


Abstract
HIV protease inhibitor ABT-378 (ABT-378) was metabolized very extensively and rapidly by liver microsomes from mouse, rat, dog, monkey, and humans. The rates of NADPH-dependent metabolism of ABT-378 ranged from 2.39 to 9.80 nmol·mg microsomal protein−1·min−1, with monkey liver microsomes exhibiting the highest rates of metabolism. ABT-378 was metabolized to 12 metabolites (M-1 to M-12), which were characterized by mass and NMR spectroscopy. The metabolite profile of ABT-378 in liver microsomes from all five species was similar, except that the mouse liver microsomes did not form M-9, a minor secondary metabolite. The predominant site of metabolism was the cyclic urea moiety of ABT-378. In all five species, the major metabolites were M-1 (4-oxo-ABT-378) and M-3 and M-4 (4-hydroxy-ABT-378). Metabolite M-2 (6-hydroxy-ABT-378) was formed by rodents at a faster rate than by dog, monkey, and human liver microsomes. Metabolites M-5 to M-8 were identified as monohydroxylated derivatives of ABT-378. Metabolites M-9 and M-10 were identified as hydroxylated products of M-1. Metabolites M-11 and M-12 were identified as dihydroxylated derivatives of ABT-378. The metabolite profile in human hepatocytes and liver slices was similar to that of human liver microsomes. The results of the current study indicate that ABT-378 is highly susceptible to oxidative metabolism in vitro, and possibly in vivo, in humans.
Several HIV-1 protease inhibitors have been developed recently for the treatment of HIV-1 infection (Roberts et al., 1990; Vacca et al., 1994; Kempf et al., 1995; Patick et al., 1996; Vacca and Condra, 1997). Therapy with ritonavir (Fig. 1), a potent inhibitor of HIV-1 protease, produced a rapid and sustained decline of plasma viral RNA and a concomitant elevation of CD4 cells in HIV-infected individuals (Danner et al., 1995). Because of the high replication and mutation rates of HIV, sustained high plasma concentrations of the inhibitory drugs have been shown to be necessary to prevent emergence of resistant virus (Molla et al., 1996). Because of limited oral bioavailability, low area under the plasma concentration-time curve (AUC)1 values, and short plasma half-lives of many of the currently used protease inhibitors, new efforts are being made to design more potent protease inhibitors with improved pharmacokinetic properties.

Structures of HIV protease inhibitors ABT-378 and ritonavir.
ABT-378 (Abbott-157378, A-157378; Fig. 1), an analog of ritonavir, was designed to avoid interaction with valine-82 of the HIV protease, which initially mutates in response to ritonavir therapy (Molla et al., 1996;Sham et al., 1997). ABT-378 is a potent inhibitor of wild-type and mutant HIV protease (Ki = 1.3–28 pM) and is also active against mutant HIV selected by ritonavir in vivo (EC50 ≤ 0.06 μM). ABT-378 is currently being developed for coadministration with ritonavir as a therapeutic treatment for HIV-1 infection. The purpose of this study was to determine the in vitro metabolism of ABT-378 in hepatic preparations from animals and humans. The results of this in vitro study combined with in vivo studies in animal models will help predict the potential in vivo human metabolic pathways.
Materials and Methods
Materials.
ABT-378, [14C]ABT-378 (53.8 mCi/mmol) and metabolites M-1, M-3, and M-4 were synthesized at Abbott Laboratories (Abbott Park, IL). ABT-378 was uniformly labeled with 14C in the carbonyl carbon β to the 2,6-dimethylphenoxy group of the molecule (Fig. 1) and was found to be >98% radiochemically pure. All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO). Mouse (∼30 g adult male Crl:CD-1 BR), rat (∼250 g adult male Sprague-Dawley), dog (∼10 kg adult male beagle), and monkey (∼3 kg adult male cynomolgus) liver microsomes were prepared by differential centrifugation. Human liver was obtained from the International Institute for the Advancement of Medicine (Exton, PA) and came from a 66-year-old Caucasian male donor (subject identification no.: IEN016). The liver was rapidly chopped into small pieces and finely minced with a hand-held razor blade. The minced tissue was homogenized in ice-cold 1.15% potassium chloride containing 10 mM potassium phosphate buffer (pH 7.4) with a tissue homogenizer. Microsomes were prepared by differential centrifugation and were stored at −70°C in 0.1 M potassium phosphate buffer (pH 7.4) containing 20% (v/v) glycerol and 1.0 mM EDTA. Microsomal protein concentration was determined using a bicinchoninic acid assay kit procedure (Pierce Chemical Co., Rockford, IL), with bovine serum albumin as the standard.
Microsomal Incubations.
Individual incubations (final volume 0.25 ml) consisted of 0.1 mg/ml microsomal protein in 100 mM phosphate buffer (pH 7.4) with final concentrations of 5 mM magnesium chloride, 5 mM glucose 6-phosphate, 1 mM β-NADP+, and 1 U/ml glucose 6-phosphate dehydrogenase. The drug (25 μM; 0.4 μCi/ml), buffer, and microsomes were mixed and kept at 37°C for 5 min and the reaction was started by adding the NADPH-generating system. Incubations were conducted at 37°C. For control incubations, buffer was substituted for the NADPH-generating system. The metabolic reaction was stopped by adding 0.25 ml of acetonitrile and vortexing.
Liver Slice Incubations.
For preparation of liver slices, human liver was obtained from the International Institute for the Advancement of Medicine and came from a 44-year-old African American male donor (subject identification no.: 416961). Cylindrical tissue cores (∼10 mm in length) were taken from the liver using a sterile 8-mm biopsy punch. Following the preparation of several uniform liver cores, slices of approximately 250 μm were obtained using a Krumdieck Tissue Slicer (Alabama Research and Development, Munford, AL). During preparation of the liver cores and slices, the tissues were constantly maintained in cold tissue culture medium. Two slices were floated onto a stainless steel mesh half-cylinder and loaded horizontally into glass scintillation vials containing 1.7 ml of oxygenated (5% O2:5% CO2) tissue culture medium. The vials were gently placed on a temperature-controlled (37°C) vial roller at 4 rpm for 1 h. After preincubation, the cylinders containing the liver slices were placed in fresh, oxygenated medium containing ABT-378 (final concentration 3 μM; 0.3 μCi/incubation) or positive control 7-ethoxycoumarin (final concentration 20 μM, 0.3 μCi/incubation). The cylinders were incubated for various times up to 24 h. The metabolic reactions were terminated by transferring the medium to separate tubes and immediately freezing them. The liver slices used in this study were found to be metabolically active as evidenced by oxidative and conjugative metabolism of [14C]7-ethoxycoumarin.
Hepatocyte Incubations.
Cryopreserved human hepatocytes were obtained from the International Institute for the Advancement of Medicine and came from a 50-year-old Caucasian female donor (subject identification no.: 807951). The hepatocytes were rapidly thawed under warm water and immediately mixed with 20 ml of prewarmed Waymouth’s MB 752/1 medium withl-glutamine (GIBCO-BRL Life Technologies, Gaithersburg, MD) in a conical centrifuge tube. The tube was inverted several times to ensure resuspension of hepatocytes and centrifuged at 1000 rpm for 4 min. The supernatant was decanted without disturbing the pellet. Another 20 ml of prewarmed medium was then added and the hepatocytes were dispersed by gentle tapping and inverting. The centrifugation, decantation, and resuspension in 11 ml prewarmed medium were repeated. The viable cell count was determined by the trypan blue exclusion method and was found to be ∼44% and ∼40% at the beginning and the end of a 2-h incubation, respectively. One milliliter of medium containing hepatocytes (∼0.5 million viable hepatocytes/ml) was then placed in each well of a 24-well tissue culture plate (Multiwell; Becton Dickinson Labware, Lincoln Park, NJ) and ABT-378 (5 μM, 0.3 μCi/ml) in 3 μL of methanol or positive control 7-ethoxycoumarin (20 μM, 0.3 μCi/ml) was added. Cell-free control incubations contained substrate in the medium without the hepatocytes. The culture plates were then incubated at 37°C with gentle shaking under 5% carbon dioxide and 95% air atmosphere for 1, 2, and 4 h. The metabolic reaction was stopped by transferring the contents of the well into a vial containing 1 ml of acetonitrile and then vortexing. The hepatocytes used in this study were found to be metabolically active as evidenced by oxidative and conjugative metabolism of [14C]7-ethoxycoumarin.
High-Performance Liquid Chromatography (HPLC).
All analyses were performed using an Hewlett-Packard 1050 liquid chromatography system (Hewlett-Packard, Wilmington, DE) consisting of a quaternary pump, an autosampler, and a diode array detector operated at 220 nm. Separations were achieved at ambient temperature on a Beckman Ultrasphere C18 column (5 μm 4.6 × 250 mm) (Beckman Instruments, Palo Alto, CA). A linear gradient of 25% to 55% acetonitrile in buffer (25 mM ammonium acetate, pH adjusted to 4.8 with formic acid) over 57 min was used as column eluent at a flow rate of 1 ml/min. Radioactivity in the column effluent was monitored with a Flo-One/Beta model A-500 radioactivity flow detector (Packard Instruments, Meriden, CT).
Metabolite Isolation.
A scaled-up incubation was performed as described above for isolation of metabolites for structural identification. Separations were achieved at ambient temperature on a Beckman Ultrasphere C18 column (5 μm 10 × 150 mm) and at a flow rate of 2.8 ml/min. The HPLC effluent corresponding to individual metabolite peaks was collected and the mobile phase was evaporated under nitrogen. Each metabolite residue was redissolved in methanol, filtered to remove inorganic salts, and evaporated to dryness. For NMR spectroscopy, the metabolites were purified by rechromatographing them with a mobile phase that did not contain any added buffer. The respective metabolites, after evaporation, were reconstituted in deuterated chloroform or acetonitrile.
Liquid Chromatography-Mass Spectrometry.
The liquid chromatography mass spectrometric analyses were performed on a Perkin-Elmer Sciex API 300 Tandem Mass Spectrometer (Sciex Instruments, Toronto, Canada) equipped with an pneumatically assisted ion spray source and interfaced with a Hewlett-Packard Series 1050 module consisting of a quaternary liquid chromatography pump and a Rheodyne model 8125 manual injector (Rheodyne Instruments, Cotati, CA). Separations were achieved at ambient temperature with a Beckman Ultrasphere C18, 5 μm, 2.0 × 250 mm column. A linear gradient of 30 to 80% acetonitrile in 10 mM ammonium acetate (adjusted to pH 4.6 with formic acid) over 15 min, followed by 80% acetonitrile in buffer for 5 min was used at a flow rate of 200 μl/min. The HPLC effluent was split 4:1 such that a 50 μl/min flow was directed into the mass spectrometer. The ion energy used was 90 electron volts and the mass spectrometer was operated in positive ion mode.
NMR Spectroscopy.
NMR spectra were collected at ambient temperature using a Varian Unity 500 NMR spectrophotometer operating at 499.7 MHz for proton. The carbon-observed spectra were collected using a dedicated 3-mm carbon probe. The proton-observed spectra, including all of the twodimensional spectra, were collected using a 3-mm indirect detection probe with a z-axis pulsed field gradient coil.
Results
ABT-378 was converted to several metabolites in an NADPH-dependent manner in liver microsomes from all five species (Table1 and Fig.2). The rates of metabolism of ABT-378 ranged from 2.39 to 9.80 nmol of substrate metabolized·mg microsomal protein−1·min−1. Twelve metabolites, named M-1 to M-12, were chromatographically resolved and structurally identified. MetabolitesM-1 (range, 0.17–1.43 nmol·mg protein−1·min−1),M-3 (range, 0.41–1.80 nmol·mg protein−1·min−1) andM-4 (range, 1.02–4.33 nmol·mg protein−1·min−1) were the major metabolites in all five species. Metabolite M-2was formed by mouse and rat microsomes at faster rates than dog, monkey, and human liver microsomes. Of all five species tested, the rate of metabolism of ABT-378 was highest in monkey liver microsomes. Both human liver slices and human hepatocytes converted ABT-378 to several metabolites. In all cases, C-4 oxidation productsM-1, M-3, and M-4 were the major metabolites.
Distribution of in vitro metabolites of ABT-378 in male mouse, rat, dog, monkey, and human liver microsomal incubations

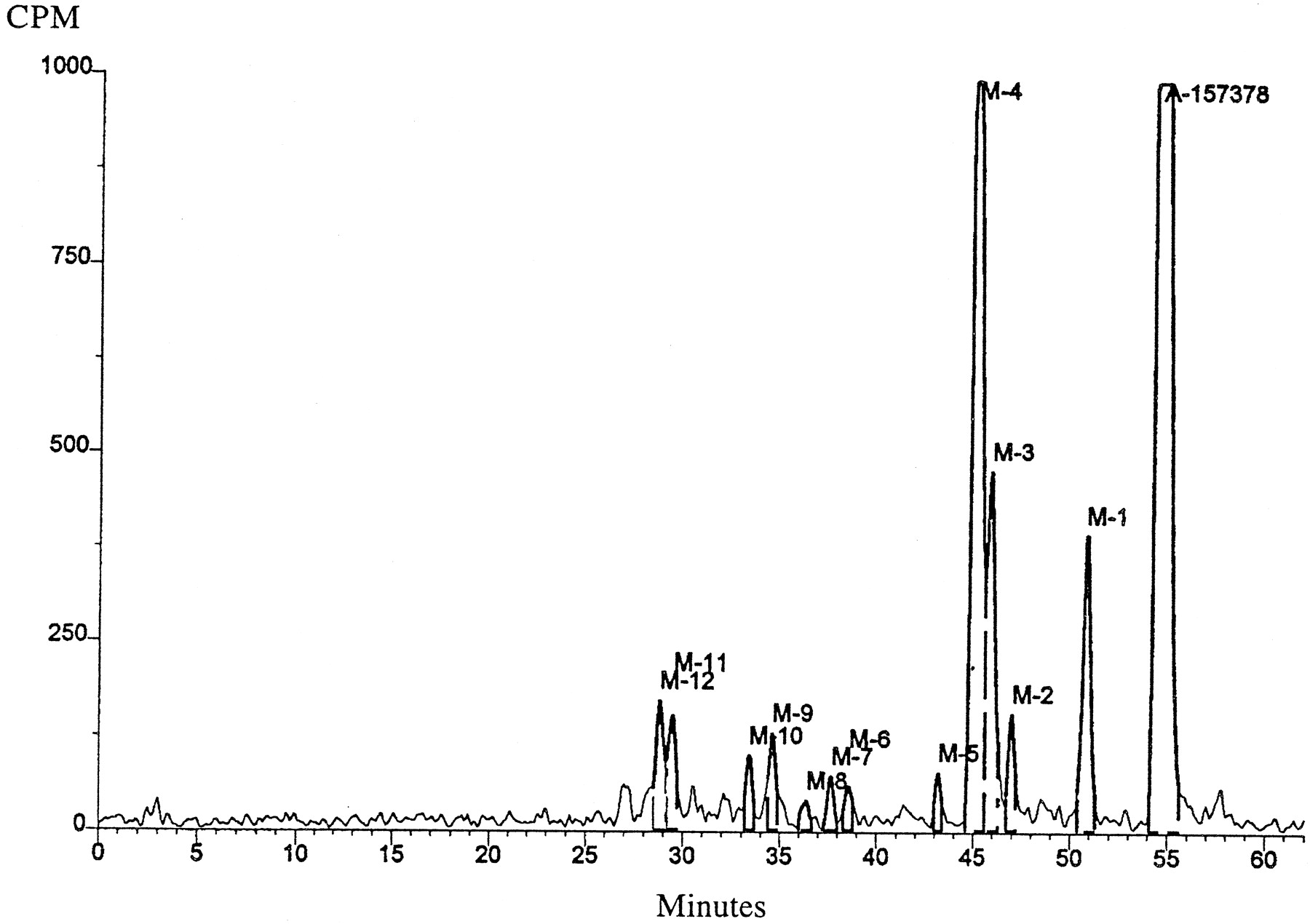
Reversed phase HPLC chromatogram of a 30-min incubation of [14C]ABT-378 (A-157378) with male human liver microsomes.
The mass spectral fragmentation pattern of ABT-378 and its metabolites is summarized in Table 2. The key1H-NMR spectral data for ABT-378 and its major metabolites are presented in Table3.
Mass spectral fragmentation pattern of ABT-378 and its liver microsomal metabolites
Proton chemical shift assignments of ABT-378 and its liver microsomal metabolites
ABT-378.
The protonated molecular ion of ABT-378 was observed at m/z629. The major fragment ions obtained from ABT-378 were m/z611 (MH+- H2O),m/z 183/447 (fragmentation at amide bond next to valine moiety, fragments B and C), and m/z 155 (isobutyl cyclic urea moiety, fragment A). Further fragmentation of the m/z447 fragment ion yielded two fragment ions at m/z 120 (2,6-dimethylphenoxy moiety, fragment E) and m/z 310 (dibenzyl core moiety, fragment D).
M-1.
The protonated molecular ion of M-1 was observed atm/z 643. The major fragment ions obtained fromM-1 were m/z 197/447 (fragments B and C) andm/z 169 (fragment A). Further fragmentation of them/z 447 fragment ion yielded two fragment ions atm/z 120 (fragment E) and m/z 310 (fragment D), which are similar to those obtained from ABT-378, indicating that this part of the molecule is unchanged in M-1. The fragment ion corresponding to the valine-cyclic urea moiety (m/z 197) is 14 atomic mass units (amu) greater than the fragment ion obtained from ABT-378 (m/z 183), indicating that it could be an oxidation product of ABT-378. The distinct difference in the NMR spectrum ofM-1 from that of ABT-378 is the disappearance of two methylene protons assigned to one of the carbons on the cyclic urea moiety of ABT-378. Four mutually coupled proton resonances were found at 2.18, 2.42, 2.76, and 3.11 ppm, suggesting a pair of adjacent methylenes. These protons were assigned to the protons of carbon-5 and carbon-6. These findings suggest that M-1 is the 4-oxo derivative of ABT-378. This was further confirmed by comparison of HPLC retention time with that of an authentic standard.
M-2.
The protonated molecular ion of M-2 was observed atm/z 645. The major fragment ions obtained fromM-2 were m/z 199 and 447 (fragments B and C, respectively) and m/z 171 (fragment A). Further fragmentation of the m/z 447 fragment ion yielded two fragment ions at m/z 120 (fragment E) and m/z 310 (fragment D), which were similar to those obtained from ABT-378, indicating that this part of the molecule is unchanged inM-2. The fragment ion corresponding to the valine-cyclic urea moiety (m/z 199) is 16 amu greater than the fragment ion obtained from ABT-378 (m/z 183), indicating thatM-2 is a hydroxylated product of ABT-378. The proton NMR spectrum of M-2 differed from that of ABT-378 in the disappearance of two of the methylene proton resonances and the concomitant appearance of two new resonances well down field at 4.96 and 4.58 ppm. Homonuclear decoupling and D2O exchange NMR experiments showed that the new resonances should be assigned to proton-6 and 6-OH, respectively. Thus metaboliteM-2 was identified as 6-hydroxy ABT-378. Further work is needed to establish the stereochemistry of the 6-hydroxy group ofM-2.
M-3 and M-4.
The mass spectral data for both M-3 and M-4 were identical. The protonated molecular ion of M-3 andM-4 was observed at m/z 645. The major fragment ions obtained from M-3 and M-4 werem/z 199/447 (fragments B and C) and m/z 171 (fragment A). Further fragmentation of the m/z 447 fragment ion yielded two fragment ions at m/z 120 (fragment E) andm/z 310 (fragment D), which were similar to those obtained from ABT-378, indicating that this part of the molecule is unchanged in M-3 and M-4. The fragment ion corresponding to the valine-cyclic urea moiety (m/z 199) was 16 amu greater than the corresponding fragment ion obtained from ABT-378 (m/z 183), indicating that both M-3 andM-4 are hydroxylated products of ABT-378. The proton NMR spectrum of both M-3 and M-4 differed from that of ABT-378 in the disappearance of two of the methylene proton resonances and the concomitant appearance of two new resonances between 4.9 and 3.6 ppm. Homonuclear and heteronuclear two-dimensional NMR experiments showed that these new resonances should be assigned to proton-4 and 4-OH. This indicated that the hydroxyl group in bothM-3 and M-4 is located on carbon-4 of the cyclic urea moiety (Fig. 3). Thus, metabolitesM-3 and M-4, which eluted very closely during HPLC analysis, are possibly epimers of 4-hydroxy-ABT-378. The HPLC analysis of M-4 after NMR spectroscopy, which was initially pure as determined by HPLC, indicated the presence of a small amount ofM-3, further suggesting that M-3 andM-4 are probably interconverting forms of the same molecule. Furthermore, chemical reduction of metabolite M-1(4-oxo-ABT-378) yielded a mixture of two 4-hydroxylated compounds that were chromatographically identical with metabolites M-3 andM-4. Based on the relative rates of formation, withM-4 forming at an ∼2-fold higher rate than M-3, it can be concluded that the 4-hydroxyl group is in a more thermodynamically favorable conformation in M-4 than inM-3.

Proposed in vitro metabolic pathway for ABT-378 in mouse, rat, dog, monkey, and human liver microsomes.
Metabolites M-1, M-2 (rodents only).M-3 and M-4 are major metabolites.
M-5.
The protonated molecular ion of M-5 was observed atm/z 645, indicating that it is a hydroxylated product of ABT-378. The major fragment ions obtained from M-5 werem/z 183/463 (fragments B and C) and m/z 155 (fragment A). Further fragmentation of the m/z 463 fragment ion yielded two fragment ions at m/z 310 (fragment D) andm/z 136 (fragment E). The fragment ion for the 2,6-dimethylphenoxy moiety obtained from M-5 is 16 amu greater than that obtained from ABT-378 (m/z 120), indicating that the site of hydroxylation in M-5 is on the 2,6-dimethylphenoxy moiety.
M-6, M-7, and M-8.
The protonated molecular ions of metabolites M-6,M-7, and M-8 were observed at m/z 645, indicating that they are hydroxylated products of ABT-378. The major fragment ions obtained from all three metabolites were m/z183/463 (fragments B and C) and m/z 155 (fragment A). Further fragmentation of the m/z 463 fragment ion yielded two fragment ions at m/z 326 (fragment D) and m/z120 (fragment E). The fragment ion for the dibenzyl core moiety (m/z 326) obtained from M-6, M-7, andM-8 is 16 amu greater than that obtained from ABT-378 (m/z 310), indicating that the site of hydroxylation inM-6, M-7, and M-8 is on the dibenzyl core moiety of ABT-378.
M-9 and M-10.
The protonated molecular ions of metabolites M-9 andM-10 were observed at m/z 659, indicating that they are possibly secondary metabolites of ABT-378. The major fragment ions obtained from both metabolites were at m/z 197/463 (fragments B and C) and m/z 169 (fragment A). Further fragmentation of the m/z 463 fragment ion yielded two fragment ions at m/z 326 (fragment D) and m/z 120 (fragment E). The fragment ion for the dibenzyl core moiety (m/z 327) obtained from M-9 and M-10is 16 amu greater than that obtained from ABT-378 (m/z 310), indicating that one of the substituents is a hydroxyl group on the dibenzyl core moiety of ABT-378. The fragment ion for the isobutyl cyclic urea moiety (m/z 169) was the same as that obtained from M-1, indicating that M-9 and M-10are hydroxylated products of 4-oxo-ABT-378 (M-1), with the hydroxyl group located on the dibenzyl core moiety of the molecule.
M-11 and M-12.
The protonated molecular ions of metabolites M-11 andM-12 were observed at m/z 661, which is 32 amu greater than the molecular ion of ABT-378, indicating that they are possibly dihydroxylated metabolites of ABT-378. The major fragment ions obtained from both metabolites were at m/z 199/463 (fragments B and C) and m/z 171 (fragment A). Further fragmentation of the m/z 463 fragment ion yielded two fragment ions at m/z 326 (fragment D) and m/z 120 (fragment E). The fragment ion for the dibenzyl core moiety (m/z 326) obtained from M-11 and M-12was 16 amu greater than that obtained from ABT-378 (m/z310), indicating that one of the substituents is a hydroxyl group on the dibenzyl core moiety of ABT-378. The fragment ion for the isobutyl cyclic urea moiety (m/z 171) was the same as that obtained from M-2, M-3, or M-4, indicating thatM-11 and M-12 are hydroxylated products of 4-hydroxy- or 6-hydroxy-ABT-378, with the second hydroxyl group located on the dibenzyl core moiety of the molecule.
Species Comparison of ABT-378 Metabolism.
Mouse liver microsomes metabolized ABT-378 to at least 11 metabolites (Table 1). Metabolite M-9, a 4-oxo-hydroxy secondary metabolite, was not formed by mouse liver microsomes. Rat liver microsomes metabolized ABT-378 to at least 12 metabolites. MetabolitesM-9 and M-10, 4-oxo-hydroxy secondary metabolites, were formed less efficiently by rat liver microsomes. Dog, monkey, and human liver microsomes metabolized ABT-378 to at least 12 metabolites. Of all the species liver microsomes examined, monkey liver microsomes exhibited the highest rates of ABT-378 metabolism.
Metabolism by Human Liver Slices.
Human liver slices (subject identification no.: 416961) efficiently metabolized ABT-378 to several metabolites (Table 1). After 24 h of incubation, ∼87% of [14C]ABT-378 was metabolized. After shorter incubations (3 and 6 h) the primary metabolites M-3 and M-4 were the major components. After longer incubation periods (12 and 24 h), a majority of the radioactivity was present as secondary metabolites and also as polar unknown metabolites.
Metabolism by Human Hepatocytes.
Human hepatocytes (subject identification number: 807951) efficiently metabolized ABT-378 to several metabolites (Table 1). The metabolism was linear up to 2 h of incubation. Further incubation up to 4 h did not increase the metabolism. The metabolite pattern obtained with human hepatocytes was similar to that obtained with human liver microsomes. The predominant metabolites were M-1,M-3, and M-4. In longer incubations (2 and 4 h) smaller quantities of M-2, M-5,M-6, M-8, M-9, M-10,M-11, and M-12 were also observed.
Discussion
ABT-378 was metabolized very extensively and rapidly by hepatic microsomes from all five species examined in this study. The rates of ABT-378 metabolism ranged from 2.39 to 9.80 nmol ABT-378 metabolized·mg microsomal protein−1·min−1, with monkey liver microsomes exhibiting the highest rate of metabolism. ABT-378 was metabolized to 12 metabolites (M-1 toM-12). The metabolite profiles of ABT-378 in liver microsomes from all five species were similar, except mouse liver microsomes did not form M-9, a minor secondary metabolite. In all five species, the major metabolites were M-1,M-3, and M-4. Metabolite M-2 was formed by rodents at a faster rate than dog, monkey, and human liver microsomes. In vivo small amounts of M-1 and M-4, with an unchanged parent compound as the predominant component, were found to be present in the plasma of rats and dogs administered ABT-378 in combination with ritonavir (G. Kumar, unpublished results). The metabolism of ABT-378 is essentially a deactivation reaction, because the major metabolites (M-1, M-3, andM-4) are less potent inhibitors of the HIV protease than ABT-378. The antiviral activity of the minor metabolites has not been studied yet.
The proposed metabolic pathway for ABT-378 is summarized in Fig. 3. The predominant route of metabolism of ABT-378 was oxidation at carbon-4 of the cyclic urea group of the molecule. Oxidation at C-6 was also a major biotransformation pathway in rat and mouse. On longer incubation, the primary metabolites were found to undergo hydroxylation on the phenyl groups of the central core moiety. Overall, of all five species tested, the rate of metabolism was highest by monkey liver microsomes. It is interesting to note that the plasma protein binding of ABT-378 was lower in monkey compared with other species (G. Kumar, unpublished results). It has been shown that the in vitro drug metabolizing activities in the cynomolgus monkey are severalfold higher than in dogs and humans (Sharer et al., 1995). For example, erythromycinN-demethylase activity in cynomolgus monkey microsomes was found to be 2.9 nmol·mg−1·min−1compared with 0.15 and 0.88 nmol·mg−1·min−1 in dog and human liver microsomes, respectively (Sharer et al., 1995). The total cytochrome (CYP) P450 content in the liver microsomes of cynomolgus monkeys has been reported to be 3-fold higher than in human liver microsomes (Shimada et al., 1997). Thus, the higher content of drug-metabolizing enzymes may be responsible for the faster rate of metabolism of ABT-378 by monkey liver microsomes compared with microsomes from other species. The comparatively lower protein binding combined with the faster rate of in vitro metabolism of ABT-378 by monkey liver microsomes suggests the possibility of higher clearance of ABT-378 in monkeys compared with other species. When ABT-378 was administered either alone or in combination with ritonavir, AUC values were lower in monkeys compared with rats and dogs (Marsh et al., 1997).
The metabolite profiles obtained with human liver slices and hepatocytes after short incubation periods were similar to those obtained with human liver microsomes. However, on prolonged incubation, both liver slices and hepatocytes produced several secondary metabolites and other polar unknown metabolites, which are probably tertiary metabolites of ABT-378. This type of metabolite profile was similar to that obtained in rat bile after administration of i.v. ABT-378 (G. Kumar, unpublished results). These results suggest that humans will also probably extensively metabolize ABT-378.
Structurally, the central core of ABT-378 is identical to that of ritonavir (Fig. 1). However, the terminal groups flanking the central core are different between the two inhibitors. The two termini of ritonavir contain a 2-isopropyl-4-thiazolyl group, the primary site of metabolism (Kumar et al., 1996; Denissen et al., 1997) and an unsubstituted 5-thiazolyl group. Recent studies demonstrated that the 5-thiazolyl group of ritonavir interacts with the heme of CYP P450 to produce a Type II spectral perturbation (Kempf et al., 1997). Consequently, ritonavir potently inhibits CYP3A4 and enhances the plasma levels of other protease inhibitors (Kempf et al., 1997). The replacement of the 5-thiazolyl group to yield ABT-378 abolishes the potent interaction with CYP and makes ABT-378 much more susceptible to oxidative metabolism than ritonavir. Thus, the rate of in vitro metabolism of ABT-378 was 3.85, 2.39, and 2.72 nmol·mg−1·min−1 in rat, dog, and human liver microsomes, respectively, compared with 0.036, 0.051, and 0.060 nmol·mg−1·min−1, respectively, found for ritonavir. The extremely high rate of in vitro metabolism for ABT-378 is consistent with its poor pharmacokinetic profile when administered alone in animals (Marsh et al., 1997) or humans (Lal et al., 1997). However, when coadministered with low doses of ritonavir, high plasma levels of ABT-378 are maintained both in animals and humans and lead to a >50-fold increase in AUC of ABT-378 in humans (Marsh et al., 1997; Lal et al., 1998). In vitro studies with human liver microsomes have indicated that this pharmacokinetic enhancement is because of potent inhibition of CYP3A-dependent ABT-378 metabolism by ritonavir (Kumar et al., 1997).
In summary, ABT-378 was extensively and rapidly metabolized by liver microsomes from mouse, rat, dog, monkey, and humans, with monkey exhibiting the highest rates of metabolism. The metabolite profile was similar across all five species examined. Seven hydroxylated primary metabolites and five secondary metabolites of ABT-378 were characterized. Human liver slices and hepatocytes produced a similar metabolic profile as that of human liver microsomes.
Acknowledgments
We thank Drs. Eugene Sun and John Leonard for helpful discussions. We thank Drs. Kennan Marsh and Stanley Roberts for reviewing this manuscript.
Footnotes
-
Send reprint requests to: Gondi N. Kumar, D46V, AP9, Abbott Laboratories, Abbott Park, IL 60064-3500. E-mail:gondi.kumar{at}abbott.com
-
This work was presented as a poster at the 8th North American ISSX Meeting, Hilton Head, SC, Abstract 94.
- Abbreviations used are::
- AUC
- area under the plasma concentration-time curve
- HPLC
- high-performance liquid chromatography
- CYP
- cytochrome
- Received May 7, 1998.
- Accepted August 12, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}