dt = 0,d[ES1]/dt = 0 andd[S2ES1]/dt= 0). We assumed also that all the equilibria between enzyme and substrate(s) are very rapid as compared with the rate at which the enzyme-substrate complex breaks down. Therefore, velocity equations were derived to generate dissociation (KS1and KS2) and rate constants (kp).
Interaction of BFC with Testosterone.
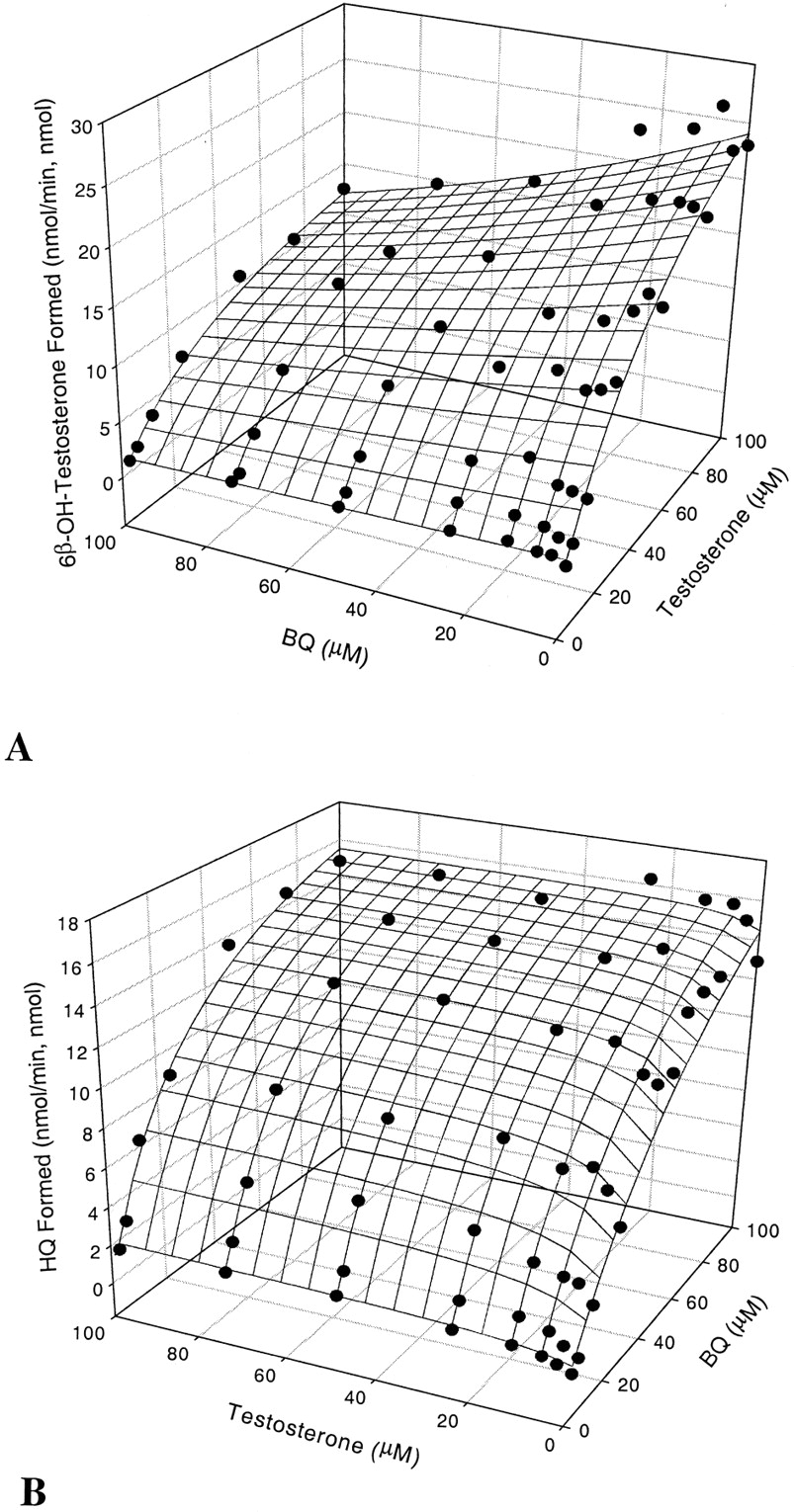
Eight concentrations each of BFC and testosterone (0–100 μM) were used to evaluate product formation, providing 64 incubation data points corresponding to various combinations of concentrations. Each incubation was divided into two aliquots that were analyzed by HPLC and LC-MS/MS for quantitation of BFC and testosterone metabolites, respectively. In the absence of effector,Km and Vmaxvalues were determined to be 49.6 μM and 29.6 min−1 for testosterone and 48.5 μM and 49.3 min−1 for BFC, respectively (Table 1). The effect of one substrate on the metabolism of another substrate is shown in Fig. 1, A and B. The scatter plots indicate actual measurements of the reaction rate (z-axis, vertical) of a given substrate (x-axis, lateral) in the presence of the second substrate (y-axis, horizontal). Velocity equations were used to fit experimental data, and the meshed plots represent predicted results, which yielded estimates of kinetic parameters (Table 4). As seen in Table 1, the addition of BFC (3.13–100 μM) had a negligible effect on the apparent Km of testosterone and resulted in a decrease (50%) in Vmax. Similarly, testosterone had no effect on the apparentKm but slightly decreasedVmax values for BFCO-dealkylation. Hence, the pattern of the kinetic interaction of the two substrates did not conform to competitive inhibition in which no change in Vmax and an increase in Km should be observed. The results suggest that the two substrates have access to different binding domains within the active site. Thus, the two-site model was adapted to fit the experimental observations (Scheme FS1) according to the observed kinetic interactions. Since the substrate interaction altered both apparent Km andVmax, factors a, α, and β, which represent changes in dissociation and rate constants for one substrate in the presence of another, were taken into consideration. Surface plots (Fig. 1, A and B) and calculated kinetic values (Table 4) were used to assess the goodness of fit of predicted results with statistics (e.g., standard error, R2, and RSS). KS1,KS2, Vmax1, andVmax2 are dissociation constants and maximal velocities for singly bound ES1(S1 = BFC) and S2E (S2 = testosterone), respectively. These values are consistent with apparent Km andVmax values measured in the absence of the second substrate (Table 1). However, when the second molecule was present in the enzyme reaction ([E]total = [S2ES1]), the changes forKS and Vmaxwere found to decrease by 51 and 28% forKS (a = 0.49 and 0.72 from the two respective equations; Table 4) and by 75% forVmax1 (testosterone, α = 0.25) and 41% for Vmax2 (BFC, β = 0.59), respectively. These results suggest that the two substrate molecules can bind simultaneously to two separate domains at the active site of the enzyme and subsequently interact with each other, leading to partial inhibition and changes in apparent kinetic constants (for example, decrease in both Km andVmax).
Kinetic parameters for the in vitro interaction between BFC and testosterone

Kinetic interaction between BFC and testosterone over the concentration range from 3.125 to 100 μM.
A, effect of BFC on CYP3A4-mediated testosterone 6β-hydroxylation; B, effect of testosterone on CYP3A4-catalyzed BFCO-dealkylation.
Predicted kinetic parameters
Interaction of BQ with Testosterone.
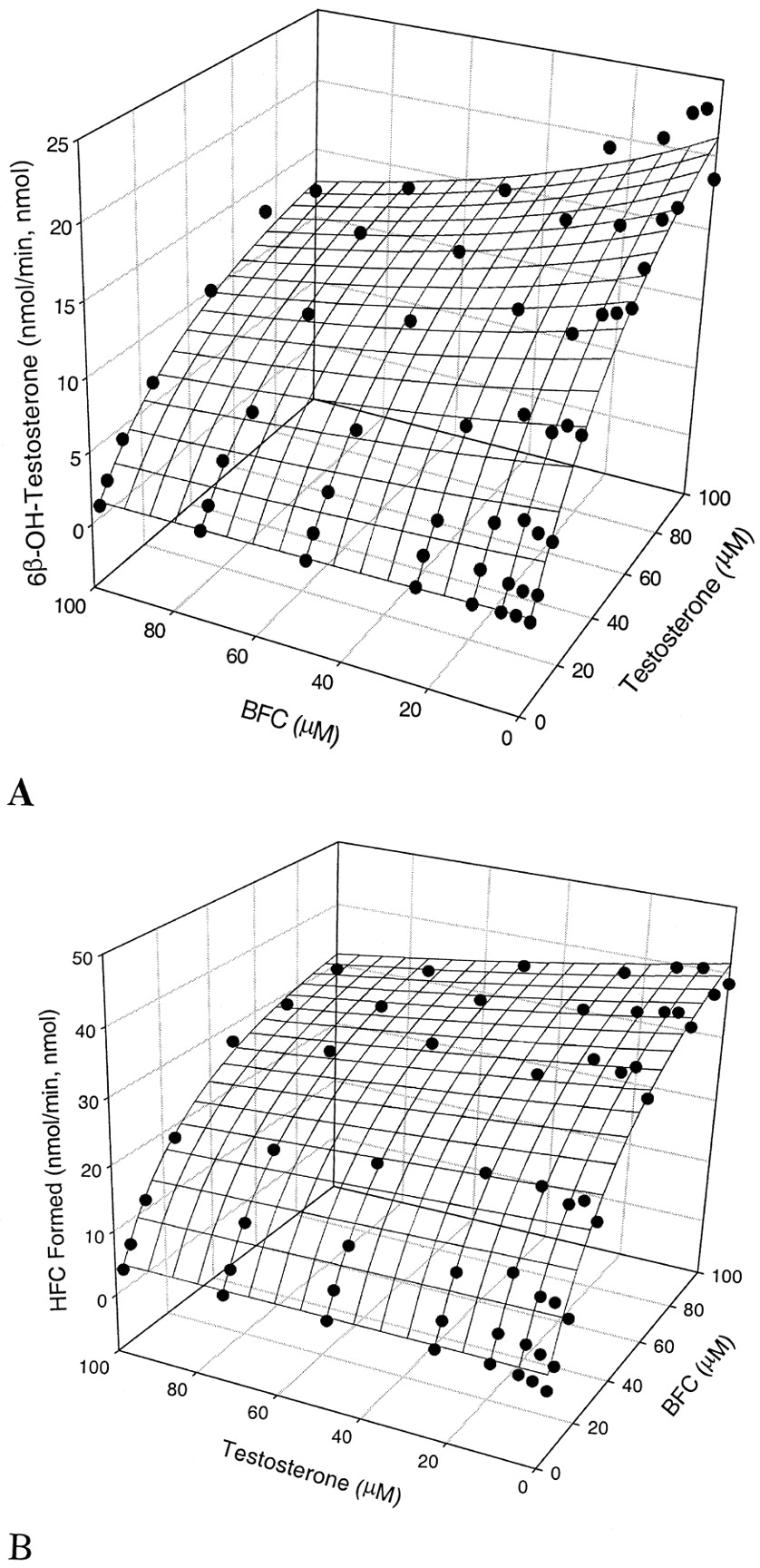
Figure 2, A and B, shows the results of kinetic interaction studies between BQ and testosterone. In the absence of effector, the Km values for testosterone and BQ were 58.6 and 76.2 μM, and theVmax values were 35.9 and 25.2 min−1, respectively (Table 2). In the presence of BQ, Km andVmax for testosterone decreased slightly. On the other hand, testosterone decreasedKm and had no significant change onVmax. The inhibition was neither competitive nor noncompetitive. These findings indicated that the two substrate molecules must bind differentially to the two domains as in the case of BFC and testosterone. The predicted results (meshed plots) and the calculated parameters are shown in Fig. 2, A and B, and Table4, respectively. The goodness-of-fit of the two velocity equations was evaluated by RSS (19.4 and 95.2) andR2 (0.987 and 0.967). As a consequence, factor a values that define a maximal decrease in apparent Km in the presence of the effector were calculated by the two equations to be 0.58 and 0.63, respectively. These imply that the inclusion of the second molecule can decrease the apparent Km for the first substrate by up to 37 to 42%. Similarly, factors α and β, which represent the changes in Vmax1 andVmax2, when the second substrate is bound completely, were estimated to be 0.75 and 0.48, respectively, resulting in a maximal inhibition of 25% for Vmax1in BQ O-dealkylation and 52% forVmax2 in testosterone 6β-hydroxylation.
Kinetic interaction of BQ and testosterone over the concentration range from 3.125 to 100 μM.
A, effect of BQ on CYP3A4-mediated testosterone 6β-hydroxylation; B, effect of testosterone on the CYP3A4-catalyzed BQO-dealkylation.
Kinetic parameters for the in vitro interaction between BQ and testosterone
Interaction of BQ and BFC.
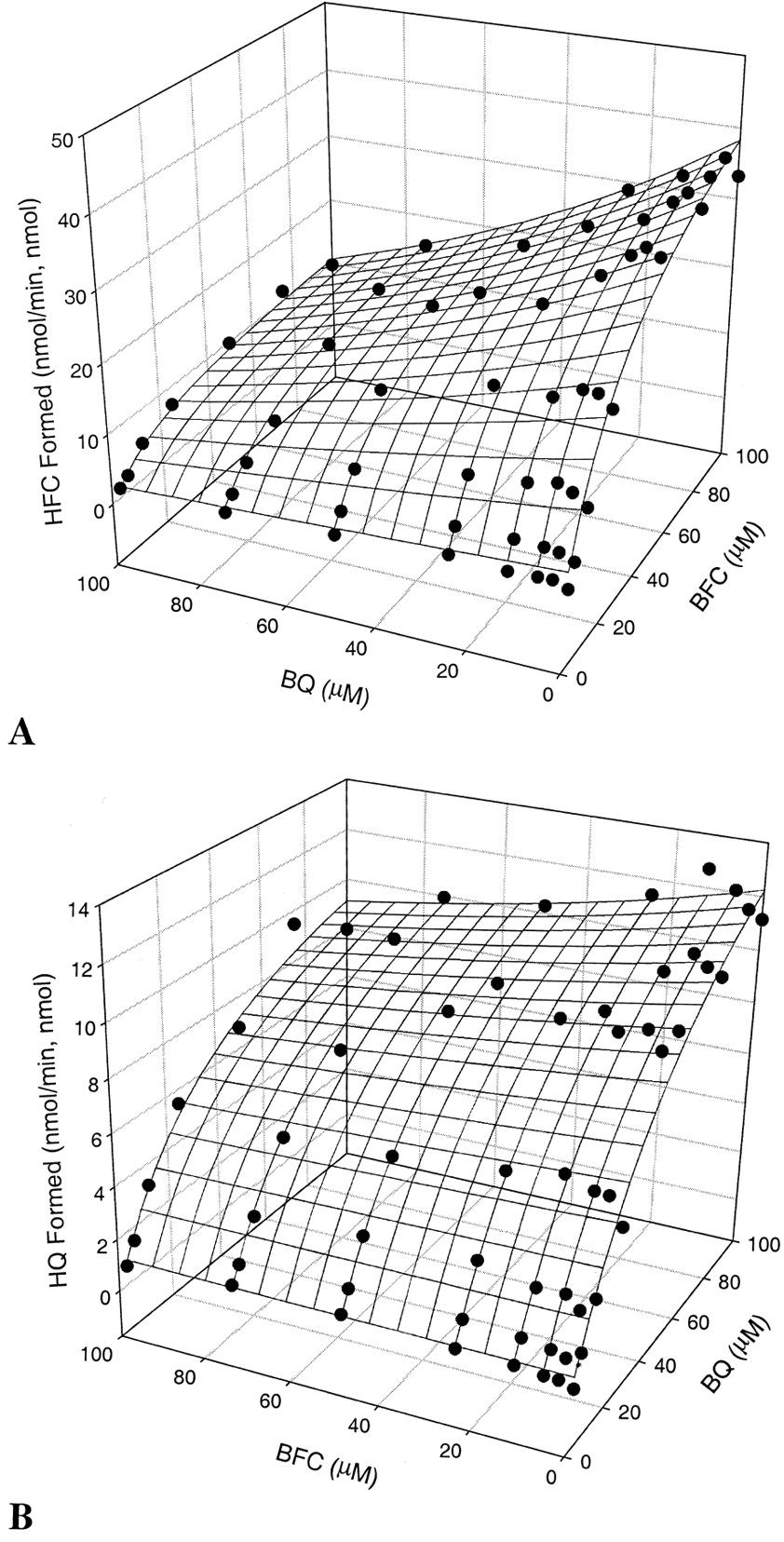
The metabolism of BQ or BFC generated fluorescentO-dealkylation products that were detected by their fluorescence following an HPLC separation. BQ (3.13–100 μM) decreased significantly both the apparentKm and Vmaxvalues for BFC from 42 to 21 μM (Km) and from 52 to 14 min−1(Vmax). Similarly, when BFC was used as an effector, apparent Km andVmax values for BQ decreased from 63 to 32 μM and from 19.2 to 11.8 min−1, respectively (Table 3). Hence, the interaction between the two substrates was not competitive. The two-site model was used to fit the data, resulting in three-dimensional surface plots (Fig. 3, A and B) and a solution of the predicted kinetic parameters (Table 4). Factor a values (0.3 and 0.21, respectively, from the two equations) indicate that one substrate can interact with another by diminishing apparent Km by as much as 70 to 79%. Similarly, the apparent Vmax for BQ decreased significantly by 91 (α = 0.09) or 77% for BFC (β = 0.21) in the presence of the respective effector.
Kinetic parameters for the in vitro interaction between BQ and BFC

Kinetic interaction between BQ and BFC over the concentration range from 3.125 to 100 μM.
A, effect of BQ on CYP3A4-catalyzed BFC O-dealkylation; B, effect of BFC on the CYP3A4-mediated BQO-dealkylation.
Discussion
Inhibition of a drug-metabolizing enzyme, typically a cytochrome P450 isoform, is a common cause of adverse clinical interactions between coadministered therapeutic agents. Consequently, high-throughput screening assays for P450 inhibition and drug-drug interactions have now become an integral part of drug discovery and development programs, where they serve to eliminate drug candidates at an early stage. Appropriate substrates that are used to probe a particular P450 enzyme can be selected to provide a rapid estimation of IC50 values and, thus, an initial determination of the inhibitory capability for the NCEs. Because the majority of drugs metabolized by P450 are substrates for CYP3A4, this isoform has been the subject of the most intensive screening efforts.
Numerous investigators have developed assays for CYP3A4 activity with different probe substrates that are markers for this isoform. Unfortunately, recent experience has revealed that 1) poor correlations often are obtained with varying degrees of inhibition when different CYP3A4 probe substrates are used (Kenworthy et al., 1999; Wang et al., 2000a,b); 2) the presence of one substrate may result in partial inhibition of another substrate and vice versa (Kenworthy et al., 1999;Wang et al., 2000a); and 3) IC50 values for a given inhibitor may differ widely between two CYP3A4 substrates (Wang et al., 1997; unpublished data). These issues are of particular concern with respect to the current trend for adopting high-throughput screening approaches for assessment of enzyme inhibition. The present study provides evidence that the multiplicity and complexity of CYP3A4-mediated kinetics is due to the existence of multiple binding domains at the active site of the enzyme. Accordingly, an inhibitor that inhibits the metabolism of one substrate (competitive inhibition) may not inhibit that of other substrates (multiple binding domains). Thus, in vitro inhibitory screens may be inconsistent and lead to erroneous prediction of drug-drug interaction potential.
In the present study, drug-drug interaction between two of three CYP3A4 substrates was studied using a wide range of substrate concentrations that varied from approximately 0.1 to twice theKm for each substrate. The concentrations for both interacting substrates were selected to cover all possible concentration-related drug-drug interactions. Thus, an entire kinetic analysis was conducted with a sufficiently large number of data points to obtain reliable estimates of kinetic parameters. The key finding from the present study is that the interaction between two CYP3A4 substrates did not conform to Michaelis-Menten kinetics (i.e., the interaction was not competitive within the active site of CYP3A4), suggesting that the two substrates can coexist and also interact with each other at the active site. This is not surprising because there is abundant evidence that CYP3A4 exhibits non-Michaelis-Menten kinetics. Thus, there are particular kinetic features for several CYP3A4 substrates that cannot be interpreted within the context of the Michaelis-Menten model and that require the adoption of an enzyme model with multiple binding sites to explain the observed drug interactions.
Overall, the model proposed here accounts for all possible interactions between two substrates over a wide range of concentrations. The resulting kinetic estimates are listed in Table 4, and the predictedKS and Vmaxvalues for each of the substrates are consistent with the corresponding values observed in the absence of the second substrate. Factor a, which represents a change inKS in the presence of the second substrate, generally was less than unity. This suggested that the addition of the second substrate could result in a binding affinity greater than that for the singly bound complex (a = 0.21–0.72). Factors α and β, which reflect changes in Vmaxfor the two singly bound complexes when the second substrate is introduced, also were predicted and found to range from 0.09 to 0.75, meaning that interaction between the two substrates can decrease apparent Vmax by as much as 25–91%. It is clear that our study made use of sufficient data points to allow detailed examination of the effects of atypical kinetics. Comprehensive data sets are particularly important for substrates showing atypical kinetics and ensure a high degree of confidence in data fitting that leads to a precise solution of kinetic parameters in the model.
Our study indicates that all kinetic interactions observed between the two substrates resulted, to varying degrees, in a decrease in both apparent Km andVmax values, suggesting that the two substrate molecules may bind in close proximity and thereby partially inhibit the metabolism of one another. Taken together with the results of previous studies (Kenworthy et al., 1999; Houston and Kenworthy, 2000; Wang et al., 2000a; Shou et al., 2001a,b), the findings of the present investigation support the view that the physicochemical properties of different CYP3A4 substrates give rise to their unique interactions (KS) with particular amino acid residues within the CYP3A4 active site. As a result, the three substrates examined here (BQ, BFC, and testosterone), which did not compete with each other in combinations of any two substrates, probably occupy their own binding domains at the active site of the enzyme. The three domains allow for the simultaneous existence of at least two substrates for metabolism. With regard to the change in apparent kinetic constants (Km andVmax), the binding of one substrate may interfere with that of another substrate. This probably is a result of steric restrictions at the active site accommodating the two substrate molecules. However, it would be difficult to demonstrate, kinetically, the simultaneous occupancy of binding domains by more than two substrate molecules due to the complexity of the kinetic expression of the multiple-site model and a poor solution of numerous variables in the resulting equations.
Although interaction of any pair of the substrates can be described by the observed kinetics, more complexity of enzyme kinetics (i.e., allosterism of steroids) has to also be taken into consideration. It has been reported that CYP3A4-mediated steroid and BFC oxidations were shown to be sigmoidal characteristics. Allosteric mechanism of the former was understood by mutagenesis study (Harlow and Halpert, 1998;Domanski et al., 2000), in that the sigmoidicity of steroid hydroxylation can be shifted to a hyperbolic kinetics when replacing residues Leu-211 and Asp-214 with the larger Phe and Glu (L211F & D214E), respectively. This suggests that an additional site (effector binding site), for which the residues 211 and 214 are responsible, may be required to conduct cooperativity of the enzyme. Thus, more complicated situations can exist, which probably involve two sites for the steroid responsible for the sigmoidicity of steroid metabolism and one site for either BFC or BQ, although an interaction between the effector (steroid) site and BFC (BQ) site has not been elucidated kinetically.
When both domains are occupied by the respective substrates, complexity of drug-drug interaction within the CYP3A4 active site can occur and can be expressed by the factors, such as a, α, and β, in a manner different from that of the singly bound site. Thus, evaluation of the test compound as a CYP3A4 inhibitor can be inconsistent with use of different probe substrates. Although there is no simple or alternative solution for accurately predicting from one substrate to other due to the multiple binding, it is necessary to categorize CYP3A4 substrates by a fully kinetic analysis of drug-drug interaction. The rationale includes 1) grouping of the CYP3A4 substrates to ensure the substrates in a given group are competitive in nature, suggesting that the substrates bind to the same site; and 2) probe substrates from each group, therefore, can be selected for screening of NCEs for P450 inhibition, which may correlate well between substrates in the same group but may vary with the substrates between the different groups. In addition, care should be taken when atypical kinetics occur in terms of test compounds used. The interaction can be both substrate- and inhibitor-dependent due to the multiple bindings. Therefore, at present, it forces the choice of investigation of as many CYP3A4 substrates as possible in inhibition screens, particularly for the leads, until the mechanism of the unusual drug-drug interactions is fully understood, and thorough data for the rationale are completely convinced.
Acknowledgments
We gratefully acknowledge Dr. Paul G. Pearson for valuable discussions.
Footnotes
- Abbreviations used are::
- P450
- cytochrome P450
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- BQ
- 7-benzyloxyquinoline
- BFC
- 7-benzyloxy-4-trifluoromethyl-coumarin
- OR
- cytochrome P450 oxidoreductase
- HPLC
- high-pressure liquid chromatography
- NCE
- new chemical entities
- RSS
- residual sum of squares
- Received June 19, 2001.
- Accepted August 6, 2001.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}