


Abstract
The efficiency and interindividual variability in bioactivation of prasugrel and clopidogrel were quantitatively compared and the mechanisms involved were elucidated using 20 individual human liver microsomes. Prasugrel and clopidogrel are converted to their thiol-containing active metabolites through corresponding thiolactone metabolites. The formation rate of clopidogrel active metabolite was much lower and more variable [0.164 ± 0.196 μl/min/mg protein, coefficient of variation (CV) = 120%] compared with the formation of prasugrel active metabolite (8.68 ± 6.64 μl/min/mg protein, CV = 76%). This result was most likely attributable to the less efficient and less consistent formation of clopidogrel thiolactone metabolite (2.24 ± 1.00 μl/min/mg protein, CV = 45%) compared with the formation of prasugrel thiolactone metabolite (55.2 ± 15.4 μl/min/mg protein, CV = 28%). These differences may be attributed to the following factors. Clopidogrel was largely hydrolyzed to an inactive acid metabolite (approximately 90% of total metabolites analyzed), and the clopidogrel concentrations consumed were correlated to human carboxylesterase 1 activity in each source of liver microsomes. In addition, 48% of the clopidogrel thiolactone metabolite formed was converted to an inactive thiolactone acid metabolite. The oxidation of clopidogrel to its thiolactone metabolite correlated with variable activities of CYP1A2, CYP2B6, and CYP2C19. In conclusion, the active metabolite of clopidogrel was formed with less efficiency and higher variability than that of prasugrel. This difference in thiolactone formation was attributed to hydrolysis of clopidogrel and its thiolactone metabolite to inactive acid metabolites and to variability in cytochrome P450-mediated oxidation of clopidogrel to its thiolactone metabolite, which may contribute to the poorer and more variable active metabolite formation for clopidogrel than prasugrel.
- prasugrel, 2-acetoxy-5-(α-cyclopropylcarbonyl-2-fluorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine
- P450, cytochrome P450
- hCE, human carboxylesterase
- R-95913, 2-[2-oxo-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl]-1-cyclopropyl-2-(2-fluorophenyl)ethanone
- R-138727, 2-[1-[2-cyclopropyl-1-(2-fluorophenyl)-2-oxoethyl]-4-mercapto-3-piperidinylidene]acetic acid
- CV, coefficient of variation
- HLM, human liver microsomes.
Prasugrel and clopidogrel are thienopyridine antiplatelet agents. Prasugrel is shown to reduce the rate of thrombotic cardiovascular events and stent thrombosis in patients with acute coronary syndrome and those to be managed with percutaneous coronary intervention (Wiviott et al., 2007). Likewise, clopidogrel is used for the management of patients after percutaneous coronary intervention and stent placement (Braunwald et al., 2002; Schulman, 2004). Thienopyridines are prodrugs that are converted in vivo to pharmacologically active metabolites through corresponding thiolactone intermediates. The thiol-containing active metabolites inhibit platelet function by irreversibly binding to the platelet P2Y12 ADP receptor (Niitsu et al., 2005; Savi and Herbert, 2005; Algaier et al., 2008).
In animal models and aspirin-treated patients with stable coronary artery disease, prasugrel, a new thienopyridine P2Y12 receptor antagonist, showed more potent antiplatelet activity with more rapid onset and significantly reduced the cardiovascular events compared with clopidogrel (Sugidachi et al., 2000; Niitsu et al., 2005; Jernberg et al., 2006; Wiviott et al., 2007). This potent effect of prasugrel can be explained by higher exposure to the active metabolite of prasugrel than to the active metabolite of clopidogrel (Brandt et al., 2007; Payne et al., 2007; Sugidachi et al., 2007), leading to a better pharmacodynamic response (Ernest et al., 2008; Farid et al., 2009).
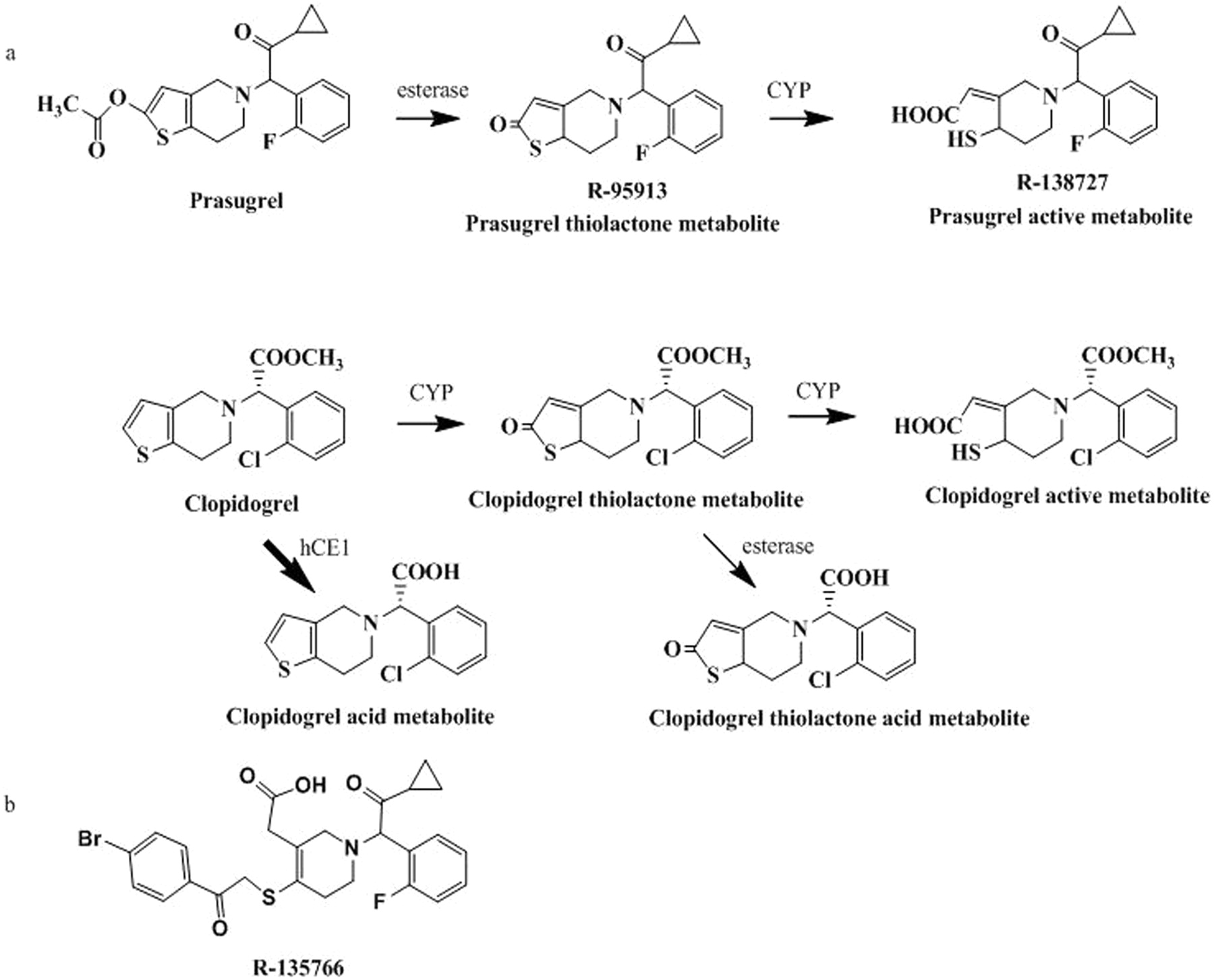
The pharmacologically active metabolites of thienopyridines have been shown to be produced through cytochrome P450 (P450)-mediated oxidation of their corresponding thiolactones as shown in Fig. 1 (Kurihara et al., 2005; Rehmel et al., 2006). Clopidogrel has two competing metabolic pathways (Fig. 1), with the primary pathway leading to the formation of the inactive clopidogrel acid metabolite (Caplain et al., 1999). The clopidogrel inactive acid metabolite is formed through ester hydrolysis by hepatic human carboxylesterase (hCE) 1 (Tang et al., 2006). The minor pathway in clopidogrel metabolism that yields the active metabolite requires two sequential P450-dependent steps. The first is the formation of a thiolactone from clopidogrel by CYP1A2, CYP2B6, and CYP2C19 and the second is the subsequent formation of the active metabolite from the thiolactone by CYP2B6, CYP2C9, CYP2C19, and CYP3A4 (Kurihara et al., 2005). On the other hand, the bioactivation of prasugrel to its active metabolite requires the hydrolysis of the ester by carboxylesterases, which occurs during the absorption process, to form the thiolactone followed by oxidation by CYP2B6, CYP2C9, CYP2C19, and CYP3A4 (Rehmel et al., 2006; Farid et al., 2007b; Williams et al., 2008) (Fig. 1) to form the active metabolite of prasugrel. The hydrolysis step of prasugrel mediated by hCE1 and hCE2 is very rapid, such that prasugrel is not detected in human or animal plasma after oral administration even at early time points (Farid et al., 2007b; Smith et al., 2007; Williams et al., 2008; Hagihara et al., 2009).
This study was performed to directly compare the efficiency and variability in the formation of the active metabolites from prasugrel and clopidogrel and to elucidate the detailed mechanisms of the differences using individual human liver microsomes.
Materials and Methods
Materials.
Prasugrel (Effient in the United States and Efient in the European Union), prasugrel thiolactone metabolite (R-95913), prasugrel active metabolite (R-138727), clopidogrel (Plavix, Iscover), clopidogrel acid, and the internal standard R-135766, shown in Fig. 1b, were synthesized by Ube Industries, Ltd. (Ube, Japan). Clopidogrel thiolactone metabolite, clopidogrel thiolactone acid, and clopidogrel active metabolite were synthesized by Daiichi Sankyo Co., Ltd (Tokyo, Japan). The purity of these compounds ranged from 89 to 100%. Individual human liver microsomes were purchased from the Human and Animal Bridging Research Organization (Tokyo, Japan), where each activity of P450 or hCE1 was determined. A derivatizing reagent, 3′-methoxyphenacyl bromide was obtained from Tokyo Kasei Kogyo Co., Ltd. (Tokyo, Japan).

Metabolic pathways of prasugrel and clopidogrel studied (a) and chemical structure of the internal standard R-135766 (b).
Metabolism of Prasugrel, Clopidogrel Thiolactone Metabolite, and Clopidogrel in Human Liver Microsomes.
Each mixture (total volume 99 μl) in duplicate contained potassium phosphate buffer (29.5 mM, pH 7.4), 2.5 mM β-NADP, 25 mM glucose 6-phosphate, 0.5 unit/ml glucose-6-phosphate dehydrogenase, 10 mM MgCl2, 5 mM glutathione, and human liver microsome (1 mg of protein/ml). Each mixture was preincubated at 37°C for 5 min, and 1 μl of prasugrel, clopidogrel thiolactone metabolite, or clopidogrel (1, 1, or 50 μM, respectively, as final concentrations) was added to the respective mixture, which was incubated at 37°C for 15 min for prasugrel and clopidogrel thiolactone metabolite or 30 min for clopidogrel. At each time point, 100 μl of the incubation mixture was mixed with 200 μl of acetonitrile, 2 μl of 3′-methoxyphenacyl bromide acetonitrile solution (500 mM), and 100 μl of R-135766 acetonitrile solution (100 ng/ml) to stop the reaction and derivatize the thiol moiety of prasugrel active metabolite or clopidogrel active metabolite. After storage at room temperature for 10 min, the mixture was centrifuged (15,000g, 3 min, 4°C). A 10-μl aliquot of the supernatant fraction was subjected to liquid chromatography-tandem mass spectrometry analysis.
Assay Procedure for Prasugrel and Clopidogrel Metabolites.
The assays were performed following the methods reported previously (Takahashi et al., 2006; Farid et al., 2007a). The analytes were separated by high-performance liquid chromatography using an Alliance2690 Separations Module (Waters, Milford, MA). Mass spectra were obtained using a Quattro liquid chromatography-tandem mass spectrometry system (Micromass Ltd., Milford, MA) in the positive ion detection mode using an electrospray ionization interface. The transitions m/z 498 → 348 for derivatized prasugrel active metabolite, m/z 548 → 206 for R-135766, m/z 374 → 206 for prasugrel, m/z 332 → 149 for prasugrel thiolactone metabolite, m/z 504 → 354 for derivatized clopidogrel active metabolite, m/z 322 → 212 for clopidogrel, m/z 338 → 183 for clopidogrel thiolactone metabolite, m/z 308 → 198 for clopidogrel acid metabolite, and m/z 324 → 278 for clopidogrel thiolactone acid metabolite were monitored using the multiple reaction monitoring mode. The lower limit of quantification for each compound was 1.6 nM. Data acquisition and analysis were performed using MassLynx software (version 4.0; Micromass Ltd.).
Multiple Linear Regression Analysis.
Multiple linear regression analyses for each enzyme activity were performed using Microsoft Office Excel 2003 (SP2; Microsoft, Richmond, WA) and JMP 6.0.3 (SAS Institute Japan Co., Ltd., Tokyo, Japan). Each estimate and its 95% confidence interval of the intercept (a) and slope (bi) were calculated by the least-squares method. Y = a + bi × Xi, where Y is an estimate of the metabolite concentration, bi is the partial regression coefficient for each enzyme, and Xi is each enzyme activity. P ≤ 0.05 obtained from a t test in which each slope was assumed to be zero was considered statistically significant.
Calculation of Metabolite Formation Ratio and Formation Rate Divided by Substrate Concentration (V/S).
The metabolite formation ratio was calculated by dividing concentrations of the metabolites by those of the total metabolites analyzed and is expressed as a percentage. The formation rate (V) was calculated by dividing the metabolite concentration by the incubation time and protein concentrations and is expressed in units of picomoles per minute per milligram of protein. The formation rate divided by the substrate concentration (V/S) is expressed in units of microliters per minute per milligram of protein. These values are expressed as the mean ± S.D. throughout Results.
Statistical Analyses.
Variation and magnitude of the V/S values for the formation of thiolactones and active metabolites from prasugrel and clopidogrel were compared by F test statistic and Welch test statistic using SAS system release 8.2 software. Level of significance was set at 0.05.
Results
P450s and hCE1 Activities in Individual Human Liver Microsomes.
Variable activities of cytochrome P450 isoforms and hCE1 in individual human liver microsomes (n = 20) used in this experiment are shown in Table 1. The CV values of the enzyme activities ranged from 36 to 148% (Table 1).
Activities of P450 isoforms and hCE1 in individual human liver microsomes
The P450 activities were determined by measuring the following activities: CYP1A2, phenacetin deethylation; CYP2B6, 7-benzyloxyresorufin debenzylation; CYP2C9, tolbutamide methylhydroxylation; CYP2C19, S-mephenytoin 4′-hydroxylation; CYP2D6, debrisoquine 4-hydroxylation; CYP2E1, chlorzoxazone 6-hydroxylation; CYP3A4, diazepam 3-hydroxylation. The hCE1 activity was determined by analyzing hydrolysis of temocapril. n = 20.
Comparison of V/S for the Formation of Active Metabolites from Prasugrel and Clopidogrel.
The efficiency of the formation of the active metabolites between prasugrel and clopidogrel was compared (Fig. 2). Prasugrel active metabolite was formed with lower individual variability for most of the enzyme activities. The V/S values for prasugrel active metabolite formation (8.68 ± 6.64 μl/min/mg protein) were significantly higher than those for the formation of clopidogrel active metabolite (0.164 ± 0.196 μl/min/mg protein) (P < 0.0001, Welch test). The mean V/S for the formation of prasugrel active metabolite was approximately 53-fold higher than that for the formation of clopidogrel active metabolite. Variability in the formation of prasugrel active metabolite (CV = 76%) was similar to those (CV = 86%) observed in previous experiment (Rehmel et al., 2006) and was significantly less than that for the formation of the active metabolite of clopidogrel (CV = 120%) (P < 0.0001, F test). The formation of both prasugrel active metabolite and clopidogrel active metabolite was not detected in one human liver microsomal sample (HLM-009-1), and this was considered to be due to the lower P450 activities in this sample than those in the other samples.

Comparison of V/S for the formation of the active metabolites from prasugrel (1 μM) and clopidogrel (50 μM), respectively, in individual human liver microsomes (n = 20). Prasugrel active metabolite and clopidogrel active metabolite were not detected in HLM-009-1. V/S, formation rates divided by substrate concentrations. P < 0.0001, prasugrel active metabolite versus clopidogrel active metabolite, F test and Welch test.
Comparison of V/S for the Formation of Thiolactone Intermediates from Prasugrel and Clopidogrel.
The efficiencies of the formation of the thiolactone intermediates of prasugrel and clopidogrel were compared (Fig. 3). Because the thiolactone intermediates are subsequently metabolized to the active metabolites (for prasugrel and clopidogrel), the sum of the thiolactone and the active and inactive thiolactone acid (for clopidogrel only) metabolites were used to calculate V/S of prasugrel thiolactone metabolite and clopidogrel thiolactone metabolite. The V/S for the formation of prasugrel thiolactone metabolite (55.2 ± 15.4 μl/min/mg protein) was significantly higher than that for the formation of clopidogrel thiolactone metabolite (2.24 ± 1.00 μl/min/mg protein) (P < 0.0001, Welch test). The mean V/S for the formation of prasugrel thiolactone metabolite was approximately 25-fold higher than that of clopidogrel thiolactone metabolite. Variation was significantly higher in the formation of clopidogrel thiolactone metabolite (CV = 45%) compared with that of prasugrel thiolactone metabolite (CV = 28%) (P < 0.0001, F test).

Comparison of V/S for the formation of the thiolactone metabolites from prasugrel (1 μM) and clopidogrel (50 μM), respectively, in individual human liver microsomes (n = 20). V/S, formation rates divided by substrate concentrations. P < 0.0001, prasugrel thiolactone metabolite versus clopidogrel thiolactone metabolite, F test and Welch test.
Metabolism of Clopidogrel in Individual Human Liver Microsomes.
The V values of clopidogrel acid, clopidogrel thiolactone metabolite, clopidogrel thiolactone acid, and clopidogrel active metabolite generated from clopidogrel are shown in Fig. 4. Clopidogrel was largely hydrolyzed to the carboxylic acid metabolite (metabolite formation ratio of 92.19 ± 3.81%). The substrate concentrations consumed after incubation of clopidogrel in human liver microsomes were best correlated with hCE1 activity (R2 = 0.4028) (Fig. 5). The results show that high hCE1 activity possibly contributes to lower efficiency of the formation of clopidogrel thiolactone metabolite and subsequent active metabolite because this activity would divert the compound to an alternate inactive pathway.

Formation of clopidogrel acid, clopidogrel thiolactone metabolite, clopidogrel thiolactone acid, and clopidogrel active metabolite from clopidogrel (50 μM) in individual human liver microsomes (n = 20). The data are expressed as the mean of 20 results ± S.D.

Correlation between each enzyme activity and the clopidogrel concentrations consumed in individual human liver microsomes (n = 20).
Metabolite Formation from Clopidogrel Thiolactone Metabolite in Individual Human Liver Microsomes.
The active metabolite formation from clopidogrel thiolactone metabolite was evaluated because clopidogrel undergoes two sequential steps of P450 oxidation to form the active metabolite via a thiolactone intermediate. The results demonstrated that approximately 48% of the clopidogrel thiolactone metabolite was also hydrolyzed to an inactive thiolactone acid as shown in Table 2.
Formation ratio of clopidogrel active metabolite and clopidogrel thiolactone acid from clopidogrel thiolactone (1 μM) in individual human liver microsomes (n = 20)
Correlation between P450 Activities and Concentrations of Clopidogrel Thiolactone Metabolite Formed.
The correlation between the activities of P450 isoforms (Kurihara et al., 2005), and clopidogrel thiolactone metabolite formation rates was investigated. The coefficients of correlation of thiolactone metabolite formation from clopidogrel, calculated as the sum of thiolactone, thiolactone acid, and active metabolite, with the activities of CYP1A2, CYP2B6, and CYP2C19 were 0.4058, 0.3380, and 0.1300, respectively (Fig. 6).
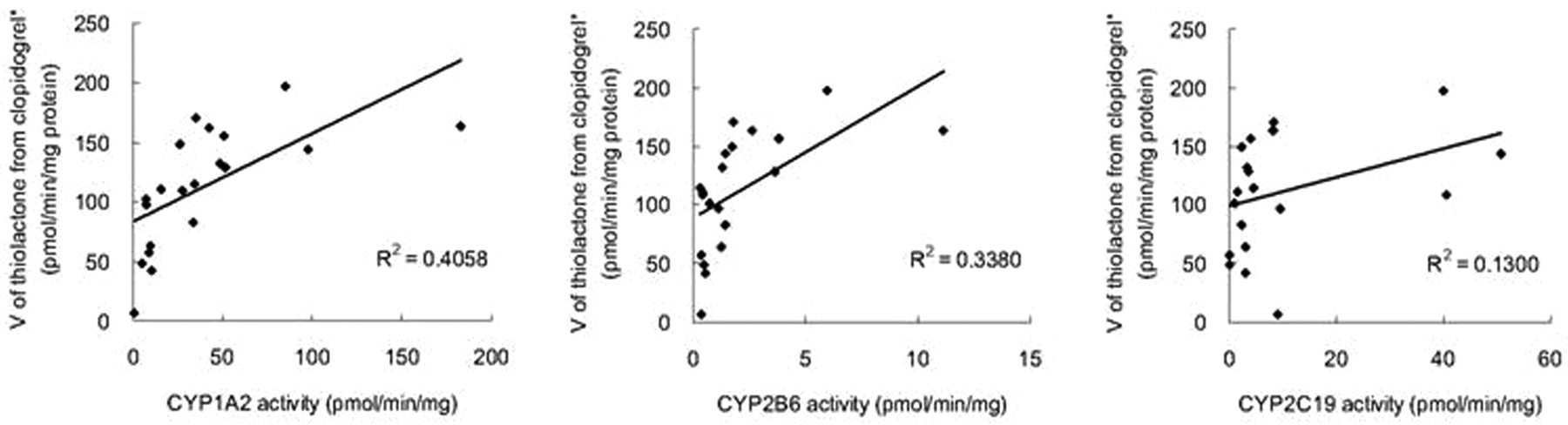
Correlation between the activities of CYP1A2, CYP2B6, and CYP2C19 and thiolactone metabolite formation rates from clopidogrel in individual human liver microsomes (n = 20). *, sum of V for the formation of thiolactone, thiolactone acid, and active metabolite.
Multiple Linear Regression Analyses for Clopidogrel Thiolactone Metabolite Formation.
To confirm that clopidogrel thiolactone metabolite formation is mediated by three P450 isoforms, CYP1A2, CYP2B6, and CYP2C19, in 20 individual liver microsomes used in this experiment, we evaluated whether the concentration of clopidogrel thiolactone metabolite formed in each liver microsome can be estimated by their respective three P450 isoform activities. With the values of the observed concentration of clopidogrel thiolactone metabolite (sum of clopidogrel thiolactone, clopidogrel thiolactone acid, and clopidogrel active metabolite) and P450 activities (CYP1A2, CYP2B6, and CYP2C19) in each liver microsome sample, coefficients for each P450 isoform activity and an intercept best for estimating the concentration of clopidogrel thiolactone metabolite were calculated by multiple linear regression analyses. The multiple regression equation and the determination coefficient r2 were as follows: Y (estimate of clopidogrel thiolactone metabolite concentration) = 2117.91 + 15.907 × CYP1A2 activity + 243.369 × CYP2B6 activity + 12.397 × CYP2C19 activity, r2 = 0.5003. By using the equation above, the observed clopidogrel thiolactone metabolite concentrations were well correlated with the estimated concentrations (coefficient of correlation by single linear regression between observed and estimated concentrations, R2 = 0.5792) (Fig. 7), demonstrating that CYP1A2, CYP2B6, and CYP2C19 contributed to clopidogrel thiolactone metabolite formation from clopidogrel in each human liver microsome used in this experimental system.

Correlation between estimated and observed clopidogrel thiolactone metabolite formation in individual human liver microsomes (n = 20). P450-mediated metabolites are the sum of thiolactone, thiolactone acid, and active metabolite. The estimated metabolite formation was calculated by the following multiple regression equation: 2117.91 + 15.907 × CYP1A2 activity + 243.369 × CYP2B6 activity + 12.397 × CYP2C19 activity. R2, coefficient of correlation by simple linear regression.
Correlation between P450 Activities and Prasugrel Active Metabolite Formation.
The correlation between the activities of P450 isoforms involved (Rehmel et al., 2006) and prasugrel active metabolite formation was investigated. The coefficients of correlation with the activities of CYP2B6, CYP2C9, CYP2C19, and CYP3A4 were 0.3734, 0.4884, 0.2716, and 0.1497, respectively (Fig. 8). The coefficients were not so high for each P450 isoform probably because of its multi-isoform-mediated reaction. On the other hand, according to multiple linear regression analysis, the prasugrel active metabolite concentrations observed were well correlated with the concentrations estimated by the CYP2B6, CYP2C9, CYP2C19, and CYP3A4 activities (coefficient of correlation by single linear regression between observed and estimated concentrations, R2 = 0.8563) (Fig. 9), demonstrating that CYP2B6, CYP2C9, CYP2C19, and CYP3A4 contributed to prasugrel active metabolite formation in individual human liver microsomes used in this experimental system.

Correlation between the activities of CYP2B6, CYP2C9, CYP2C19, and CYP3A4 and active metabolite formation rates from prasugrel in individual human liver microsomes (n = 20).

Correlation between estimated and observed prasugrel active metabolite in individual human liver microsomes (n = 20). The estimated prasugrel active metabolite formation was calculated by the following multiple regression equation: 15.978 + 0.0091 × CYP3A4 + 23.862 × CYP2B6 + 1.121 × CYP2C9 + 1.764 × CYP2C19 activity. R2, coefficient of correlation by simple linear regression.
Discussion
This study was performed to directly and quantitatively compare the efficiency and variability in the formation of active metabolites from thienopyridines, prasugrel, and clopidogrel and to clarify the detailed mechanisms of the differences using human liver microsomes. In this study, the substrate concentration of clopidogrel was set at a relatively high level (50 μM) in view of the analytical sensitivity of clopidogrel active metabolite. This approach may cause an underestimate of the intrinsic clearance of clopidogrel active metabolite formation. However, the V/S values (0.164 ± 0.196 μl/min/mg protein) obtained in this experimental system corresponded to the Vmax/Km value (0.27 μl/min/mg protein and Km = 44 μM) reported for pooled (n = 10) human liver microsomes (Kurihara et al., 2005). The V/S value for prasugrel active metabolite formation (8.68 ± 6.64 μl/min/mg protein) from 1 μM prasugrel was also consistent with the Vmax/Km values reported previously (5.8–12 μl/min/mg protein) (Rehmel et al., 2006). Accordingly, it was considered reasonable to compare the V/S values for metabolite formation from 1 μM prasugrel and 50 μM clopidogrel. In addition, the time points and protein concentrations selected were also considered to be appropriate. In this study, prasugrel and clopidogrel were incubated for 15 and 30 min, respectively, in human liver microsomes with a protein concentration of 1 mg/ml. Our preliminary results showed that formation of active metabolites from prasugrel and clopidogrel was linear up to 30 and 60 min, respectively, and up to 1 and 2 mg/ml, respectively, of protein concentrations in human liver microsomes (data not shown).
Consistent with the clinical observations (Payne et al., 2007; Wallentin et al., 2008), prasugrel active metabolite was generated more efficiently and less variably than clopidogrel active metabolite in individual human liver microsomes. We expected that these differences arose from those in their thiolactone formation because it has been reported that prasugrel thiolactone metabolite is formed by hydrolysis, whereas clopidogrel needs P450 oxidation to form its thiolactone intermediate (Kazui et al., 2005; Kurihara et al., 2005; Williams et al., 2008). According to the results, the prasugrel thiolactone metabolite formation level was higher with more consistency compared with that of clopidogrel thiolactone metabolite. In the in vivo situation, prasugrel is wholly converted to prasugrel thiolactone metabolite by esterases including hCE1 and hCE2 and in part subsequently to prasugrel active metabolite in the intestine, and therefore prasugrel thiolactone metabolite and prasugrel active metabolite, not prasugrel itself, flow into the liver, whereas clopidogrel is not metabolized to its thiolactone metabolite in the intestine (Kazui et al., 2005; Williams et al., 2008; Hagihara et al., 2009). In this study, prasugrel was not completely converted to prasugrel thiolactone metabolite in human liver microsomes probably because of the lower expression level of hCE2 protein compared with that in the intestine (Imai, 2006; Taketani et al., 2007). This result could indicate that more efficient formation of prasugrel thiolactone metabolite may occur in vivo than in the in vitro experimental system.
To clarify the mechanism of the less efficient and more variable formation of the thiolactone metabolite from clopidogrel, the metabolism was investigated using individual human liver microsome samples. Reflecting the previous clinical results (Caplain et al., 1999), clopidogrel was largely converted to its inactive acid metabolite by hCE1, which competes with the oxidation of clopidogrel to form the thiolactone metabolite. Because it has been reported that there is relatively large interindividual variability in hCE1 activity for some esterified drugs in human liver microsomes (Takahashi et al., 2008; Yang et al., 2009), the clopidogrel thiolactone metabolite formation level might change, depending on individual hCE1 activities. In addition, clopidogrel thiolactone metabolite was hydrolyzed to an inactive thiolactone acid (48%), which also contributes to the lower concentrations of clopidogrel thiolactone metabolite available to be metabolized to clopidogrel active metabolite. The esterases involved in this inactivation pathway have not been identified yet.
According to the previous studies using expressed P450 isoforms (Kurihara et al., 2005), clopidogrel is metabolized by CYP1A2, CYP2B6, and CYP2C19 to form clopidogrel thiolactone metabolite. To confirm involvement of these P450 isoforms in individual human liver microsomes, a correlation study and multiple linear regression analysis were performed. The results showed a positive correlation between clopidogrel thiolactone metabolite formation and the activities of CYP1A2, CYP2B6, and CYP2C19. Variability in clopidogrel thiolactone metabolite formation in vivo may be related to changes in these enzyme activities associated with intrinsic and/or extrinsic factors. It has recently been reported that cigarette smoking is associated with enhanced platelet inhibition by clopidogrel (Bliden et al., 2008). Because smoking is generally known to induce CYP1A activity, this clinical observation is consistent with our results in this report. Moreover, a large number of clinical studies indicate involvement of CYP2C19 in the formation of clopidogrel active metabolite and in efficacy of clopidogrel (Hulot et al., 2006; Brandt et al., 2007; Frere et al., 2008; Trenk et al., 2008; Umemura et al., 2008; Collet et al., 2009; Mega et al., 2009). Our report showing a contribution of CYP2C19 in clopidogrel thiolactone metabolite formation could support these clinical findings. Prasugrel active metabolite formation was well correlated with the activities of CYP2B6, CYP2C9, CYP2C19, and CYP3A4, which is consistent with the previous report (Rehmel et al., 2006).
In conclusion, the formation of the active metabolite of clopidogrel was less efficient and less consistent than the formation of prasugrel active metabolite. This difference was attributed to decreased clopidogrel thiolactone metabolite concentrations because of the large hydrolytic inactivation to acid metabolites of both clopidogrel and its thiolactone metabolite and the variable P450 isoform-mediated oxidation of clopidogrel. The hydrolysis pathways occur in the liver and compete directly with both the thiolactone metabolite formation and the formation of clopidogrel active metabolite from the thiolactone. In contrast, hydrolysis of prasugrel to its corresponding thiolactone occurs in the intestine during the absorption process, and thiolactone is metabolized to active metabolite in the intestine and in the liver in a single P450-dependent step. These differences may help to explain the poorer and more variable response to clopidogrel compared with that to prasugrel in patients treated for acute coronary syndrome.
Acknowledgments.
The activities of some P450 isoforms and hCEs were kindly analyzed by Dr. Satoshi Suzuki and colleagues in Human and Animal Bridging Research Organization. We thank Drs. Mary Pat Knadler and Steven A. Wrighton of Eli Lilly and Company and Dr. Takashi Izumi of Daiichi Sankyo Co., Ltd. for helpful comments and discussion on this study. N.A.F. is currently retired from Eli Lilly and Company.
Footnotes
-
Parts of this work were previously presented as follows: Hagihara K, Kazui M, Yoshiike M, Honda K, Farid NA, and Kurihara A (2008) American Heart Association Scientific Session 2008; 2008 Nov 8–12; New Orleans, LA. AstraZeneca, London, United Kingdom.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.109.028498
- Received May 15, 2009.
- Accepted August 17, 2009.
- Copyright © 2009 by The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}