


Abstract
(R,S)-Oxazepam is a 1,4-benzodiazepine anxiolytic drug that is metabolized primarily by hepatic glucuronidation. In previous studies, S-oxazepam (but not R-oxazepam) was shown to be polymorphically glucuronidated in humans. The aim of the present study was to identify UDP-glucuronosyltransferase (UGT) isoforms mediating R- and S-oxazepam glucuronidation in human liver, with the long term objective of elucidating the molecular genetic basis for this drug metabolism polymorphism. All available recombinant UGT isoforms were screened for R- and S-oxazepam glucuronidation activities. Enzyme kinetic parameters were then determined in representative human liver microsomes (HLMs) and in UGTs that showed significant activity. Of 12 different UGTs evaluated, only UGT2B15 showed significant S-oxazepam glucuronidation. Furthermore, the apparent Km for UGT2B15 (29–35 μM) was similar to values determined for HLMs (43–60 μM). In contrast, R-oxazepam was glucuronidated by UGT1A9 and UGT2B7. Although apparent Km values for HLMs (256–303 μM) were most similar to UGT2B7 (333 μM) rather than UGT1A9 (12 μM), intrinsic clearance values for UGT1A9 were 10 times higher than for UGT2B7. A common genetic variation results in aspartate (UGT2B15*1) or tyrosine (UGT2B15*2) at position 85 of the UGT2B15 protein. Microsomes from human embryonic kidney (HEK)-293 cells overexpressing UGT2B15*1 showed 5 times higherS-oxazepam glucuronidation activity than did UGT2B15*2 microsomes. Similar results were obtained for other substrates, including eugenol, naringenin, 4-methylumbelliferone, and androstane-3α-diol. In conclusion, S-oxazepam is stereoselectively glucuronidated by UGT2B15, whereasR-oxazepam is glucuronidated by multiple UGT isoforms. Allelic variation associated with the UGT2B15 gene may explain polymorphic S-oxazepam glucuronidation in humans.
Oxazepam is a 1,4-benzodiazepine derivative that is used in clinical practice for its anxiolytic, sedative, and anticonvulsant effects (Greenblatt et al., 1980, 1981). In humans, this drug is cleared from the body almost exclusively by hepatic glucuronidation, followed by urinary excretion (Abernethy et al., 1983). Oxazepam is formulated as a racemic preparation of S- and R-stereoisomers although the S-enantiomer is thought to be much more active as a benzodiazepine receptor agonist compared with theR-enantiomer (Mohler et al., 1978). Conjugation occurs via the hydroxyl group attached to the asymmetric 3-carbon position yielding diastereomeric glucuronides that are readily separated by routine high pressure liquid chromatography (HPLC1) (Mascher et al., 1984; Patel et al., 1995a).
Interindividual variability in the pharmacokinetics and metabolism of (R,S)-oxazepam have been investigated in human volunteers (Patel et al., 1995a). S-Oxazepam glucuronide was found to be formed preferentially over R-oxazepam glucuronide with S/R glucuronide diastereomeric ratios in the plasma and urine of volunteers averaging 3.5 ± 0.6 and 3.9 ± 0.8, respectively. Interestingly, in 2 of 11 subjects (18%), the S/R ratio in the urine was relatively low (<1.9). Since the plasma clearance of oxazepam in these individuals was also very low (<0.6 ml/min/kg) compared with other individuals (0.9–1.4 ml/min/kg), it was concluded that these differences probably were the result of slower S-oxazepam clearance by glucuronidation in a significant minority of the study population (i.e., a “slow metabolizer” phenotype). Although pharmacodynamic measurements were not made, the relatively slow elimination of oxazepam would be expected to result in prolonged sedation in these individuals.
In vitro studies using human liver microsomes (HLMs) showed a similar picture in that S-oxazepam glucuronide was the predominant metabolite (S/R ratios averaging 4.0), and 4 of 37 livers displayed relatively slow oxazepam glucuronidation activities coinciding with low S/R metabolite ratios (<2.0) (Patel et al., 1995a). Enzyme kinetic analysis showed that the low glucuronidation activity was associated with higher apparentKm values and lowerVmax values for S-oxazepam glucuronidation in the four atypical livers compared with the other livers. A genetic polymorphism in the gene encoding for the enzyme mediating this biotransformation was proposed to explain this phenomenon.
UGT isoforms that mediate stereoselective glucuronidation of either S- or R-oxazepam in human liver have not yet been identified. S- and R-oxazepam glucuronide formation by HLMs was shown to be differentially inhibited by a range of compounds suggesting that different enzymes catalyze the formation of the 2 metabolites (Patel et al., 1995b).S-Oxazepam glucuronidation was selectively inhibited by ketoprofen and morphine, which are known to be glucuronidated by UGT2B7, suggesting involvement of this isoform. However, the selectivity of these compounds as inhibitors of UGT2B7 has not been verified. Two other studies have investigated the possible role of UGT2B7 in oxazepam glucuronidation using recombinant enzyme (Jin et al., 1993; Coffman et al., 1998). In both instances very low oxazepam glucuronidation activities were reported, and bothR-oxazepam and S-oxazepam were glucuronidated to an equal extent. Together these findings indicate that UGT2B7 may be involved in oxazepam glucuronidation but is not responsible for stereoselective conjugation of either of S- orR-oxazepam.
The aim of the present study was to identify UGT isoforms mediating stereoselective glucuronidation of R- andS-oxazepam in liver using recombinant enzymes and HLMs. Recombinant allelic variants (UGT2B15*1 and UGT2B15*2) of UGT2B15, the principal isoform found to mediate S-oxazepam glucuronidation, were also used to evaluate whether this highly prevalent polymorphism could account for interindividual variability inS-oxazepam glucuronidation.
Materials and Methods
Reagents.
Unless otherwise indicated, most chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile was from Fisher Scientific Co. (Fairlawn, NJ). Oxazepam and oxazepam glucuronide were gifts from Wyeth Pharmaceuticals (Philadelphia, PA). The oxazepam was a racemic mixture of R- and S-enantiomers as is found in clinical preparations. The oxazepam glucuronide was also a mixture of the two enantiomers that contained 86%S-oxazepam glucuronide and 14% R-oxazepam glucuronide by HPLC analysis.
Recombinant UGTs.
Homogenates from baculovirus-insect cell expressed UGTs 1A1, 1A3, 1A4, 1A6, 1A9, 2B7, 2B15*1-BV and were obtained from BD Gentest (Woburn, MA), whereas UGTs 1A7 and 1A10 were from PanVera Corp. (Madison, WI). UGTs 2B4, 2B10, 2B17, and two UGT2B15 allelic variants (UGT2B15*1-HK and UGT2B15*2-HK) were obtained by stable expression in HEK-293 cells and membrane fractions prepared as previously described (Guillemette et al., 2000). Briefly, cells were harvested using trypsin-EDTA treatment to release the cell monolayer, collected in centrifuge tubes, centrifuged at 500g, and then resuspended in ice-cold phosphate-buffered saline. After repeating the wash step, the cell pellets were resuspended in 0.25 M sucrose, homogenized using a Potter-Elvehjem glass homogenizer, and centrifuged for 20 min at 5,000g to remove nuclei and other particulates. The membrane fractions were then collected by sedimentation at 100,000gfor 1 h, followed by resuspension of the membrane pellet in buffer containing 0.1 M potassium phosphate (pH 7.5), 1 mM EDTA, and 20% glycerol. Protein concentrations were determined by the bicinchoninic acid method (Smith et al., 1985) using 0.5% SDS as the assay diluent.
The glucuronidation activity of each of the expressed UGTs were substantiated by the respective source using the following substrates: bilirubin and estradiol (UGT1A1), 7-hydroxy-4-trifluoromethylcoumarin (UGTs 1A3, 1A8, 1A10, and 2B15*1-BV), trifluoperazine (UGT1A4), 1-napthol (UGTs 1A6 and 1A10), propofol (UGT1A9), morphine (UGT2B7), octyl-gallate (UGT1A7), and eugenol (UGTs 2B4, 2B10, 2B17, 2B15*1-HK, and 2B15*2-HK). In addition, immunoblotting conducted in this laboratory with an antibody specific for the conserved C-terminal region of all UGT1A isoforms (WB-UGT1A; BD Gentest), as well as an antibody that recognizes UGT1A and UGT2B isoforms (RAL; a gift from Dr. Brian Burchell, Dundee, Scotland), showed that the content of immunodetectable UGT protein was qualitatively similar for all of the expressed UGT1A isoforms and for UGT2B7 and UGT2B15*1-BV.
Relative UGT2B15 protein content of UGT2B15*1 and UGT2B15*2 in membrane fractions from HEK-293 cells was determined by semiquantitative immunoblotting using the anti-human UGT2B antibody (EL-93), as previously described (Guillemette et al., 2000). Briefly, membrane proteins were resolved by electrophoresis through SDS-10% polyacrylamide gels and electroblotted onto nitrocellulose membranes. After the blocking step, blots were incubated at room temperature in UGT2B antisera diluted 1:2,000 in Tris-buffered saline with 0.2% Tween 20 (TBS-T) containing 5% dry milk. Blots were washed and then incubated overnight in a solution containing the secondary antibody (horseradish peroxidase-conjugated rabbit antimouse IgG; Amersham Biosciences Inc., Piscataway, NJ) diluted 1:20,000 in TBS-T containing 5% blocking reagent. Blots were washed extensively with several changes of TBS-T prior to detection of horseradish peroxidase. Immunocomplexes were visualized using enhanced chemiluminescence (Amersham Biosciences Inc.), exposed on hyperfilm for 30 s (Eastman Kodak Co., Rochester, NY), and quantified by BioImage Visage 110s (Genomic Solution Inc., Ann Arbor, MI). Blots were also probed for a second endoplasmic reticulum resident protein (Calnexin) to ascertain equal loading efficiency as described previously (Guillemette et al., 2000).
Human Liver Microsomes.
Liver tissue from three donors were randomly selected from frozen banks maintained at the Division of Clinical Pharmacology, Department of Pharmacology and Experimental Therapeutics, Tufts University School of Medicine, Boston. Donors included a 36-year-old white male (LV12), a 74-year-old white male (LV21) and a 66-year-old white male (LV24). Tissues were from livers that had been donated for transplantation but had failed to match (LV12) or were from apparently normal tissue adjacent to tumors that were being surgically removed (LV21 and LV24). The donor of LV21 had a history of cigarette smoking, whereas the donor of LV12 had received morphine. Pooled liver microsomes were also obtained by combining equal amounts of protein from HLMs prepared from 55 different donors.
Microsomes were prepared by differential centrifugation as previously described (Court and Greenblatt, 1997b). The resultant pellet was reconstituted in 20% glycerol/phosphate buffer, aliquoted, and stored at −80°C. Protein concentrations were measured by the bicinchoninic acid assay method (Smith et al., 1985). Frozen microsomes were thawed once only immediately prior to use. The quality of the liver samples used was ascertained by reference to at least 10 other UGT and cytochrome P450 enzyme activities measured in this laboratory using the same set of liver samples. Livers with activities that were consistently less than 50% that of the median activity value for the entire liver set were excluded from the study.
Oxazepam Glucuronidation Assay.
An in vitro UGT activity assay using oxazepam as substrate was developed based on methods previously used in this laboratory (von Moltke et al., 1993; Court and Greenblatt, 1997a,b). Incubations (250 μl for HLMs, 100 μl for expressed UGTs) were performed at 37°C in 50 mM phosphate buffer (pH 7.5) with 5 mM MgCl2, 5 mM UDPGA, and (R,S)-oxazepam (10–1000 μM). Alamethicin was also included in incubations in an amount (50 μg of alamethicin/mg microsomal protein) determined in preliminary experiments using HLMs and UGT2B15 to result in maximal activation (100–200% increase in activity for HLMs; 50–100% increase for UGT2B15). Incubation time (up to 360 min) and protein concentration (up to 0.8 mg/ml) were also established in preliminary studies with both HLMs and expressed UGTs to be within initial linear rate conditions. Typically, protein concentration was 0.5 mg/ml, and incubation time was 180 min. Incubations were terminated by addition of acetonitrile (50% incubation volume), vortexed, and placed on ice. After addition of internal standard (1–2 μg of phenacetin), samples were centrifuged, and the supernatants were analyzed by HPLC.
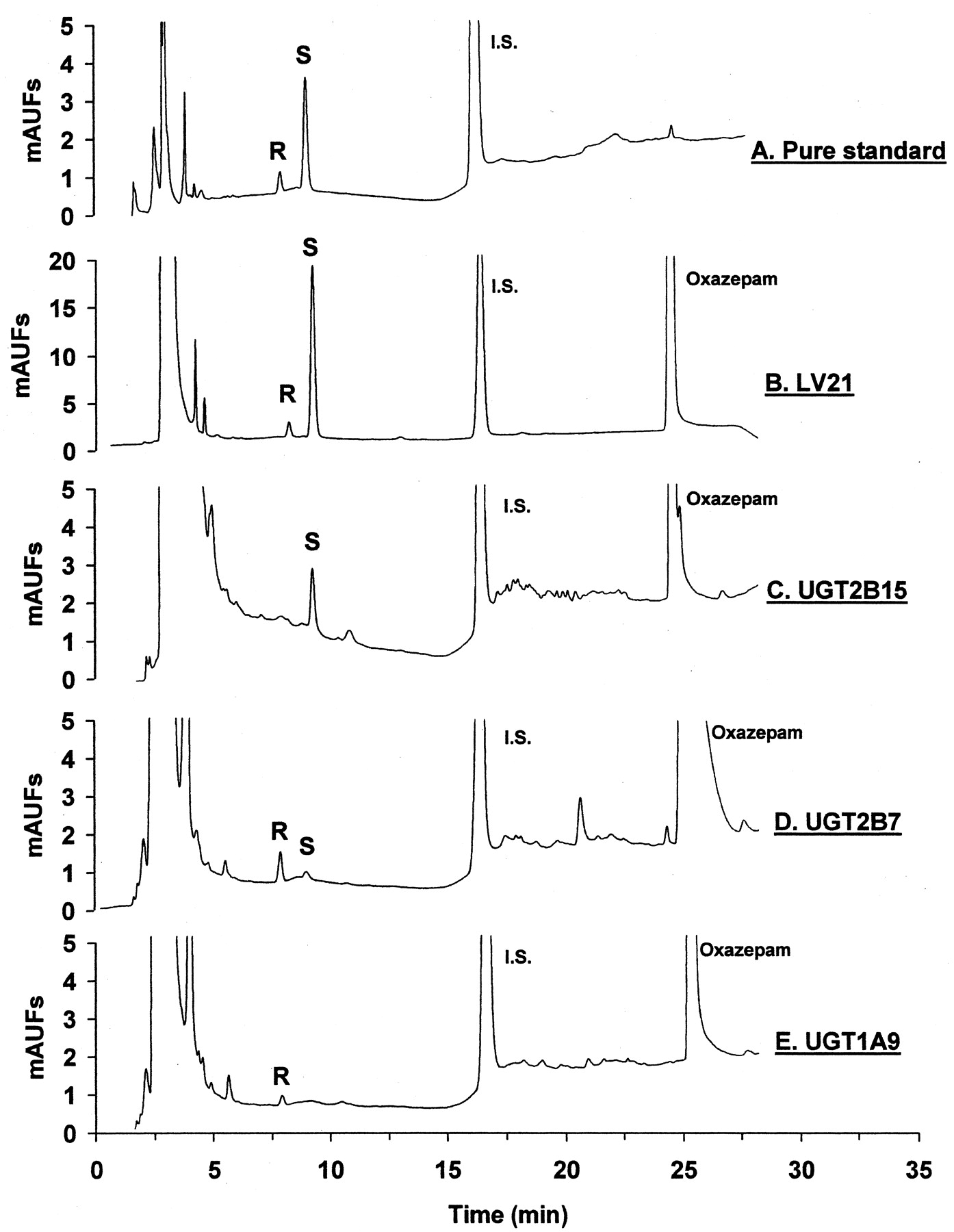
HPLC apparatus (model 1100; Agilent, Palo Alto, CA) consisted of an autoinjector, binary pump, column, and UV absorbance detector set at a wavelength of 214 nm. Chromatographic separation of the oxazepam glucuronide enantiomers was possible using a C18column (4.6 mm × 25 cm, 10 μm, Luna; Phenomenex, Torrance, CA). The mobile phase was 20 mM, pH 4.5, potassium phosphate buffer in water (solution A) and acetonitrile (solution B). The solvent program consisted of an initial isocratic mobile phase mix (25% solution B) for 15 min, followed by a linear gradient from 25 to 60% solution B over 10 min. Approximate analyte retention times were 8 min forR-oxazepam glucuronide, 9 min for S-oxazepam glucuronide, 17 min for phenacetin, and 25 min for oxazepam. Glucuronide enantiomer peaks were identified by relative retention time (R-eluting before S-oxazepam glucuronide) and relative peak size (S-approximately 5 times larger than theR-oxazepam glucuronide peak) in HLM incubates, as previously described (Fig. 1) (Patel et al., 1995a,b). Metabolite concentrations in the incubate were determined using a standard curve of peak area ratios (normalized to the internal standard) and generated by HPLC analysis of a series of known concentrations (0.1–50 ng/ml) of purified R- andS-oxazepam glucuronide dissolved in incubation buffer. Enzyme activities were then calculated by dividing metabolite concentration by protein concentration and incubation time and expressed as picomoles per minutes per milligram of protein. Intra- and interassay variability was less than 10 and 15%, respectively. The lower limit of quantitation for this assay was 0.1 pmol/min/mg of protein (signal to noise ratio of 4:1). All experiments were conducted in duplicate and results given as a mean value, except for comparisons of expressed UGT2B15*1 and UGT2B15*2, which were conducted in triplicate and results given as a mean ± standard deviation.

HPLC chromatograms of R-oxazepam glucuronide (R) and S-oxazepam glucuronide (S).
Panel A shows a typical chromatogram of the purified glucuronide standards used for quantitation. The remaining panels show chromatograms of injectates from incubations performed in the presence of UDPGA, MgCl2, oxazepam, and either HLMs from LV21 (panel B), UGT2B15*1-BV (panel C), UGT2B7 (panel D), and UGT1A9 (panel E). (R,S)-Oxazepam concentration was 100 μM except for UGT2B7 (panel D) in which it was 1000 μM (R,S)-oxazepam. Note the highery-axis scale on panel B. Column effluent was monitored with a UV absorbance detector set at 214 nm. Phenacetin was used as the internal standard. Other details are given underMaterials and Methods. mAUFs, arbitrary UV absorbance units.
Enzyme Kinetic Analysis.
For enzyme kinetic studies of oxazepam glucuronidation, substrate concentration (S) and velocity (V) data were fitted to the appropriate model (Venkatakrishnan et al., 2001) by nonlinear least-squares regression. The models were chosen initially based on the appearance of Michaelis-Menten and Eadie-Hofstee plots. These included the simple Michaelis-Menten model (eq. 1), the substrate activation model (eq. 2), and the uncompetitive substrate inhibition model (eq. 3): Equation 1
Equation 1
Other Glucuronidation Activities.
Activities were also measured with microsomal fractions prepared from UGT2B15 allelic variants expressed in HEK-293 cells and pooled human liver microsomes using eugenol, naringenin, 4-methylumbelliferone, dihydrotestosterone, and androstane-3α-diol as substrates. Reactions typically contained 50 mM Tris, pH 7.5; 6 mM MgCl2; 6 mM d-saccharo-1,4-lactone; 500 μM UDPGA containing 0.2 mCi 14C-UDPGA (PerkinElmer Life Sciences, Boston, MA); 0.4 to 0.6 mg/ml microsomal protein; and 200 μM aglycone concentration. Reaction mixtures were incubated with microsomes for 2 h at 37°C. Assays were terminated by addition of 100 μl of methanol. After centrifugation of samples, 100 μl was applied onto thin layer chromatography plates (0.25-mm-thick silica gel; Whatman, Maidstone, UK) and chromatographed in a mixture of toluene/methanol/acetic acid (7:3:1). Thin layer chromatography plates were exposed for 24 h, and the extent of glucuronidation was assessed by PhosphorImager (Molecular Dynamics, Sunnyvale, CA). The lower limit of quantitation for this assay was approximately 1 pmol/min/mg of protein. Assays were conducted in triplicate and results given as a mean ± standard deviation.
Results
Oxazepam Glucuronidation by Expressed UGTs.
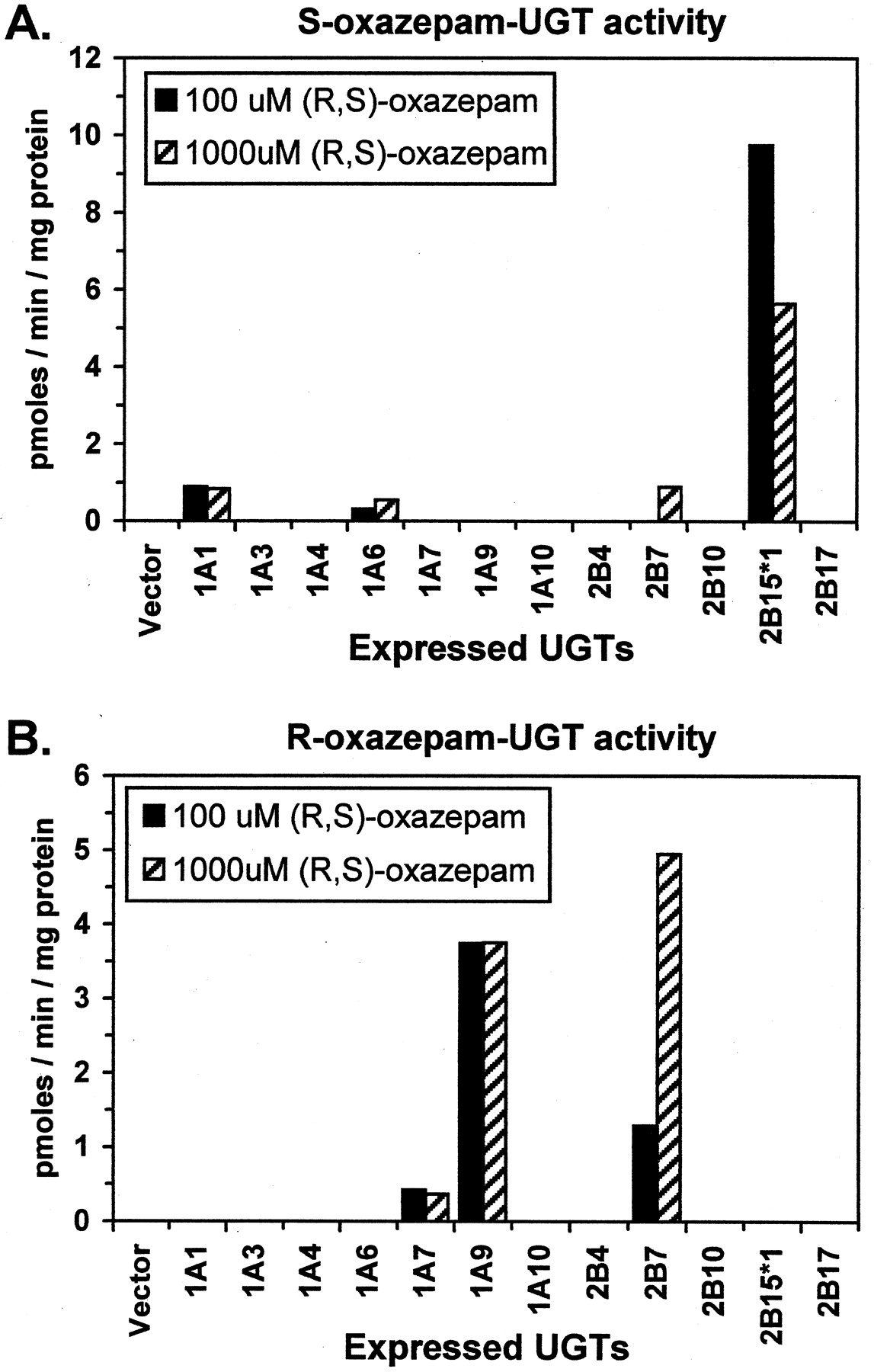
All available expressed UGTs were screened for oxazepam-UGT activity using 100 μM and 1 mM substrate concentration (Fig. 1). At 100 μM oxazepam concentration UGT2B15 (*1-BV) was the predominant isoform mediating S-oxazepam glucuronidation with more than 10 times greater activity than any other isoform (Fig.2A). Relatively low activities were observed for UGT1A1 and 1A6 but not for any other isoform. Although UGT2B15 mediated S-oxazepam glucuronidation activity appeared to be lower at 1000 μM compared with 100 μM oxazepam concentration, it was still over 6 times more active than any other isoform assayed. In addition to UGT1A1 and UGT1A6, UGT2B7 also showed a small amount of activity at this high concentration.

S-oxazepam (panel A) and R-oxazepam (panel B) glucuronidation activities measured at 100 and 1000 μM (R,S)-oxazepam concentration using UGT isoforms expressed in either stably transfected HEK-293 cells (UGTs 2B4, 2B10 and 2B17) or baculovirus-infected insect cells (all other isoforms and vector control); UGT2B15*1 was the D85 variant expressed in baculovirus-infected insect cells.
In contrast, both UGT1A9 and UGT2B7 were found to mediateR-oxazepam glucuronidation (Fig. 2B). UGT1A9 showed relatively high activity at both of the substrate concentrations examined, whereas UGT2B7 was primarily active at the higher substrate concentration. All of the remaining UGTs showed no activity except for UGT1A7, which had low although measurable activities at both substrate concentrations (Fig. 2B).
Enzyme Kinetics of Expressed UGTs Compared with HLMs.
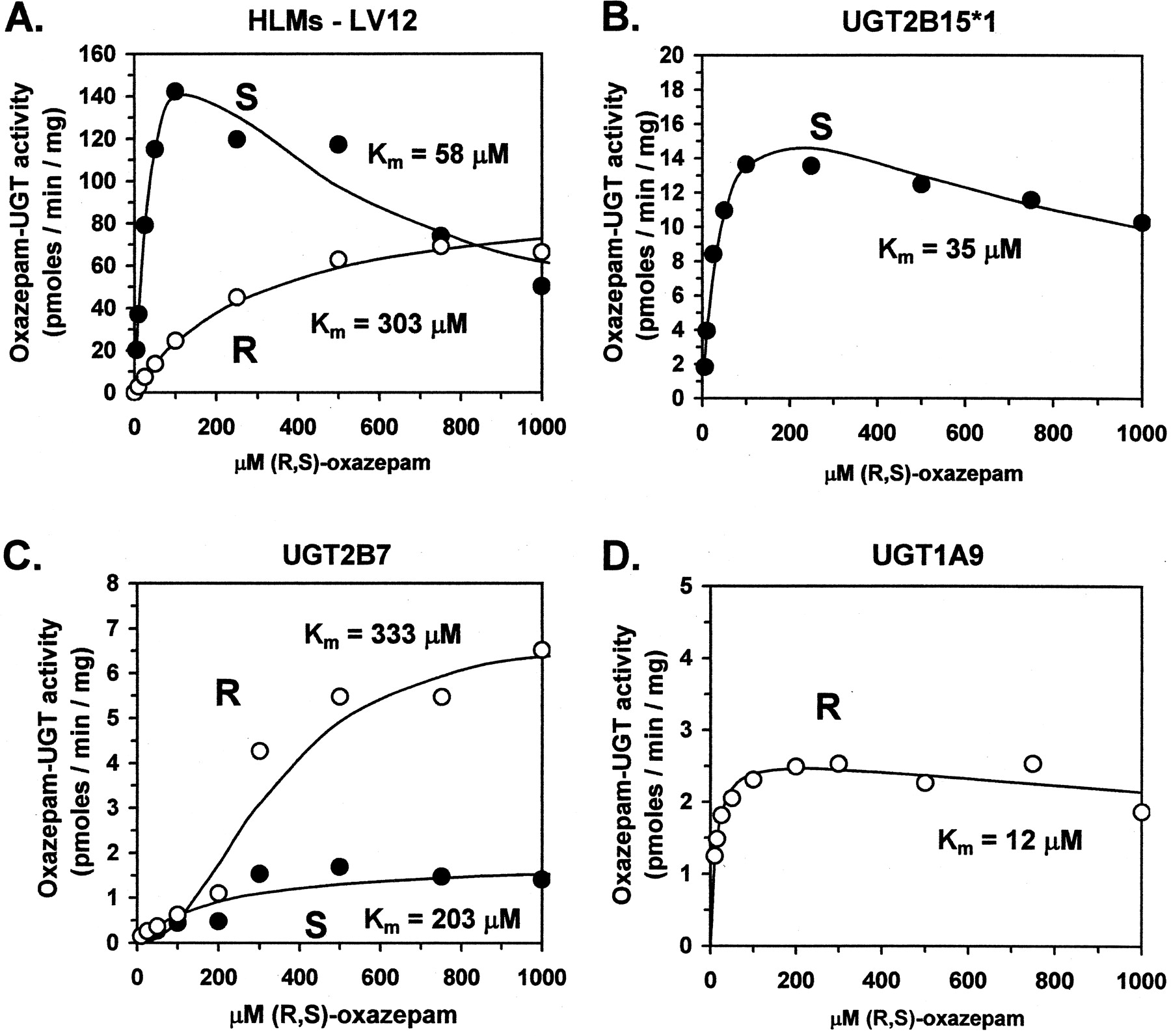
Enzyme kinetic studies were then performed using UGTs 2B15 (*1-BV), 1A9, and 2B7 as well as HLMs from three individuals. Kinetic data forS-oxazepam glucuronidation were best described by a single enzyme kinetic model with substrate inhibition for both HLMs and UGT2B15 (eq. 3; Fig. 3, A and B, respectively), whereas the simple Michaelis-Menten model best described UGT2B7 data (eq. 1; Fig. 3C). Similarly, apparentKm values for S-oxazepam glucuronidation by HLMs (43, 58, and 60 μM) were most similar to UGT2B15 (35 μM), rather than UGT2B7 (203 μM) (Table1). In addition, the calculated intrinsic clearance (Vmax/Km) was over 50 times higher for UGT2B15 (0.49 ml/min/kg) compared with UGT2B7 (0.0091 ml/min/kg) (Table 1).

Enyme kinetics of S-oxazepam (closed circles) and R-oxazepam (open circles) glucuronidation by representative HLMs (LV12; panel A), UGT2B15*1 (panel B), UGT2B7 (panel C), and UGT1A9 (panel D).
Shown are individual data points as well as curves representing best-fit estimates of these data determined by nonlinear curve fit to either eq. 1 (HLMs, R-oxazepam; UGT2B7,S-oxazepam), eq. 2 (UGT2B7, R-oxazepam), eq. 3 (HLMs, S-oxazepam; UGT2B15*1; UGT1A9). Also shown are the apparent Km estimates for each fit. UGT2B15*1 was the D85 variant expressed in baculovirus-infected insect cells. R-oxazepam-UGT activity was not detected for UGT2B15*1, whereasS-oxazepam-UGT activity was not detected for UGT1A9.
Enzyme kinetic parameters for oxazepam glucuronidation by HLMs and expressed UGTs
R-Oxazepam glucuronidation kinetic data were best described by the simple Michaelis-Menten model for liver microsomes and UGT1A9 (Fig. 3, A and D, respectively), while UGT2B7 data were fitted using the substrate activation kinetic model (eq. 2; Fig. 3C). ApparentKm values for R-oxazepam glucuronidation by HLMs (256, 273, and 303 μM) were most similar to UGT2B7 (333 μM) rather than UGT1A9 (12 μM) (Table 1). However, the intrinsic clearance was over 10 times higher for UGT1A9 (0.22 ml/min/kg) compared with UGT2B7 (0.021 ml/min/kg) (Table 1). NoR-oxazepam glucuronidation activity was observed for UGT2B15.
Comparative Activities of UGT2B15 Allelic Variants and HLMs.
Allelic variants of UGT2B15 (UGT2B15*1 and UGT2B15*2) were stably expressed in HEK-293 cells and microsomes prepared for comparison of activities using oxazepam, eugenol, naringenin, 4-methylumbelliferone, dihydrotestosterone, and androstane-3α-diol as substrates. Immunoblotting of these preparations showed an average 3.3 times higher immunoreactive UGT2B protein in the UGT2B15*1 microsomes compared with the UGT2B15*2 microsomes (Fig. 4, A and B). No R-oxazepam glucuronidation activity was detected in any of the UGT2B15 preparations assayed. Enzyme kinetic analysis ofS-oxazepam glucuronidation showed a similar apparentKm value for HEK-293-expressed UGT2B15*1 (29 μM; Fig. 4C and Table 1) compared with baculovirus-insect cell expressed UGT2B15*1 (35 μM; Fig. 3B and Table1). However Vmax values were about 3 times higher in baculovirus-insect cell expressed UGT2B15*1 compared with HEK-293-expressed UGT2B15*1 (17 versus 5 pmol/min/mg of protein, respectively; Table 1). S-Oxazepam glucuronidation activities measured with UGT2B15*2 over the same substrate concentrations (5–1000 μM) was only 3 to 8% (6 ± 2%) that of UGT2B15*1 (Fig. 4C). When corrected for immunoreactive UGT2B15 protein content, activities with UGT2B15*2 were still only 10 to 28% (20 ± 5%) that of UGT2B15*1. Unfortunately these low activities with UGT2B15*2 precluded determination of apparentKm values.

S-oxazepam glucuronidation by UGT2B15 allelic variants.
UGT2B15*1 and UGT2B15*2 microsomes were prepared from stably transfected HEK-293 cells. Immunoblotting with a UGT2B specific antibody (panel A, upper blot) showed 3.3 times higher immunoreactive protein in UGT2B15*1 microsomes compared with UGT2B15*2 microsomes (panel B). Similar loading efficiency was ascertained by probing with an antibody specific for another endoplasmic reticulum resident protein (Calnexin; panel A, lower blot). S-oxazepam glucuronidation activities were then measured using these microsomes over a range of (R,S)-oxazepam concentrations (5–1000 μM). In addition to individual data points, the enzyme kinetic curve derived by fitting UGT2B15*1 data to eq. 3 are shown (panel C). Activities for UGT2B15*2 at (R,S)-oxazepam concentrations less than 50 μM were too low to allow kinetic analysis.
Substantial differences between these allelic variant preparations in activities were also observed for other substrates assayed. After correction for relative immunoreactive UGT2B15 protein content, activities for UGT2B15*2 were from 8 ± 3% (for naringenin) to 29 ± 6% (for 4-methylumbelliferone) that of UGT2B15*1 (Table2). An exception was dihydrotestosterone activity in UGT2B15*2, which was 67 ± 13% that of UGT2B15*1. It should be acknowledged that naringenin, androstane-3α-diol, and dihydrotestosterone glucuronidation activities for UGT2B15*2 were so low that measured values were near to the lower limit of quantitation for the assay used. On the other hand, eugenol and 4-methylumbelliferone glucuronidation activities for UGT2B15*2 were at least 10 times higher, whereas oxazepam glucuronidation activities were 2 to 3 times higher, than the lower limit of quantitation for the respective assays.
Comparative glucuronidation activities for UGT2B15*1 and UGT2B15*2
Activities for all substrates were also measured using pooled HLMs and compared with activities for UGT2B15*1 (Table 2). Differences between expressed UGT2B15*1 and pooled HLMs were smallest forS-oxazepam-UGT activities in that UGT2B15*1 averaged 10% of the S-oxazepam-UGT activity of pooled HLMs. In contrast, UGT2B15*1 activities ranged from 3 to 5% that of pooled HLMs activities for the other five UGT substrates evaluated.
Discussion
To our knowledge, this is the first study to show that UGT2B15 stereoselectively glucuronidates S-oxazepam and is most likely the principal human UGT isoform mediating this activity. Evidence in support of these conclusions include the markedly higherS-oxazepam glucuronidation activity displayed by expressed UGT2B15 compared with all other isoforms studied, lack of detectableR-oxazepam glucuronidation activity by UGT2B15, and the remarkable similarities between expressed UGT2B15 and HLMs in enzyme kinetic properties. In particular, decreasing activity at oxazepam concentrations over 100 μM, consistent with substrate inhibition, was consistently observed for both HLMs and UGT2B15 but not for any other UGT isoform evaluated.
Although a previous study (Patel et al., 1995b) indicated that UGT2B7 could be the major enzyme responsible for S-oxazepam glucuronidation in HLMs, this is unlikely since activities and intrinsic clearance values for expressed UGT2B7 were very low, and apparent Km values were relatively high compared with UGT2B15. Furthermore, we found that UGT2B7 preferentially glucuronidated R-oxazepam overS-oxazepam, with more than 5 times higher activities for theR-stereoisomer, which is not consistent with the pattern of glucuronidation observed in HLMs (S-glucuronidation ≫R-glucuronidation). This conclusion is also supported by results from a previous study, which showed that UGT2B7 poorly glucuronidated both of the oxazepam stereoisomers (Coffman et al., 1998).
R-Oxazepam glucuronidation was catalyzed by both UGT1A9 and UGT2B7. Apparent Km values for HLMs (256–303 μM) were most similar to UGT2B7 (333 μM) rather than UGT1A9 (12 μM). Although both isoforms may contribute toR-oxazepam glucuronidation in HLMs, the substantial differences in apparent Km values indicate that UGT1A9 would be most active at relatively low substrate concentrations (<100 μM), whereas UGT2B7 would contribute primarily at relatively high concentrations (<500 μM). This was also reflected by intrinsic clearance values for R-oxazepam glucuronidation, which were over 10 times higher for UGT1A9 compared with UGT2B7. Interestingly, oxazepam has been reported to inhibit 3′-azido-3′-deoxythimidine glucuronidation by HLMs (Rajaonarison et al., 1991), a conjugation reaction that is thought to be primarily mediated by UGT2B7 (Barbier et al., 2000). However, this only occurs at relatively high oxazepam concentrations (>500 μM) consistent with a low affinity interaction.
Plasma concentrations following the recommended dose of oxazepam (15–30 mg) to human subjects range from 1 to 5 μM (300–1500 ng/ml) (Greenblatt, 1981). Although plasma concentrations do not necessarily predict enzyme substrate concentrations, such low oxazepam concentrations relative to apparent Kmvalues would tend to favor S-oxazepam glucuronidation by UGT2B15 and R-oxazepam glucuronidation by UGT1A9, and minimize involvement of UGT2B7 for either activity. This conclusion is supported by a drug-drug interaction study in human immunodeficiency virus infected patients, which showed no pharmacokinetic interaction between coadministered oxazepam and 3′-azido-3′-deoxythimidine (Mole et al., 1993).
Substantial involvement of UGT2B15 in the glucuronidation of oxazepam may provide a satisfactory mechanistic explanation for a pharmacokinetic phenomenon first described almost 20 years ago (Abernethy et al., 1983). Then it was observed that oxazepam clearance values were over 3 times higher in obese individuals compared with nonobese controls. The authors of that study (Abernethy et al., 1983) speculated that there might be enhanced extrahepatic glucuronidation of oxazepam perhaps resulting from the increase in adipose tissue mass in obese individuals. The recent finding (Tchernof et al., 1999) that UGT2B15 mRNA (but not UGT2B17mRNA) is highly expressed in adipose tissue supports this hypothesis, although further work is needed to verify that the appropriate enzyme activities are also present in this tissue.
S-Oxazepam glucuronidation activities for expressed UGT2B15 were from 5 to 20 times lower than activities for HLMs. Although all UGTs known to be expressed in human liver were screened for activity (except for UGT2B11 and UGT2B28, which were not available at the time of study), it is possible that there is another UGT isoform present in liver with a higher turnover rate for oxazepam than UGT2B15. However, it is more likely that this difference simply reflects a difference in relative abundance of enzyme between the two preparations. In support of this, we found greater differences (20 to 33 times lower activity) when comparing expressed UGT2B15*1 and pooled HLMs for each of the five other activities measured.
UGT2B15 shares amino acid sequence homology (>92% identity), tissue distribution, and substrates with UGT2B17 (Hum et al., 1999). Consequently it was surprising to find no detectable oxazepam glucuronidation activity with recombinant UGT2B17, despite evidence of activity with this preparation for other substrates. This contrasts with the other substrates identified to date for UGT2B15 (such as dihydrotestosterone and androstane-3α-diol), which are also glucuronidated by UGT2B17 (Turgeon et al., 2001). Although this finding needs to be confirmed, it indicates that S-oxazepam may have substantial utility as a selective substrate probe for UGT2B15 in both in vitro and human studies.
As discussed previously (Patel et al., 1995a,b), although racemic oxazepam is the only preparation currently available, there are limitations to the use of a racemic drug for these investigations. In particular, interpretation of our results could be confounded by either stereoselective hydrolysis of the glucuronides or inhibitory interactions. For instance, high S-oxazepam glucuronidation activity relative to R-oxazepam glucuronidation activity in HLMs could be explained by an inhibitory effect ofS-oxazepam on R-oxazepam glucuronidation by the same UGT isoform, rather than there being different UGT isoforms that preferentially conjugate one stereoisomer over the other. Although this could be resolved (theoretically) by the use of purified stereoisomers, both R- and S-oxazepam (and other 3-hydroxylated benzodiazepines) have been shown to spontaneously racemize with a half-life of less than 4 min in aqueous solution at 37°C (Yang and Lu, 1991).
The UGT2B15 gene and associated regulatory region appears to be the relevant candidate gene for investigating the molecular genetic basis of polymorphic S-oxazepam glucuronidation previously identified in human studies (Patel et al., 1995a). A single nucleotide polymorphism has been identified in the amino acid coding portion of the UGT2B15 gene, which results in either an aspartate (UGT2B15*1) or tyrosine (UGT2B15*2) at position 85 of the protein (Levesque et al., 1997). This polymorphism appears to be highly prevalent with frequencies of the UGT2B15*1 allele ranging from 0.45 in white Americans to 0.63 in Hispanic Americans (Lampe et al., 2000; Riedy et al., 2000). Furthermore, this coding difference was shown to have functional consequence, in that 2-fold higher rates of glucuronidation of dihydrotestosterone and androstane-3α-diol were measured in intact HEK-293 cells expressing UGT2B15*2 compared with UGT2B15*1 (Levesque et al., 1997). Using microsomal fractions from the same expression system, we also found an effect on S-oxazepam glucuronidation activities, however, the difference between allelic variants was substantially larger (about 5-fold), and activities were much higher with UGT2B15*1 compared with UGT2B15*2 when corrected for UGT2B15 protein content. Similarly, high activities for UGT2B15*1 compared with UGT2B15*2 were also found for most other substrates assayed, including androstane-3α-diol, naringenin, eugenol, and 4-methylumbelliferone. Although we did see substantially higher absolute dihydrotestosterone glucuronidation activities in UGT2B15*1 compared with UGT2B15*2, correction for UGT2B15 protein content essentially eliminated the difference, indicating that the disparity could be substrate dependent.
Similar results (UGT2B15*1 showing greater activity than UGT2B15*2) have been obtained independently with HEK-293 cell homogenates (E. Levesque, unpublished data). Consequently, the discrepancy in results between the current study and the previous report (Levesque et al., 1997) appears to relate to whether intact cells (with substrate added to the cell media) or disrupted cell preparations (homogenates or microsomes) are used for the glucuronidation assay. One possible explanation is that UGT2B15*2 is less stable than UGT2B15*1 under the conditions used to prepare the microsomes from the HEK-293 cells. In the present study, we found no evidence for stability differences between the variants (after microsome preparation) as indicated by a linear increase in glucuronide formation with increasing incubation time. Protein stability studies using intact cells indicate that there are substantial differences between some UGT2B isoforms, although the exact mechanism for this has not been elucidated (Turgeon et al., 2001). Interestingly, in both this study and in the previous report (Levesque et al., 1997), 3- to 4-fold more UGT2B15 protein was present in cells expressing the UGT2B15*1 variant compared with the UGT2B15*2 variant, suggesting that the single amino acid difference could affect protein stability, although it could simply reflect differences in expression plasmid copy number. Further studies using genotyped human liver microsomes and human subjects will be needed to substantiate the functional (and clinical) relevance of this genetic polymorphism.
In conclusion, we have shown that oxazepam is glucuronidated by several UGTs normally expressed in human liver. Whereas R-oxazepam is glucuronidated by multiple UGTs (UGT1A9 and 2B7),S-oxazepam is preferentially and selectively glucuronidated by UGT2B15. Consequently, S-oxazepam may be a specific substrate for UGT2B15 and have utility for pharmacogenetic studies of UGT2B15 in human subjects and tissues. Furthermore, allelic variation associated with the UGT2B15 gene is likely to explain polymorphicS-oxazepam glucuronidation in humans. Preliminary studies indicate that the UGT2B15*2 polymorphism in the coding region of the UGT2B15 gene could be responsible, although further study is needed to confirm this finding.
Footnotes
-
This work was supported by Grants GM-61834, DA-05258, MH-58435, DA-13209, DK-58496, DA-13834, AG-17880, and RR-00054 from the National Institutes of Health (Bethesda, MD).
- Abbreviations used are::
- HPLC
- high pressure liquid chromatography
- HLM
- human liver microsomes
- UGT
- UDP-glucuronosyltransferase
- HEK
- human embryonic kidney
- TBS-T
- Tris-buffered saline with 0.2% Tween 20
- UDPGA
- UDP-glucuronic acid
- Received June 4, 2002.
- Accepted August 14, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}