


Abstract
In human liver microsomes, triazolam is principally metabolized by CYP3A4 to form two metabolites, 1′-hydroxytriazolam (1′OHTz) and 4-hydroxytriazolam (4OHTz). The velocity of 1′OHTz formation was found to decrease at higher triazolam concentrations (>200 μM), indicative of “substrate inhibition”. Coincubation of [14C]triazolam with authentic metabolite standards of either 1′OHTz or 4OHTz up to 30 μM did not significantly inhibit the rate of [14C]1′OHTz formation. The effects of secondary compounds on triazolam oxidation were shown to be product-specific, producing either activation or inhibition depending on the triazolam metabolite monitored. When human liver microsomes were supplemented with exogenous human cytochrome b5, it was observed that substrate inhibition was attenuated and the resulting increase in 1′OHTz formation, relative to control (nonsupplemented) incubations, corresponded to a decrease in the ratio of 4OHTz to 1′OHTz. In contrast, when cofactor (e.g., 100 μM NADPH) was rate limiting, the metabolite ratio (4OHTz/1′OHTz) was markedly increased over the entire substrate concentration range (0.5–1000 μM). To explain these kinetic observations, a two-site binding model is proposed in which triazolam is hypothesized to bind within the CYP3A4 active site in spatially distinct orientations, which may lead to the formation of either the 1′-hydroxytriazolam or 4-hydroxytriazolam. Differential inhibition/activation is consistent with this two-site model and substrate inhibition is hypothesized to result from competition between the two sites for reactive oxygen.
The cytochrome P-450s are a superfamily of drug-metabolizing enzymes found principally in liver and intestine and are responsible for the oxidative metabolism of a wide range of xenobiotic and endobiotic compounds. In humans, P-4501 3A4 (CYP3A4) is the most abundant P-450 enzyme and is responsible for the biotransformation of the majority of drugs currently on the market (Maurel, 1996). As a consequence, there are several well documented clinical drug interactions involving compounds that are principally metabolized by CYP3A4 (Thummel and Wilkinson, 1998).
Traditionally, in vitro enzyme kinetic studies have been used to determine substrate binding affinity to a particular P-450 enzyme, and this information has then been used to predict drug metabolism in vivo (Rane et al., 1977). Unfortunately, in vitro/in vivo extrapolations are sometimes not achieved. A primary reason for the lack of CYP3A4 in vitro/in vivo correlation may in part be the variability associated with the enzyme's catalytic properties, which serve to confound the determination of accurate in vitro kinetic constants. Factors that have been demonstrated to impact CYP3A4 catalytic activity include homotropic and heterotropic activation (Schwab et al., 1988; He et al., 1997; Ueng et al., 1997; Korzekwa et al., 1998; Shou et al., 1999), modulation by ionic strength and divalent cations (Yamazaki et al., 1995; Mäenpää et al., 1998), membrane lipid composition (Imaoka et al., 1992;Ingelman-Sundberg et al., 1996), and stimulation by cytochromeb5 (Yamazaki et al., 1996b). As a consequence, CYP3A4 oxidation kinetics reflects in vitro conditions, and resulting substrate-velocity curves may not be consistent with the Michaelis-Menten model. Thus, the in vitro constants derived from assuming a Michaelis-Menten relationship may not be appropriate, producing error when in vivo pharmacokinetics and pharmacodynamics are predicted from in vitro data (Houston and Kenworthy, 2000).
The active site of CYP3A4 is generally considered to be spacious, as evidenced by its ability to oxidize large polycyclic aromatic hydrocarbons and macrolides such as cyclosporin. In deference to the large CYP3A4 active site, it has been hypothesized that two substrates may physically occupy the active site simultaneously (Shou et al., 1994). Several studies have provided evidence supporting this hypothesis, and a kinetic model has been established that rationalizes many of the anomalous kinetics associated with CYP3A4 catalytic activity (Korzekwa et al., 1998).
Recently, it was reported that the biotransformation of triazolam by CYP3A4 was characterized by substrate inhibition (Perloff et al., 2000). Moreover, midazolam, a close structural analog of triazolam, has also been shown to display the same kinetic phenomenon (Kronbach et al., 1989; Gorski et al., 1994; Perloff et al., 2000). The current study explores the hypothesis that substrate inhibition is a consequence of multiple binding of triazolam within the active site of CYP3A4. We find evidence for a model in which triazolam binds concurrently in two specific orientations (leading to separate oxidative products) that compete for activated oxygen.
Materials and Methods
Reagents.
Triazolam, [14C]triazolam, 1′-hydroxytriazolam, and 4-hydroxytriazolam were obtained from Pharmacia Corporation (Kalamazoo, MI). Human livers were obtained from the International Institute for the Advancement of Medicine (IIAM; Exton, PA). Microsomal preparation was achieved through differential centrifugation as described previously (Wienkers et al., 1995) and then stored at −80°C until use. Purified human cytochromeb5 was obtained from Panvera (Madison, WI).
Enzymatic Assays.
Kinetic experiments were done in duplicate under conditions in which less than 10% substrate depletion occurred. Velocity determinations contained 0.02 mg/ml microsomal P-450, 100 mM potassium phosphate buffer (pH 7.4), 1 mM EDTA, 1 mM NADPH, and varying concentrations of substrate and cytochrome b5 in a 0.5-ml incubation volume. Substrate was introduced to the incubation in less than 5 μl of acetonitrile. Samples were preincubated at 37°C for 5 min, and then catalysis was initiated by the introduction of NADPH. The formation of triazolam metabolites was linear for 14 min; thus, each reaction was terminated with 100 μl of acetonitrile after 10 min. The content of each incubation tube was directly transferred to a 1.5-ml microfuge tube, capped, and centrifuged at 14,000g for 10 min. The resulting supernatant was removed and transferred to 1.5-ml autoinjection vials for HPLC analysis.
HPLC Analysis.
Chromatography was performed with a HPLC system equipped with a PE410 pump (Perkin Elmer, Norwalk, CT), a PE ISS-200 autosampler (Perkin Elmer), and a Spectrafocus UV detector (Spectra-Physics, Mountain View, CA). Triazolam metabolites were eluted isocratically on a 5-μm C18 column, 4.6 × 250 mm (Zorbax, Chadds Ford, PA), at 2.0 ml/min with a total run time of 15 min. The mobile phase consisted of 50 mM phosphate buffer (pH 8.0), methanol, and acetonitrile in a 50:45:5 mixture, respectively. Metabolites were monitored by absorbance at 220 nm and quantified with standard curves generated from authentic standards.
Mathematical Derivation and Analysis.
A velocity equation was derived on the assumption of rapid equilibrium between the enzymatic species as depicted in Fig. 8.

Schematic describing the enzymatic species present during catalysis assuming separate binding orientations/regions.
Results
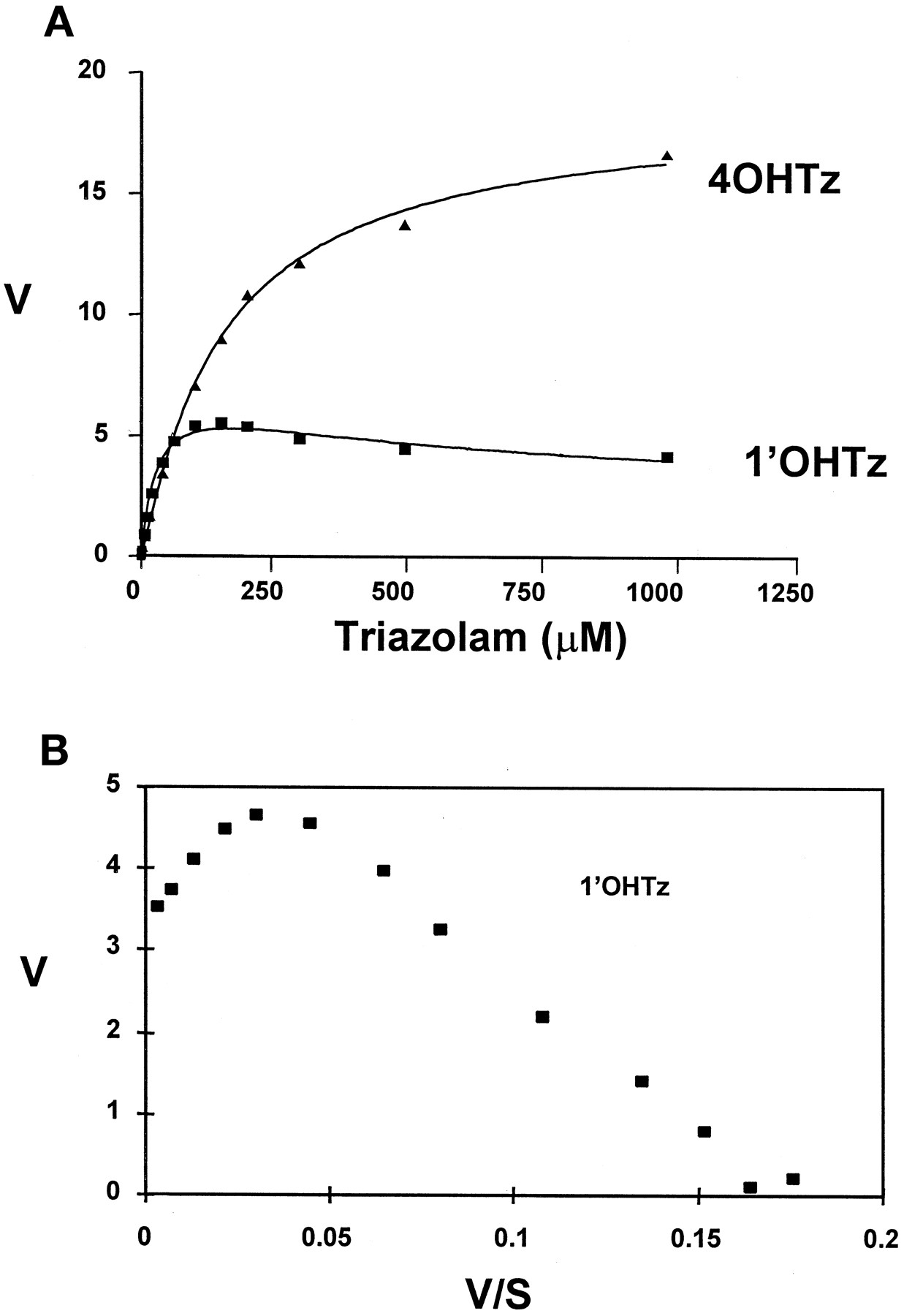
In vitro microsomal oxidation of triazolam generated both 1′-hydroxytriazolam (1′OHTz) and 4-hydroxytriazolam (4OHTz) metabolites. In preliminary microsomal kinetic studies it was observed that the substrate-velocity curve for 1′OHTz formation was nonhyperbolic at higher substrate concentrations. Figure1A shows that the rate of 1′OHTz formation was found to decline at elevated substrate concentrations, a characteristic that is commonly referred to as substrate inhibition (Segal, 1975). Figure 1B depicts the same 1′OHTz rate data replotted in the Eadie-Hofstee format. The ‘hook’ in the upper region of the Eadie-Hofstee plot is representative of substrate inhibition observed within the substrate-velocity curve.
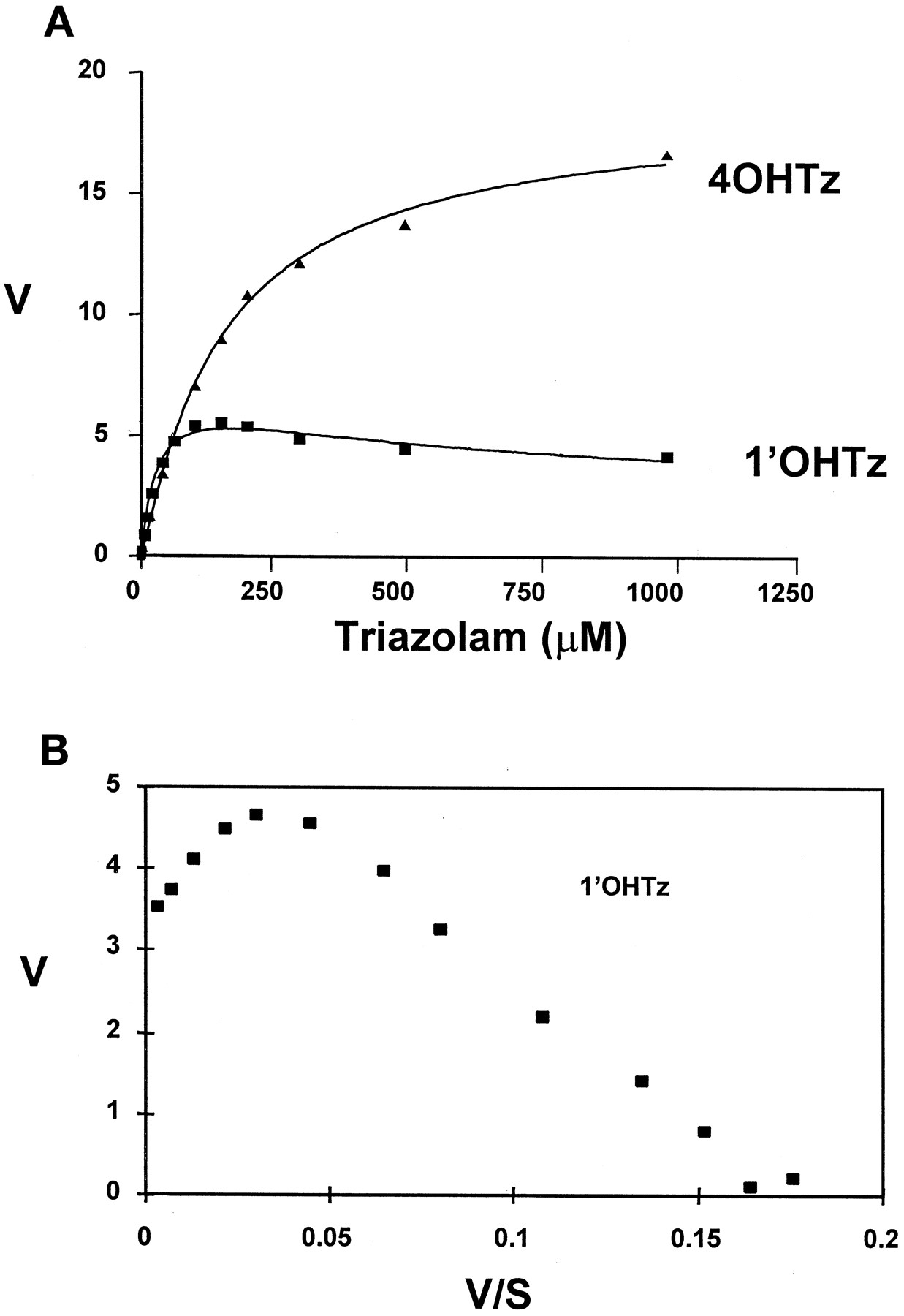
A, substrate-velocity curves for 4OHTz(▴) and 1′OHTz (▪); B, Eadie-Hofstee plot of 1′OHTz data.
The curves in A were generated from eq. 1, which yielded the following kinetic constants for 1′OHTz formation: 1′OHTz (r2 = 0.99)Vmax, 6.7 ± 0.4;Km1, 40 ± 3;Km2, 239 ± 95; α, 0.38 ±0.05. Velocity is expressed in nmol/min/mg of protein, and assays were performed in duplicate.
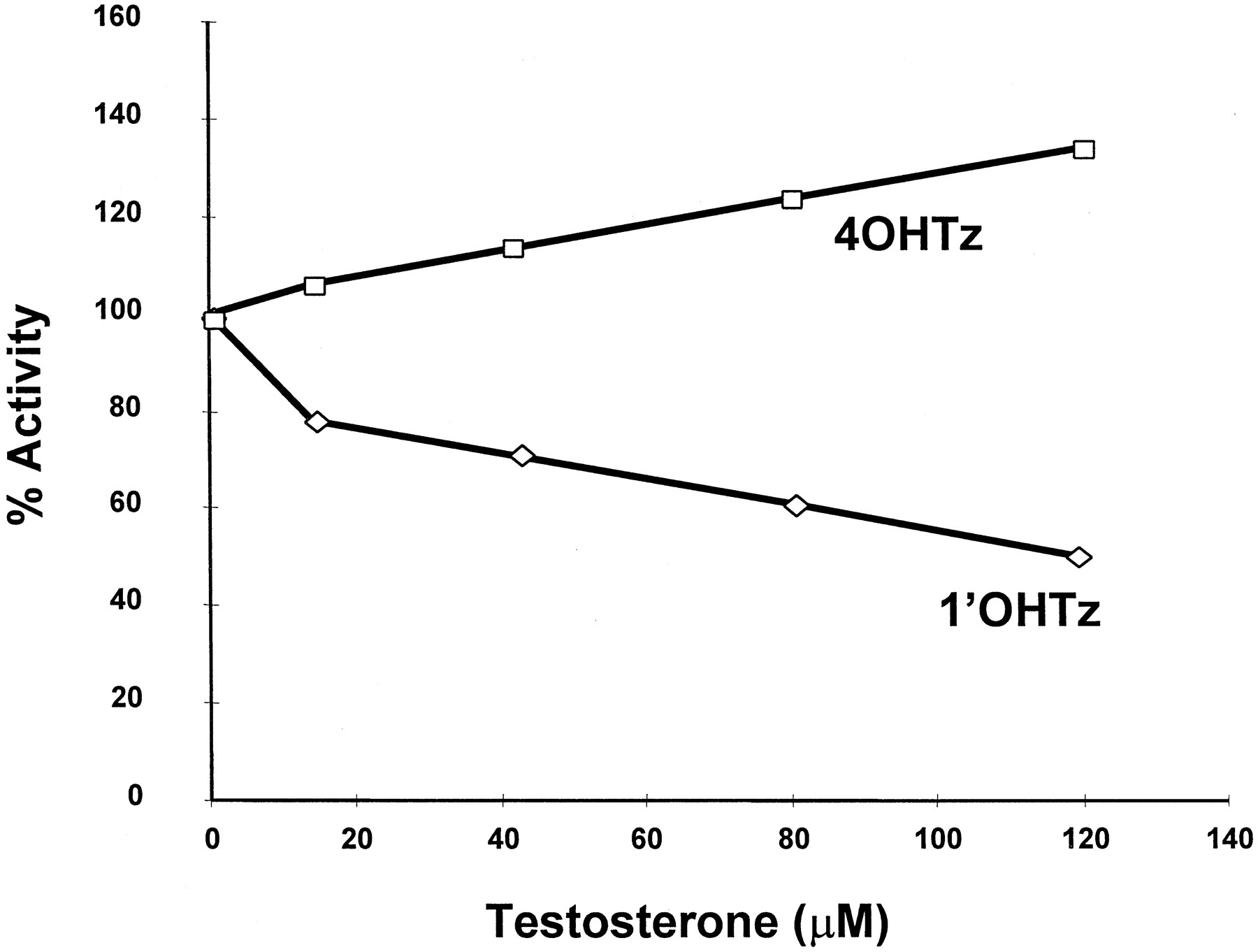
Additional kinetic experiments were conducted with the objective of identifying the mechanism behind 1′OHTz substrate inhibition. One possibility was that product inhibition could account for the decrease in 1′OHTz formation. Consequently, microsomal incubations were supplemented with nonradiolabeled authentic metabolite standards before the initiation of the reaction. The data presented in Fig.2 clearly demonstrate that the presence of both metabolites, 1-OHTz and 4OHTz up to 30 μM, had no significant effect on [14C]1′OHTz formation, indicating that product inhibition was not a likely mechanism for 1′OHTz substrate inhibition.
Inhibition of [14C]triazolam with 1′OHTz and 4OHTz.
Values on the x-axis represent the concentration of both 1′OHTz and 4OHTz. Assays were performed in triplicate.
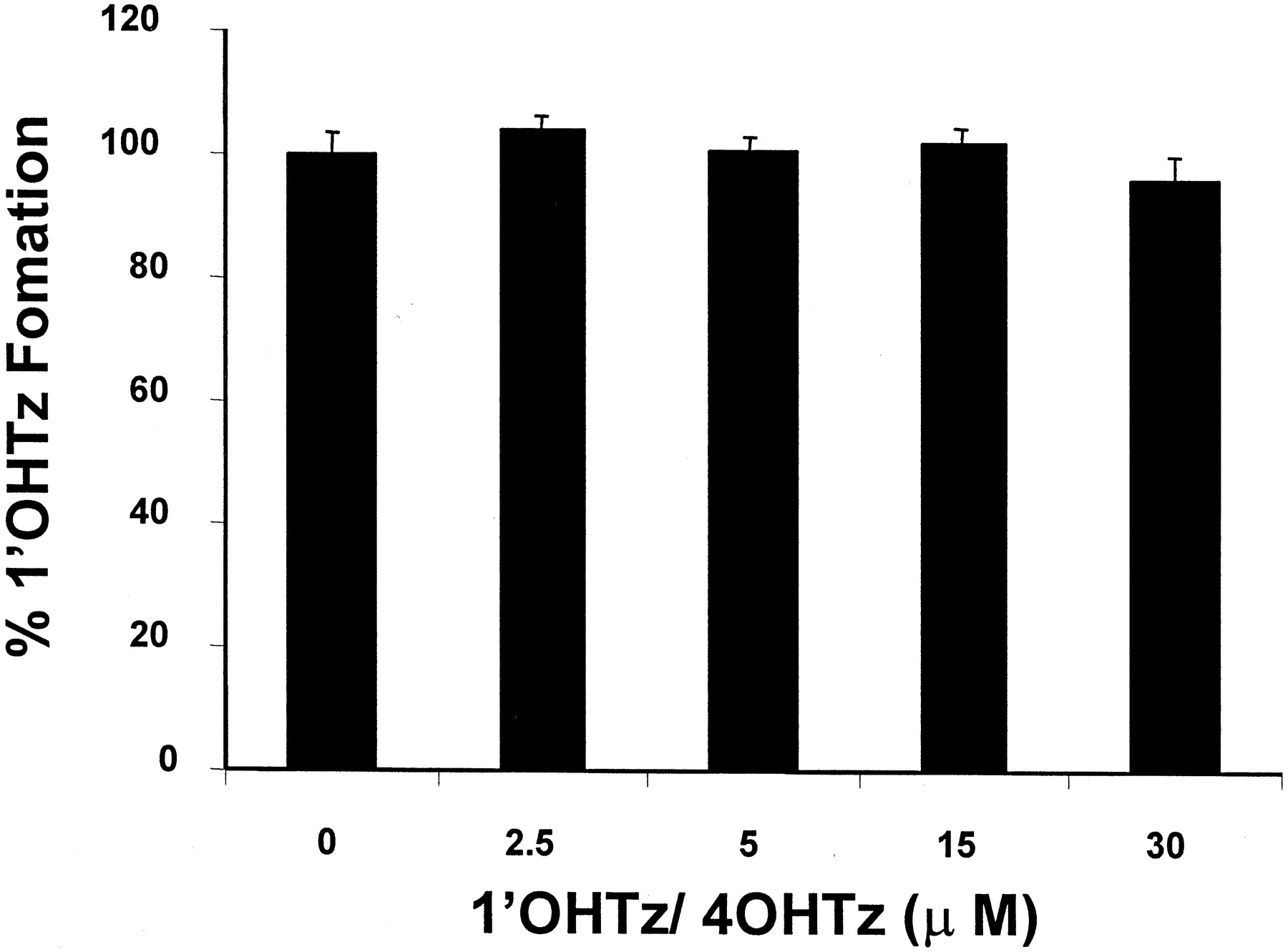
Several hypotheses have been proposed to explain unusual CYP3A kinetics, one of which is multiple binding regions within a single active site (Schwab et al., 1988; Ueng et al., 1997; Shou et al., 1999). Consequently, microsomal inhibition experiments were carried out to determine whether a panel of CYP3A4 substrates/inhibitors might differentially inhibit 1′OHTz or 4OHTz formation, thus testing the idea that triazolam oxidation could be described by a multisite mechanism. As shown in Fig. 3A, a selection of structurally similar compounds based upon the flavonoid moiety either inhibited the formation of 1′OHTz and 4OHTz to a similar extent or activated 4OHTz while inhibiting 1′OHTz. Moreover, when a survey of known substrate/inhibitors of CYP3A4 was examined, it was found that the inhibition of triazolam metabolism was not product-selective, except with progesterone (Fig. 3B). Progesterone selectively inhibited 1′OHTz formation while activating 4OHTz formation. In microsomes, testosterone (a structural analog of progesterone) also exhibited differential effects. Figure 4demonstrates that increasing concentrations of testosterone inhibited the formation of 1′OHTz, while 4OHTz production was activated. Additional studies demonstrated that the presence of testosterone had no effect on the apparent Km of 4OHTz formation (241 ± 9 μM control versus 250 ± 21 μM at 50 μM testosterone) but increased the apparentKm of 1′OHTz formation (from 42 ± 5.6 μM to 110 ± 13.6 μM). These results in combination with the data presented in Figs. 3 and 4 implicated a multisite mechanism for triazolam oxidation in which the binding at site 1 does not effect the binding kinetics of site 2; however, oxidation at site 2 is effected by the occupation of site 1.

Percentage of inhibition of 1′OHTz and 4OHTz relative to control (90 μM triazolam).
All compounds were present at a final concentration of 20 μM, and the assays were performed in triplicate.
Inhibition of 90 μM triazolam metabolism with increasing concentrations of testosterone.
The values reported are the average of duplicate experiments.
The potential of two triazolam binding sites competing for heme-bound oxygen was tested in Fig. 5. When microsomal incubations were supplemented with human cytochromeb5 (b5), the shape of the resulting 1′OHTz substrate-velocity curve was altered. Substrate inhibition displayed by 1′OHTz formation was greatly reduced, as shown by the Eadie-Hofstee plot in Fig. 5. A comparison of the Eadie-Hofstee plot in Fig. 5 with that in Fig. 1B clearly shows that the Eadie-Hofstee hook was less prominent when microsomes were supplemented with b5. Figure6 shows the concentration-dependent product ratio of 4OHTz/1′OHTz for three separate incubations: standard microsomal incubation conditions, microsomes supplemented withb5, and microsomal incubations with 100 μM NADPH (1/10 normal). Rate-limiting NADPH concentration (100 μM) was determined by decreasing the concentration of cofactor until the triazolam oxidation was 80% of control (data not shown). It is evident that under standard microsomal incubation conditions the 4OHTz/1′OHTz ratio varies in relation to the concentration of substrate. When the ratios of control versus b5-supplemented incubations were compared, the 4OHTz/1′OHTz ratio was suppressed at higher concentrations where 1′OHTz substrate inhibition was observed to occur (>200 μM). In contrast, when NADPH was rate limiting (100 μM, with no added b5), the formation of 4OHTz was increased relative to 1′OHTz, yielding a dramatically increased 4OHTz/1′OHTz ratio.

Eadie-Hofstee plot of microsomal 1′OHTz formation when supplemented with 1:1 M ratio of cytochromeb5:total microsomal P-450 content.
Velocity is expressed in nmol/min/mg of protein.
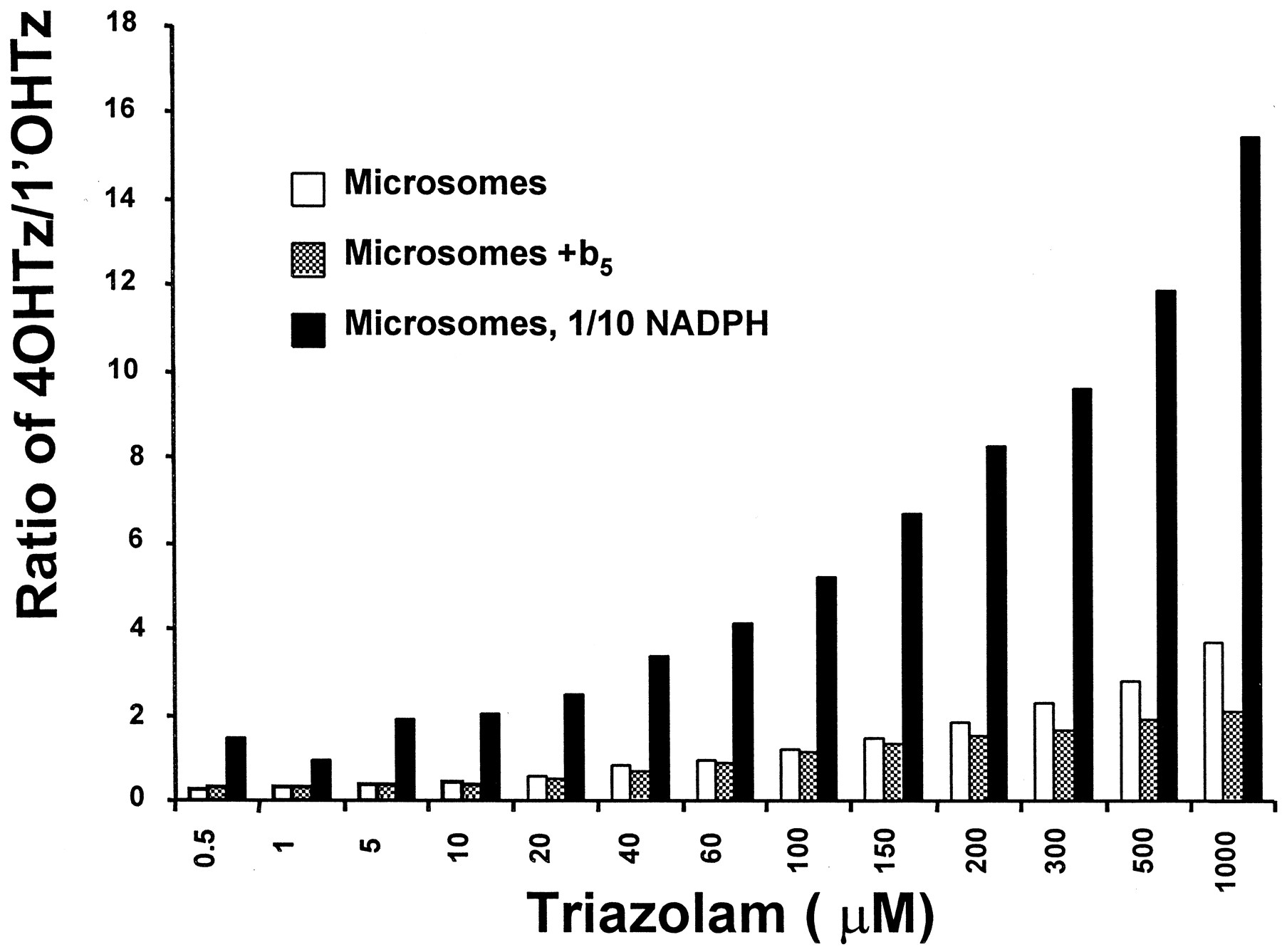
The 4OHTz/1′OHTz ratio of a control, cytochrome b5-supplemented (1:1 M ratio), and 1/10 NADPH (100 μM) incubations.
The ratios are the average of duplicate experiments.
Discussion
The oxidation of triazolam to 1′OHTz and 4OHTz is generally regarded as an index of CYP3A activity (von Moltke et al., 1996). Recently, in human liver microsomes, it has been reported that the formation of 4OHTz was characterized by a sigmoidal substrate-velocity curve, while the 1′OHTz substrate-velocity curve could be fit to the standard Michaelis-Menten model (Perloff et al., 2000). In addition, the CYP3A-mediated metabolism of midazolam (a structural analog of triazolam) to 1′-hydroxymidazolam has been found to yield substrate inhibition at higher concentrations (Kronbach et al., 1989; Gorski et al., 1994). However, it was noted that the magnitude of 1′-hydroxymidazolam substrate inhibition was variable across studies (Gorski et al., 1994). In the current study, the oxidation of triazolam to 1′OHTz was found to display substrate inhibition at higher concentrations, analogous to midazolam.
Several kinetic models have been proposed to explain the unusual kinetics sometimes obtained in P-450 metabolism, and in general, these models propose multiple substrate binding to account for substrate-velocity curves that are nonhyperbolic. Korzekwa et al. (1998) proposed a two-site model in which a substrate can occupy a single active site to produce either an ES or ESS complex. The relative orientation of each substrate in the ESS complex was not defined, but either substrate must have access to activated oxygen for oxidation to occur. The resulting mathematical derivation was found to describe a number of substrate-velocity curve shapes including sigmoidal activation and substrate inhibition. A subsequent kinetic model presented by Shou et al. (1999) proposed two distinct binding sites that were assumed to be cooperative. The basic mathematical form for either model can be represented as follows:
In addition to nonhyperbolic substrate-velocity curves associated with CYP3A4 metabolism, the enzyme may also exhibit anomalous inhibition as well. Aflatoxin, midazolam, and triazolam are three CYP3A4 substrates characterized by unusual inhibition. In this example, each substrate is metabolized in at least two positions, leading to distinct metabolites. Investigators found that 7,8-benzoflavone (α-naphthoflavone) inhibited the 3-hydroxyaflatoxin formation, while the formation of the 8,9-aflatoxin epoxide metabolite was activated (Raney et al., 1992). Recently, testosterone was found to inhibit 1′-hydroxymidazolam formation and activate the formation of 4-hydroxymidazolam (Wang et al., 2000). These results are analogous to the current study in which 3-hydroxyflavone, 6-hydroxyflavone, flavone, flavanone, progesterone, and testosterone inhibited 1′OHTz formation and activated 4OHTz formation. Furthermore, recent evidence demonstrated that nifedipine can inhibit CYP3A4 testosterone catalysis, but testosterone cannot inhibit nifedipine metabolism (Wang et al., 2000). For these substrates, differential inhibition/activation suggests that separate binding sites and/or orientations are involved in the formation of each metabolite.
The hypothesis of distinct binding orientations is also compatible with one of the two mathematical kinetic models noted above (Shou et al., 1999) and with a conceptual model presented by Harlow and Halpert (1998). In this light, the current study explored the idea that substrate inhibition might be the result of competition for reactive oxygen between the two separate triazolam binding orientations within a single CYP3A4 active site (Fig. 8). Such a hypothesis was deemed reasonable in light of isotope effect experiments, which have shown that the substrate rotational and transnational movement within a P-450 active site can be faster than the rate of the substrate oxidation (Jones et al., 1986; Korzekwa et al., 1989, 1995). Thus, it appeared likely that triazolam might occupy the active site in a single (ES) or double (ESS) binding mode and that the relatively faster time scale of binding would allow for equilibration between two (or more) binding locations within the CYP3A4 active site. Moreover, these distinct orientations could potentially compete for reactive oxygen (Fig.7).
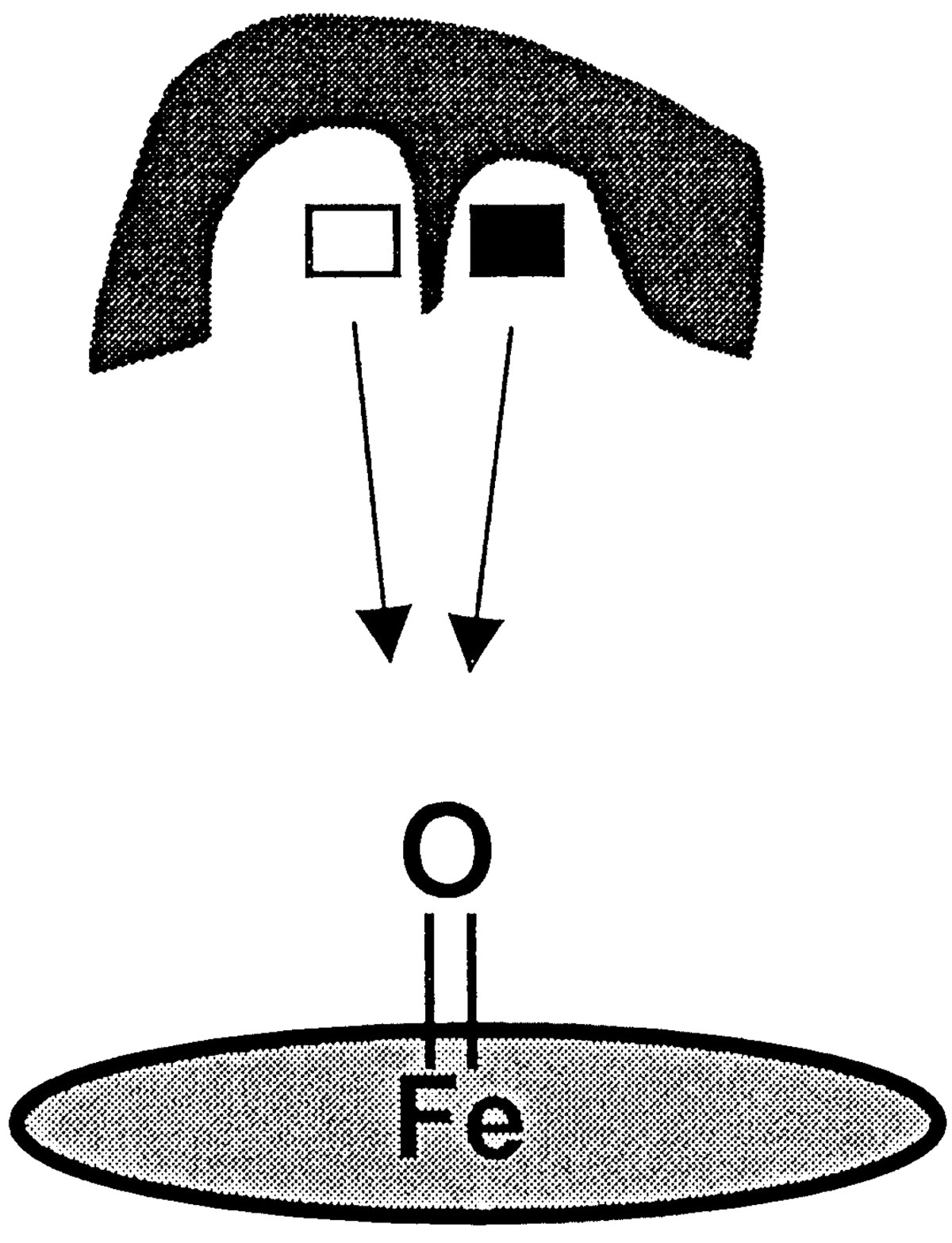
Schematic representation of competition for heme-bound oxygen by separate binding orientations/regions.
As a means to test this hypothesis, it was reasoned that the addition of cytochrome b5 would increase the efficiency of CYP3A4-reactive oxygen production and therefore enhance the availability of heme-bound oxygen for either binding orientation. Previous studies with CYP3A4 have demonstrated that cytochromeb5 can decrease the uncoupling of the catalytic cycle and increase the partition of the iron-oxygen complex toward substrate oxidation (Perret and Pompon, 1998). Additionally,Yamazaki et al. (1996a) reported that b5dramatically increased the rate of CYP3A4 reduction with a parallel increase in substrate oxidation rate. In the current study, addition of exogenous b5 stimulated triazolam oxidation (1:1 M ratio, data not shown), and it was therefore anticipated that the incorporation of exogenous cytochromeb5 in microsomal incubations would facilitate and increase the rate of heme-bound reactive oxygen production. Furthermore, the increased availability of heme-bound reactive oxygen was hypothesized to reduce the competition for reactive oxygen between the two proposed binding orientations of triazolam (i.e., decrease the 4OHTz/1′OHTz ratio at concentrations where substrate inhibition was observed).
The data presented in Figs. 5 and 6 support this interpretation. Figure5 shows that a microsomal incubation supplemented by cytochromeb5 no longer exhibits significant substrate inhibition of 1′OHTz. One potential interpretation is that at low substrate concentrations, 1′OHTz production effectively competes with 4OHTz because the 4OHTz binding orientation has not yet saturated (apparent Km = 171 μM). However, as occupation of the 4OHTz site increases, this in turn leads to increased competition with the 1′OHTz site for heme-bound reactive oxygen. The result is a 4OHTz/1′OHTz ratio that increases with higher substrate concentrations. At higher concentrations, where most of the enzyme is driven toward the doubly occupied enzyme (ESS), formation of 1′OHTz is decreased because of competition with the saturated 4OHTz site. The result is apparent substrate inhibition. The addition of cytochromeb5 increases the availability of heme-bound reactive oxygen and therefore decreases the observed substrate inhibition as visualized by an Eadie-Hofstee plot or as measured by a change in the ratio of 4OHTz/1′OHTz.
Since the interaction CYP3A4 with cytochromeb5 may also involve an enzyme conformational change, this could complicate the interpretations proposed above. In a separate experiment, triazolam substrate-velocity data were obtained in the presence of 100 μM NADPH, 1/10 the amount of cofactor typically used in an in vitro incubation. It was reasoned that by using a reduced concentration of NADPH, the presentation of heme-bound oxygen to either triazolam orientation would become limited, and hence the competition between binding orientations would be increased. The data presented in Fig. 6 confirmed this prediction, as reduced NADPH concentration greatly enhanced substrate competition and markedly increased the concentration-dependent ratio of 4OHTz/1′OHTz. The reduction of NADPH concentration would not be expected to produce a CYP3A4 conformational change. Thus, the substantial increase in the 4OHTz/1′OHTz ratio supported the conclusion that the rate of reactive oxygen formation, not conformational change, was critical in determining metabolite ratio.
Intuitively, the arguments proposed above suggest an enzymatic model in which triazolam may be bound in two distinct CYP3A4 active site binding regions and therefore would give rise to three possible catalytic species represented in Fig. 8. The resulting mathematical model is similar to that proposed by Shou et al. (1999), except that Km1 for a singly bound species (ES) was assumed to be unaltered in the doubly bound species (ESS), implying that the two binding orientations within the active site are spatially distinct and the binding of one molecule does not affect the other. This assumption was made based on the observation that 50 μM testosterone (which inhibits the 1′OHTz site) had no effect on the apparent Km for 4OHTz formation. However, testosterone did increase the rate of 4OHTz formation, and hence eq. 1 assumes that the catalytic activity of doubly bound CYP3A4 is different from the singly bound species (due to competition for reactive oxygen, or lack thereof), and a modulating factor α has been included in the equation to reflect this. When control data were fit to eq. 1, it was found that the α value equaled 0.38 ± 0.05 for 1′OHTz formation. When cofactor was rate limiting (100 μM NADPH), substrate velocity data were fit to the model, and the calculated 1′OHTz α value decreased to 0.15 ± 0.03 (data not shown). Within the context of this model, a decrease in the 1′OHTz α value was interpreted to indicate that the ESS complex was less efficient at producing 1′OHTz, reflective of increased competition with the 4OHTz binding orientation.
In conclusion, the enzymatic properties of CYP3A4 are complex, and at present, knowledge of active site dynamics is limited. Flash photolysis experiments with CYP3A4 have found that the rate of CO heme binding was indicative of a mixture of conformers that could have different affinities for substrates (Koley et al., 1996). The model presented in this manuscript is static and does not consider protein conformational changes or substrate cooperativity. As a consequence, the model presented is undoubtedly an oversimplification when applied to general CYP3A4 catalytic dynamics (other substrates or systems). However, the data presented suggest that one factor involved in CYP3A4 active site dynamics does stem in part from competition between two triazolam binding orientations for heme-bound reactive oxygen within a single CYP3A4 active site.
Footnotes
-
Send reprint requests to: Larry C. Wienkers, Pharmacia Corporation, 301 Henrietta St., 7265-300-313, Kalamazoo, MI 49007-4940. E-mail: Larry.C.Wienkers{at}am.pnu.com
- Abbreviations used are::
- P-450 or CYP
- cytochrome P-450
- b5
- cytochromeb5
- 4OHTz
- 4-hydroxytriazolam
- 1′OHTz
- 1′-hydroxytriazolam
- HPLC
- high-performance liquid chromatography
- ES
- enzyme bound with one substrate
- ESS
- enzyme bound with two substrates
- Received May 23, 2000.
- Accepted September 25, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}