


Abstract
N-(3,5-Dichloro-4-pyridyl)-3-(cyclopentyloxy)-4-methoxybenzamide (DCMB) is a known marker substrate for cytochrome P450 2B6. Based on the chemical template of DCMB, a novel terminal acetylene compound,N-(3,5-dichloro-4-pyridyl)-4-methoxy-3-(prop-2-ynyloxy)benzamide (TA) was synthesized and evaluated as a mechanism-based inactivator of P450 2B6. The pseudo first-order inactivation of expressed P450 2B6 by TA was both substrate and time-dependent. The kinetics of inhibition resulted in a maximal rate constant (kinactivation) of 0.09 min−1and an apparent KI of 5.1 μM. Incubation of expressed P450 2B6 with TA and NADPH resulted in a 68% loss in enzyme activity and a concurrent 62% loss in the formation of a reduced carbon monoxide complex, suggesting that heme destruction is the primary mode of enzyme inactivation. Enzyme inactivation of P450 2B6 was not reduced by the presence of 10 mM glutathione and was protected by incubation of excess DCMB with TA. The production of the carboxylic acid metabolite,N-(3,5-Dichloro-4-pyridyl)-3-(2-carboxyethoxy)-4-methoxybenzamide (TA-COOH), during the incubation of TA with 2B6 suggests that inactivation proceeds through a ketene intermediate. For 2B6 inactivation, the partition ratio was approximately 1.5 nmol TA-COOH formed/nmol P450 inactivated. Finally, TA was evaluated for mechanism-based inactivation of P450 3A4, 2C9, 2C19, 2D6, and 2E1 using human liver microsomes. In addition to 2B6, P450 2C forms were also found to be sensitive to TA-mediated inactivation, suggesting that subtle changes in the O-alkyl chain of the parent may be critical for the selectivity of enzyme inactivation.
The cytochrome P450 enzyme family is responsible for the metabolism of many structurally diverse xenobiotics. To determine the role of specific human P4501 forms in drug metabolism, an array of in vitro metabolism techniques including the use of P450 form-specific substrates, antibodies, and chemical inhibitors have been developed (Wrighton et al., 1993; Rodrigues, 1999). For chemical inhibition, both the mechanism and selectivity of enzyme inhibition are important characteristics for consideration. Mechanism-based enzyme inhibition, by definition, requires metabolism of a substrate to a reactive intermediate that can bind to the enzyme irreversibly, resulting in the loss of enzyme activity (Kent et al., 2001). This process of inactivation has been exploited in the design of P450 form-selective inactivators by the incorporation of dihalomethyl (Halpert et al., 1989) or acetylenic moieties (Ortiz de Montellano and Reich, 1984) at the preferred site of oxidation. Furthermore, the inactivation process can occur by heme alkylation and destruction and/or apoprotein modification (Halpert et al., 1985; Lin et al., 2002). Investigations of mechanism-based inactivation can therefore lead to detailed information on the interaction of the compound and the enzyme, i.e., the nature of the reactive intermediate formed, the efficiency of the inactivation process, and amino acid residues located within the enzyme active site (Kent et al., 2001). Regardless of the experimental objective(s), a clear understanding of the mechanism and selectivity of inhibition of a particular compound is necessary prior to application to in vitro metabolism experiments.
Many acetylenic compounds have been shown to irreversibly inactivate P450 enzymes in vitro and produce porphyria in vivo (Tebbe et al., 1999). Ortiz de Montellano and coworkers established that these molecules are converted to radical species that alkylate the P450 heme moiety (Ortiz de Montellano and Kunze, 1980; Ortiz de Montellano and Reich, 1984). As a consequence, iron is lost from the heme and abnormalN-alkylated porphyrins are produced. In addition to heme alkylation, irreversible P450 inhibition by terminal acetylenes such as 1-ethynylpyrene (Gan et al., 1984) or 10-undecynoic acid (CaJacob et al., 1988) via apoprotein modification has also been demonstrated.
Within the human P450 enzyme family, one of the less-characterized forms is 2B6. There is some dispute in the reported levels of 2B6 in human liver microsomes. Initial studies reported that 2B6 levels were only 0.2% of the total P450 content in human liver microsomes (Mimura et al., 1993; Shimada et al., 1994). However, other laboratories have recently demonstrated a greater frequency of detection and a higher percentage of 2B6 relative to total P450 content using improved immunoquantitation techniques (Ekins et al., 1998; Stresser and Kupfer, 1999). These and other investigators have also shown a wide range of interindividual variability in 2B6 protein levels and/or enzyme activity possibly due to genetic polymorphisms and/or exposure to environmental inducers and inhibitors (Heyn et al., 1996; Lang et al., 2001). Recently, P450 2B6 has gained more attention due to the demonstrated involvement in the metabolism of a number of clinically important drugs such as cyclophosphamide (Chang et al., 1993) and bupropion (Faucette et al., 2000).
N-(3,5-Dichloro-4-pyridyl)-3-(cyclopentyloxy)-4-methoxybenzamide (DCMB) has been shown to be metabolized exclusively by P450 2B6 viatrans-hydroxylation of the cyclopentyl group to yield DCMB-OH (Stevens et al., 1997). Using DCMB as a chemical template and our current understanding of mechanism-based P450 inactivation, a novel P450 2B6 inhibitor,N-(3,5-dichloro-4-pyridyl)-4-methoxy-3-(prop-2-ynyloxy)benzamide (terminal acetylene or TA), was designed and synthesized. The hypothesis was that the replacement of the cyclopentyl group of DCMB with a terminal acetylene functional group would result in a mechanism-based inactivator of P450 2B6. The objectives of this study were to therefore characterize the mechanism of P450 2B6 inhibition by TA, including kinetic and partition ratio analysis and to test the selectivity of enzyme inhibition by monitoring isoform-specific enzyme activities.
Materials and Methods
Chemicals.
Diclofenac, dextromethorphan, dextrorphan, triazolam, 1′-hydroxytriazolam, and HEPES were purchased from Sigma-Aldrich (St. Louis, MO). Chlorzoxazone, 6-hydroxychlorzoxazone, 4′-hydroxydiclofenac, (S)-(+)-mephenytoin, and 4′-hydroxymephenytoin were purchased from Ultrafine Chemicals (Manchester, UK). DCMB was obtained from Maybridge Organics (Cornwall, UK). The hydroxycyclopentyl metabolite of DCMB (DCMB-OH) was isolated from several incubations of DCMB with expressed human P450 2B6 and NADPH using preparative HPLC. All other chemicals and reagents were of the highest quality commercially available.
Enzymes.
Microsomes prepared from control or baculovirus insect cells infected with cDNA from human 2B6 supplemented with human reductase and human cytochrome b5 was purchased from BD Gentest (Woburn, MA). Pooled human liver microsomes (mixed gender,n = 15) were obtained from Xenotech (Kansas City, KS).
Synthesis ofN-(3,5-Dichloro-4-pyridyl)-4-methoxy-3-(prop-2-ynyloxy) benzamide.
To a suspension of 3-hydroxy-4-methoxybenzoic acid (1; Fig.1A) (5 g, 30 mmol) and anhydrous potassium carbonate (10.4 g, 75 mmol) in dryN,N-dimethylformamide (30 ml) was added propargyl bromide (9.64 g, 67.5 mmol, 80% solution in toluene). After heating at 65°C for 21 h, the reaction was quenched with water, extracted with diethyl ether, and concentrated. The crude product was dissolved in methanol (30 ml). To this solution, sodium hydroxide (4.32 g, 100 mmol) in water (10 ml) was added. The solution was heated at reflux for 30 min, then quenched with water, and extracted with diethyl ether. The basic aqueous layer was saved and acidified. The crude acid 4-methoxy-3-(prop-2-ynyloxy)benzoic acid (2), 2.77 g (45% yield) precipitated out of the solution. This was used in the next step without further purification. To a solution of 2(0.51 g, 2.5 mmol) in chloroform (2.5 ml) was added 2 Eq of thionyl chloride. The solution was heated to reflux under nitrogen for 4 h, and excess thionyl chloride was then removed under vacuum. The acid chloride was dissolved in toluene (1 ml) and added to a suspension of 4-amino-3,5-dichloropyridine (2.4 mmol, 0.39 g) and sodium hydride (4.8 mmol, 0.18 g, 60% dispersion in mineral oil) in dry tetrahydrofuran (3 ml). After 1 h, the reaction was quenched with water, extracted with chloroform, concentrated, and purified on a silica gel column eluted with 9.5:0.5 methylene chloride/ether. A total of 0.4 g of pure compound was isolated (47% yield).1H-NMR (d6 -acetone) δ 2.97 (t, 1H, HC≡C, J = 2.4), 3.80 (s, 3H, OCH3), 4.74 (d, 2H, C≡CCH2, J = 2.2), 7.01 (d, 1H, ArH, J = 8.3), 7.64 (m, 2H, ArH), 8.49 (s, 2H, Pyr-H), negative ion electrospray MS m/z 349 (100%) (M – H), 351 (80%) (M – H + 2).

Synthetic schemes for the preparation of TA and the carboxylic acid metabolite TA-COOH.
A, synthetic route for TA. B, synthetic route for TA-COOH.
Synthesis ofN-(3,5-Dichloro-4-pyridyl)-3-(2-carboxyethoxy)-4-methoxybenzamide (TA-COOH).
3-Hydroxy-4-methoxybenzaldehyde (3, Fig. 1B) was coupled to 3-bromopropan-1-ol in a similar manner as described above. The crude product was purified on a silica gel column eluted initially with 1:1 hexane/ethyl acetate followed by ethyl acetate. A total of 5.5 g of pure compound [3-(3-hydroxypropionoxy)-4-methoxybenzaldehyde;4] was isolated (80% yield). To a solution of 4(2.87 g, 14 mmol) in N,N-dimethylformamide (15 ml) was addedN,N-diisopropylethylamine (5.41 g, 42 mmol) andtert-butyldimethylsilyl chloride (4.65 g, 31 mmol). After stirring the reaction under nitrogen for 30 min, it was quenched with water, extracted with diethyl ether, and concentrated. The crude compound (yellowish oil) was purified on a silica gel column eluted with 9:1 hexane/ethyl acetate. A total of 4.50 g of final product [3-(3-tert-butyldimethylsilyloxypropionoxy)-4-methoxybenzaldehyde;5] (colorless oil) was isolated (99%). A solution of5 (4.74 g, 14 mmol) in acetone (100 ml) was added to a warm solution of potassium permanganate (4.74 g, 30 mmol) in water (15 ml)-acetone (75 ml) mixture. The reaction was stirred at 65°C for 45 min, and the solution was then washed thoroughly with 10% sodium metabisulfite. After washing, 2.18 g (46% yield) of a white solid [3-(3-tert-Butyldimethylsilyloxypropionoxy)-4-methoxybenzoic acid, 6] was isolated and coupled to 4-amino-3,5-dichloropyridine in a similar manner as described above. The crude compound was purified on a silica gel column eluted with 9:1 methylene chloride/ether. A total of 0.79 g of product [N-(3,5-dichloro-4-pyridyl)-3-(3-tert-butyldimethylsilyloxypropionoxy)-4-methoxybenzamide;7] was isolated (30% yield). To a solution of 7(0.40 g, 0.8 mmol) in tetrahydrofuran (5 ml) was added tetrabutylammonium fluoride (1.6 ml, 1 M solution in tetrahydrofuran), and the solution was stirred at room temperature overnight. After removing the solvent under vacuum, the crude product was purified on a silica gel column eluted with 1:1 methylene chloride/ether. A total of 0.22 g of white solid [N-(3,5-dichloro-4-pyridyl)-3-(3-hydroxypropionoxy)-4-methoxybenzamide;8] was isolated (76% yield). To a solution of 8(0.13 g, 0.35 mmol) in N,N-dimethylformamide (1.5 ml) was added pyridinium dichromate (0.38 g, 1 mmol), and the material was stirred for 4 days. The reaction mixture was washed thoroughly with sodium metabisulfite and extracted with n-butanol. The organic layer was washed with water (2 ml) and dried over anhydrous sodium sulfate. The organic solvent was removed under vacuum, and the crude greenish product was purified on a silica gel column eluted with 8:2 methylene chloride/methanol. A total of 20 mg of TA-COOH (15%) was isolated. 1H-NMR (CD3OD) δ 2.78 (t, 2H, CH2COO), 3.91 (s, 3H, OCH3), 4.35 (t, 2H, OCH2), 7.10 (d, 1H, ArH, J = 8.2), 7.68 (m, 2H, ArH), 8.00 (s, 1H, ArH) 8.64 (s, 2H, Pyr-H), negative ion electrospray MSm/z 383 (100%) (M – H), 385 (70%) (M – H + 2).
Inactivation of Expressed P450 2B6 and Human Liver P450 Forms.
Expressed P450 2B6 or pooled human liver microsomes were assayed for residual DCMB hydroxylase activity after incubation with different concentrations of the terminal acetylene inhibitor (0 to 40 μM TA). Primary incubations included either expressed P450 2B6 (75 pmol/ml) or human liver microsomes (1 mg/ml) in 50 mM HEPES buffer (pH = 7.6), 15 mM MgCl2, 0.1 mM EDTA, and 1 mM NADPH. The inhibitor, TA, was added in a final concentration of 1% acetonitrile. The final primary incubation volume was 0.5 ml. The mixture was preincubated at 37°C for 3 min, and the reactions were started by the addition of NADPH and allowed to proceed for up to 13.5 min. A 50-μl aliquot (representing a 3-fold dilution of the initial inhibitor concentration) was removed at various times and added to a secondary incubation mixture containing 50 mM HEPES buffer (pH 7.6), 15 mM MgCl2, 0.1 mM EDTA, and 50 μM DCMB to achieve a final volume of 150 μl. These reactions were incubated for 5 min for expressed P450 2B6 or 10 min for human liver microsomes and quenched with 50 μl of cold acetonitrile. Finally, the samples were centrifuged and analyzed by LC/MS/MS (Method C, Table1). Rate constants for inactivation were calculated by linear regression analysis of the natural logarithm of the residual DCMB hydroxylase activity as a function of time.
HPLC and LC/MS procedures for the identification and/or quantification of TA-COOH and various P450 marker metabolites1-a
To examine the effect of TA on enzyme activity and spectrally detectable P450 levels, separate experiments were performed using a single concentration of inhibitor and incubation time. Assay mixtures contained either expressed P450 2B6 (0.2 nmol/ml) or human liver microsomes (1 mg/ml), TA (50 μM), 50 mM HEPES buffer (pH 7.6), MgCl2 (15 mM), EDTA (0.1 mM), and NADPH (1 mM). The P450 concentration was chosen as the minimal amount needed for postinactivation analysis and the inactivation time estimates ≥50% loss of enzyme activity. The final incubation volume was 1 ml, and the final acetonitrile content was 1%. Controls were treated with solvent and NADPH or with TA but without NADPH. Samples were preincubated for 3 min at 37°C prior to the addition of NADPH and then allowed to proceed for 12 min at 37°C. The reactions were terminated by the addition of 1 ml of cold 10 mM Tris-acetate buffer (pH 7.4) containing 1 mM EDTA and 20% (v/v) glycerol. The samples were then centrifuged (180,000g) at 4°C for 1 h. The supernatant from each sample was saved for the measurement of metabolites and subsequent determination of the partition ratio associated with enzyme inactivation (described below). The protein was then resuspended in 50 mM HEPES buffer. Protein concentration, spectrally detectable P450 content, and heme levels were determined by standard methods (Lowry et al., 1951; Omura and Sato, 1964). Duplicate aliquots (50 μg of protein) of the treated microsomes were then assayed for residual DCMB hydroxylase activity as described earlier (Method C, Table 1).
For human liver microsomes, duplicate aliquots (50 μg of protein) of the reconstituted samples were also assayed for residual P450 form-selective enzyme activities. Representative P450 marker substrates were used, and their respective incubation conditions are summarized in Table 2. Protection from TA-dependent inactivation of P450 2B6 was evaluated using DCMB (molar ratios of 1:0, 1:1, and 1:2.5 of TA/DCMB in the primary reaction) or glutathione (10 mM). The concentration of TA in the DCMB protection experiment was 5 μM and in the glutathione experiment was 10 or 50 μM. All other reaction conditions and analyses were as described above (Method C, Table 1).
Summary of incubation conditions for cytochrome P450 marker assays
Identification and Derivatization of TA-COOH.
The incubation supernatants from the single time-point inactivation experiments mentioned above were individually concentrated, reconstituted in a final volume of 500 μl, and analyzed by LC/MS. To determine whether the proposed carboxylic acid metabolite (TA-COOH) was formed during microsomal incubations, two analytical approaches were used. First, the retention time of a metabolite formed during enzyme inactivation was compared with the carboxylic acid metabolite (TA-COOH) synthetic standard (Method A, Table 1). In addition, a 50-μl aliquot of the supernatant from individual samples was mixed with a carboxylic acid derivatizing agent (100 μl) prepared fresh by carefully mixing acetyl chloride (60 μl) with methanol (750 μl). After mixing, individual samples were allowed to stand at room temperature for 1 h. The solutions were then evaporated to dryness under nitrogen and reconstituted in 1:1 water/acetonitrile. A solution of the synthetic standard (TA-COOH) was treated in a similar manner and was used as the positive control. Samples were analyzed by LC/MS/MS (Method B, Table1).
Partition Ratio Determination.
The partition ratio for the inactivation of CYP2B6 by TA was determined by incubating the enzyme (0.2 nmol CYP2B6) at 37°C with a saturating concentration of TA (50 μM) and NADPH (1 mM) for 10 min. Control incubations contained enzyme and TA but not NADPH. The reactions were terminated, and the supernatant was concentrated to dryness, reconstituted in 1:1 water/acetonitrile, and analyzed by LC/MS to quantitate the amount of TA-COOH formed. The spectrally detectable P450 content of the enzyme pellet from the samples treated with NADPH was determined and compared with levels from samples in which NADPH was omitted. The partition ratio was calculated by dividing the amount of TA-COOH formed by the amount of P450 inactivated.
Results
Concentration- and Time-Dependent Inactivation of P450 2B6 by TA.
Inactivation of expressed P450 2B6 by the terminal acetylene inhibitor was found to be concentration- and time-dependent (Fig.2A). Inactivation also required NADPH, suggesting that metabolism of TA by cytochrome P450 to form a reactive intermediate was a prerequisite for enzyme inactivation. Pseudo first-order inactivation kinetics were observed with TA concentrations ranging from 0 to 40 μM at 37°C. The kinetic constants describing the inactivation of P450 2B6 with TA were determined from the double reciprocal plot shown in Fig. 2B. The maximal rate of inactivation at saturation (kinactivation) was 0.09 min−1, the concentration of TA required for half-maximal inactivation (KI) was 5.1 μM, and the time required for half of the enzyme to become inactivated (t1/2) was 7.7 min (Table3). A significant decrease in the initial enzyme activity was observed (Fig. 2A) with increasing TA concentration, suggesting that TA inhibits DCMB hydroxylase activity in a competitive manner as well. For human liver microsomes, similar pseudo first-order inactivation kinetics were observed, and the corresponding inhibition kinetic parameters are summarized in Table 3. Both expressed 2B6, and human liver microsomes exhibited the same maximal rate of inactivation (kinactivation = 0.09 min−1) but slightly differentKI values (5.1 μM for the former and 1.8 μM for the latter).

Concentration- and time-dependent inactivation of DCMB hydroxylase activity for expressed P450 2B6 by TA.
A, aliquots were removed from the primary reaction mixture at indicated time points and assayed for residual activity as described underMaterials and Methods. The concentrations of the inhibitor, TA, were 0 (■), 5 (▾), 10 (♦), 25 (●), and 40 (▪) μM. Percent enzyme activity remaining was plotted in the natural logarithmic scale determined from a single experiment. B, the corresponding double-reciprocal plot of the rates of inactivation as a function of the TA concentrations.
Apparent KI, kinactivation, and t1/2 values for the inhibition of DCMB hydroxylase activity by TA in expressed P450 2B6 and human liver microsomes
Irreversibility of P450 2B6 Inactivation by TA.
To determine the reversibility of the inactivation of P450 2B6 by TA, incubations containing either expressed 2B6 or human liver microsomes, inhibitor, and NADPH were followed by centrifugation and washing of the protein pellet. The treated microsomes were then tested for residual enzyme activity and compared with control samples that contained all components except NADPH. HPLC analysis showed that >99% of the unbound TA was removed by decanting the supernatant after centrifugation followed by a single wash of the protein pellet (data not shown). Removal of unbound TA by centrifugation did not, however, lead to recovery of the DCMB hydroxylase activity of TA-inactivated samples regardless of whether the source of the enzyme was expressed 2B6 or human liver microsomes. Incubation of expressed P450 2B6 with 50 μM TA and NADPH for 12 min resulted in a 68% decrease in DCMB hydroxylase activity, a 62% loss in spectrally detectable P450, and 38% decrease in heme content (Table 4). For human liver microsomes, similar incubation conditions resulted in a 40% decrease in DCMB hydroxylase activity and 29% loss in the ability to form a reduced carbon monoxide complex. However, heme loss was not observed for human liver microsomes, possibly due to the small percentage of CYP2B6-associated heme relative to the large amount of heme in human liver microsomes. The smaller loss of P450 content and enzyme activity in human liver microsomes compared with that for expressed enzyme is likely due to the nonspecific binding of TA to proteins other than P450 2B6. Hence, a higher concentration of free TA was available to access the expressed P450 compared with human liver microsomes. Control incubations in which TA or NADPH were omitted retained most of the enzyme activity and spectrally detectable P450. The fact that decreases in P450 2B6 enzyme activity paralleled the loss of spectrally detectable P450 content suggests that TA-mediated inactivation of P450 2B6 proceeds primarily via heme modification. However, further studies to identify the ultimate inactivated form of the enzyme are necessary to confirm the exact mechanism.
Effect of TA on P450 content and residual DCMB hydroxylase activity for expressed P450 2B6 or human liver microsomes4-a
Protection From Enzyme Inactivation by DCMB and Glutathione.
Incubations of expressed P450 2B6 with TA together with increasing concentrations of DCMB (one to 2.5-fold excess DCMB) in the primary reaction slowed the rate of TA-dependent inactivation (Fig.3A). Specifically, the addition of DCMB to the primary incubation changed the rate of inactivation by 6-fold as the apparent rate constant (slope) in the absence of DCMB was 0.06 min−1 whereas in the presence of equimolar or 2.5-fold excess DCMB, an apparent inactivation rate constant of 0.01 min−1 was observed. Also, the presence of excess glutathione (10 mM) had very little effect on the rate of inactivation (0.07 min−1 versus 0.09 min−1 without glutathione) thus suggesting that any reactive intermediate formed could not be scavenged prior to the enzyme inactivation event (Fig. 3B). Although some inconsistency in levels of competitive inhibition (corresponding to ≤30% variability) are suggested by the y-axis intercept values for these protection experiments, these values are significantly influenced by both error in the data points and estimations of the true first-order time interval.
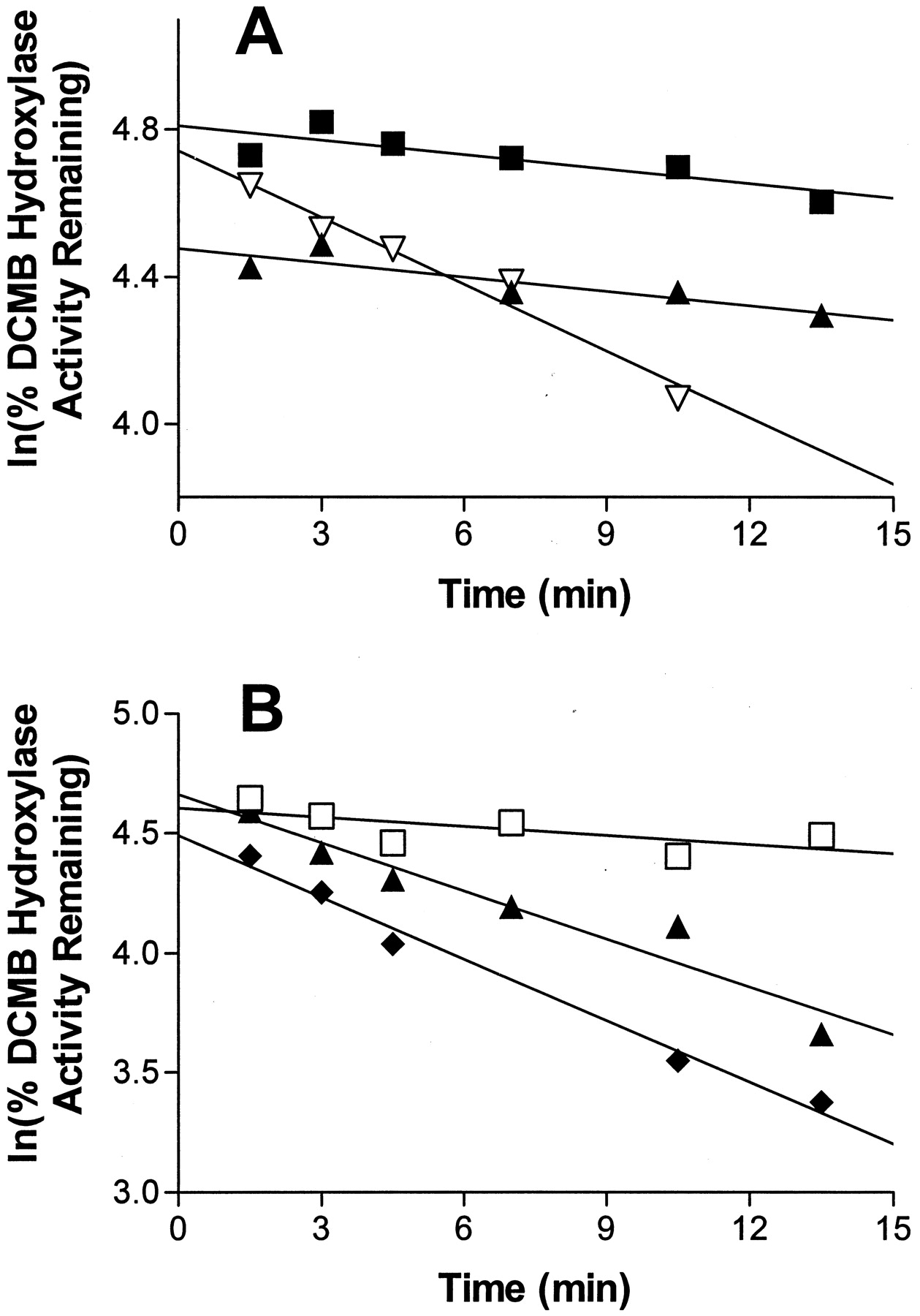
Effect of excess DCMB and glutathione on the rate of inactivation by TA for expressed P450 2B6.
Aliquots were removed from the primary reaction mixture at indicated time points and assayed for residual activity as described underMaterials and Methods. Percent enzyme activity remaining was plotted in the natural logarithmic scale determined from a single experiment. A, effect of substrate protection, primary P450 2B6 reaction mixture contained molar ratios of TA/DCMB of 1:0 (▿), 1:1 (▴), and 1:2.5 (▪). B, effect of glutathione, primary P450 2B6 reaction mixture contained no inhibitor (■), 50 μM TA with 10 mM glutathione (▴), or 50 μM TA without glutathione (♦).
P450 2B6 Inactivation and Carboxylic Acid Metabolite Formation.
There are several cases in which the production of a carboxylic acid metabolite from P450-catalyzed oxidation of terminal acetylene-containing compounds has been identified (Ortiz de Montellano and Komives, 1985; Foroozesh et al., 1997; Kent et al., 2002). The incubation of TA with expressed 2B6 in the presence of NADPH generated a new metabolite at retention time of 8.8 min (Fig.4A). This analyte was tentatively identified as the carboxylic acid metabolite of TA based on a comparison of the retention time with that of the synthetic carboxylic acid standard (TA-COOH, Fig. 4C) and HPLC coelution after samples were spiked with TA-COOH (data not shown). Characteristic fragments in MS/MS with a neutral loss produced by the cleavage of the amide bond were monitored in selected reaction monitoring (SRM) for MS/MS detection. Specifically, two SRMs were recorded (Fig. 4A) as m/z 385 → 223 (i.e., transformation of [M + H]+ ion → a nonchlorinated product ion) and m/z 387 → 223 (i.e., transformation of [(M + 2) + H]+ ion that contained one 37Cl isotopic atom → the nonchlorinated product ion). An intensity ratio of 3:2 is clearly seen (Fig. 4A), which is the characteristic pattern of molecules containing two chlorine atoms due to the natural abundance of35Cl versus 37Cl isotopes. As a result, unambiguous detection of the metabolite was accomplished. In addition, derivatizing both the metabolite and the synthetic TA-COOH with an acetyl chloride/methanol methylating reagent produced a single analyte that eluted at retention time of 10.6 min (Fig. 4B). Methylation of the carboxylic acid was concluded based on the appearance of two intense and identical signals at m/z 399 [M + H]+ and 401 [(M + 2) + H]+ in a ratio of 3:2 for both the metabolite and the synthetic standard. Finally, the molecular ions atm/z 399 and 401 each underwent typical amide bond cleavage and produced a common fragment ion at m/z 237.
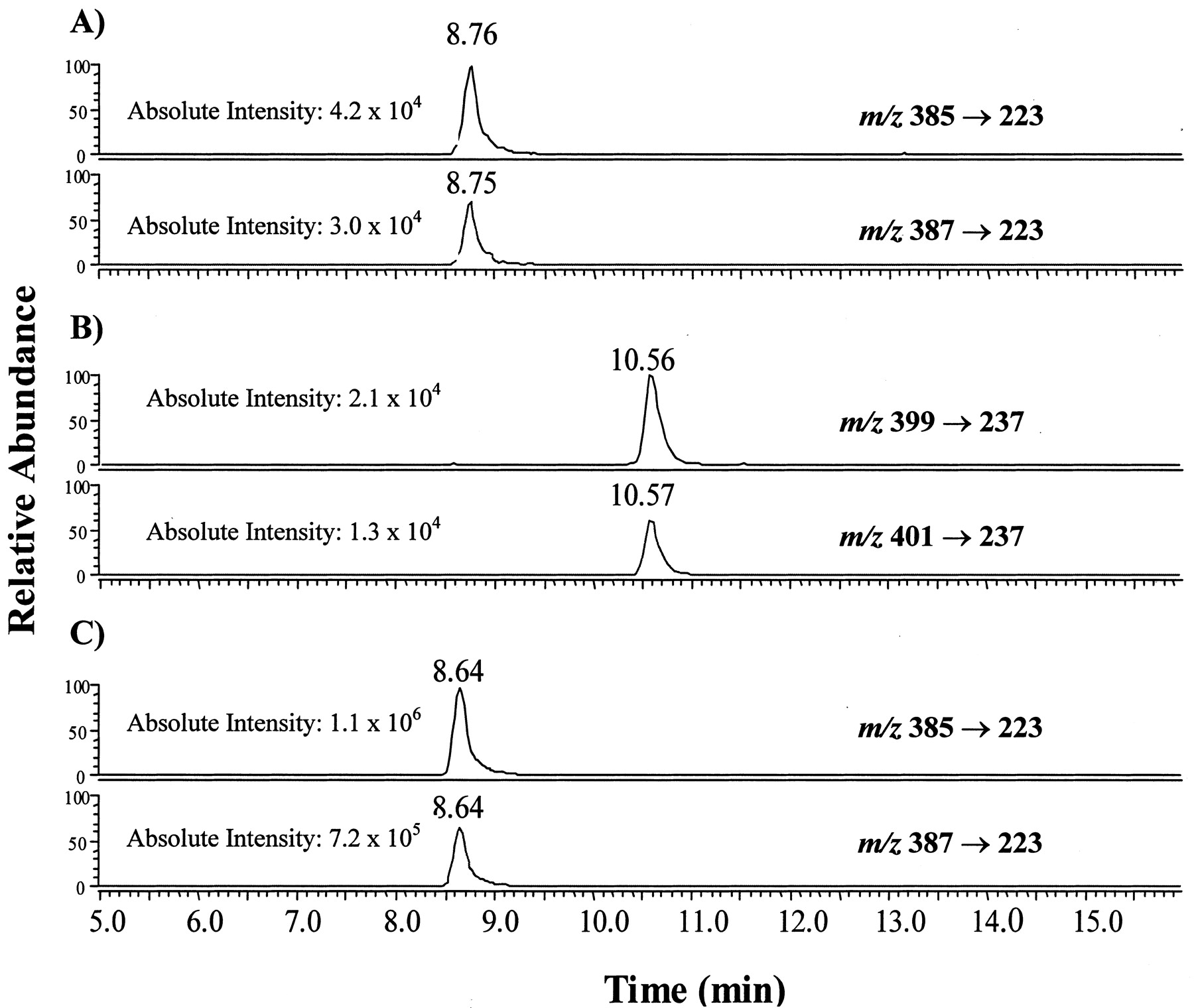
Comparison of LC/MS/MS chromatograms of TA-COOH and the methylated derivative, TA-COCH3.
A, TA-COOH generated from the incubation of P450 2B6, TA, and NADPH as described under Materials and Methods. Unambiguous detection of the metabolites was accomplished by monitoring the transition of the chlorine-containing precursor ions to the nonchlorinated product ion (m/z 223). B, methylated derivative of enzyme generated TA-COOH, TA-COCH3. SRM for the transitions of precursor 35Cl (m/z 399) and 37Cl (m/z 401) to the product ion (m/z 237) were monitored. C, TA-COOH synthetic standard. TA-COCH3, methyl ester of TA-COOH
Based on the finding of TA-COOH formation during the inactivation of CYP2B6 by TA, a partition ratio was calculated. At a saturating concentration of substrate (50 μM TA), an NADPH-dependent spectral decrease of 0.08 nmol/ml CYP2B6 was observed. In addition, a concentration of 0.121 nmol/ml TA-COOH was measured in the reaction supernatant. The resulting partition ratio of 1.5 nmol TA-COOH formed per nanomole of CYP2B6 inactivated suggests a highly efficient inactivation process.
Inhibitory Effects of TA on P450 Marker Activities in Human Liver Microsomes.
The effect of TA on human liver microsomal P450 2C9, 2C19, 2D6, and 3A4 enzyme activities was evaluated as a measure of the specificity of enzyme inactivation. TA-treated human liver microsomes showed 41% inhibition of P450 2B6 marker activity, 34% inhibition of 2C9 activity, and 45% inhibition of 2C19 activity. In contrast, only 9 and 0% inhibition were observed for 2D6 and 2E1 marker activities, respectively (Fig. 5). Finally, P450 3A4-mediated triazolam 1′-hydroxylation was reduced 9%, indicating minimal inhibition.

Irreversible inhibitory effect of TA on residual P450 marker activities in pooled human liver microsomes.
Aliquots were removed from the primary reaction mixture containing human liver microsomes and TA (50 μM) and assayed for residual P450 marker activities as described under Materials and Methods and Table 2. Representative P450 marker activities are listed as follows: P450 2B6, DCMB hydroxylation; P450 2C9, diclofenac 4′-hydroxylation; P450 2C19, mephenytoin 4′-hydroxylation; P450 2D6, dextromethorphan O-demethylation; P450 2E1, chlorzoxazone 6-hydroxylation; and P450 3A4, triazolam 1′-hydroxylation. Results are expressed as the percentage of the control velocities. Each value is the mean ± standard deviation of four different incubations.
Discussion
The use of P450 form-selective chemical inhibitors has greatly assisted the characterization of the catalytic specificities of individual P450 enzymes in animals and humans. Consequently, rich pharmacological and toxicological information pertaining to drug-drug interactions in clinical observations can be obtained. What we have demonstrated in this paper is a high affinity P450 2B6 inhibitor, which exhibited some degree of selectivity. However, the primary focus was to use TA to establish the mechanism and the partition ratio of inactivation. The characterization of numerous nonselective mechanism-based inhibitors of P450 enzymes has been reported (Foroozesh et al., 1997; Chun et al., 2000; Kent et al., 2002) as they have applications other than reaction phenotyping such as probes for the catalytic mechanism of cytochrome P450 and for identifying amino acid residues important to the function of the enzyme. A recent review of the literature has revealed that triethylenethiophosphoramide is a competitive inhibitor of P450 2B6 (Rae et al., 2002). This compound also demonstrated selectivity in that among the additional five human P450 forms evaluated, only 2B6-mediated activity was decreased ≥20% by the addition of 50 μM triethylenethiophosphoramide. However, to our knowledge, a mechanism-based inactivator specific for P450 2B6 has not been described. Such a compound would be useful as a biochemical probe in P450 reaction phenotyping experiments and evaluating structure-activity relationships for an enzyme in which steric factors have a major influence on substrate interactions and site of oxidation (Domanski et al., 1999). Toward this goal, an acetylene functional group was introduced at the anticipated site of P450 2B6-selective oxidation and the kinetics, mechanism, and selectivity of the enzyme-substrate interaction were characterized.
Several experimental approaches were used to demonstrate mechanism-based inactivation of P450 2B6 by TA. First, the loss of expressed P450 2B6 activity was shown to be concentration- and time-dependent and exhibited pseudo first-order saturation kinetics (Fig. 2A). Second, enzyme activity could not be restored upon removal of the inhibitor, suggesting covalent modification of the apoprotein and/or the prosthetic heme. The nearly identical decreases in P450 content and 2B6 enzyme activity after removal of TA suggested that heme destruction was the primary mode of inactivation. Furthermore, although TA inactivation of CYP2B6 produced a significant decrease in associated heme levels, efforts to identify a heme adduct using LC/MS/MS (time of flight) were unsuccessful. Third, coincubation of TA with excess 2B6 substrate, DCMB, slowed the rate of inactivation (Fig. 3A); however, glutathione had very little protective effect on enzyme inactivation (Fig. 3B) further suggesting that the reactive intermediate generated from the metabolism of TA was in close proximity to the active site of the enzyme.
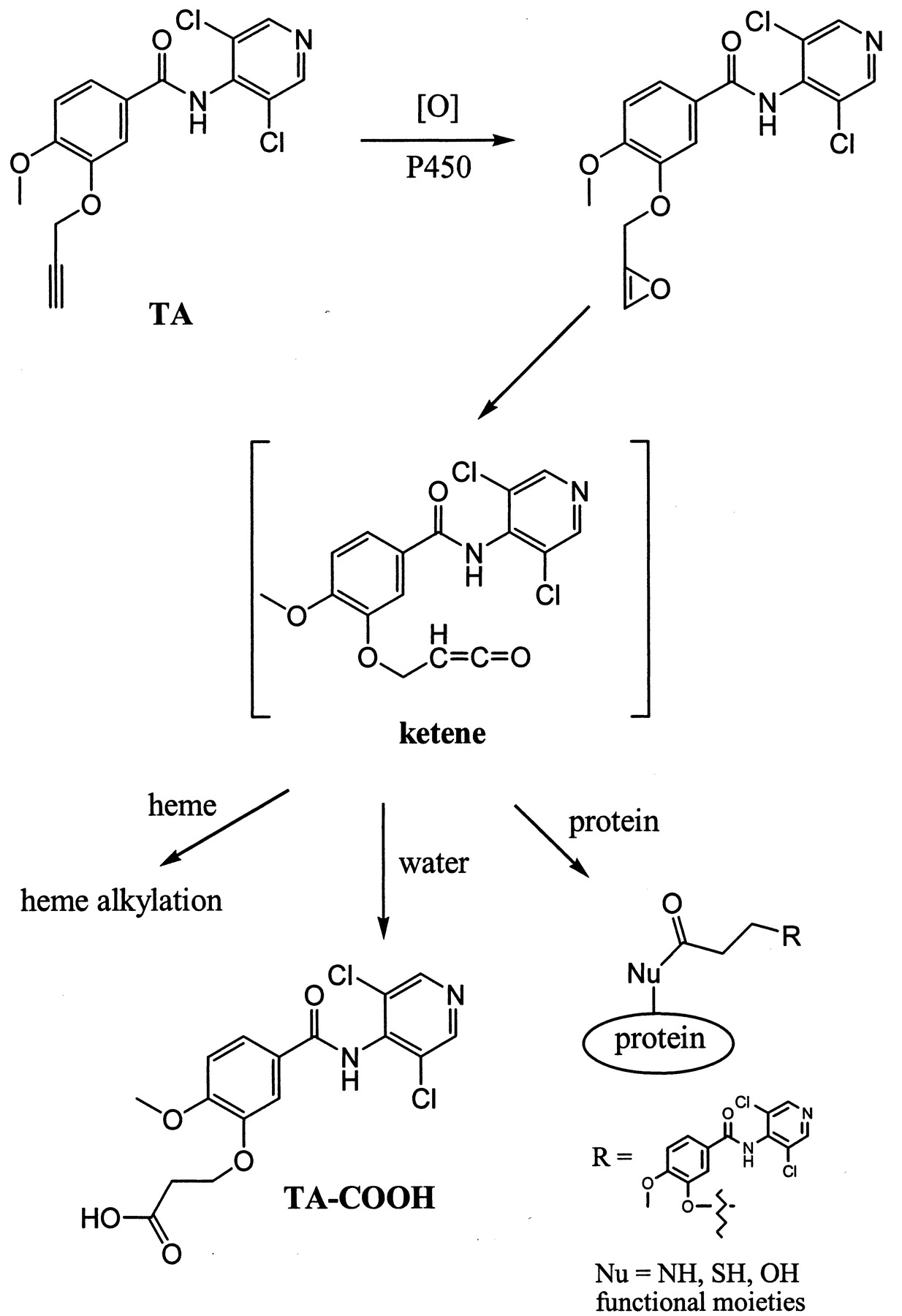
Ortiz de Montellano and others have demonstrated that terminal acetylenes can be metabolized by cytochrome P450 to form a reactive ketene intermediate by a 1,2-hydrogen shift (Ortiz de Montellano and Kunze, 1980; Ortiz de Montellano and Reich, 1984; Ortiz de Montellano and Komives, 1985). The ketene then proceeds to alkylate either the P450 heme moiety and/or apoprotein (Fig.6). Consequently, both P450 content and residual enzyme activity are irreversibly lost. Alternatively, excess ketene generated in the active site of the enzyme could react with water to form a carboxylic acid metabolite (Ortiz de Montellano and Komives, 1985; Foroozesh et al., 1997; Kent et al., 2002).
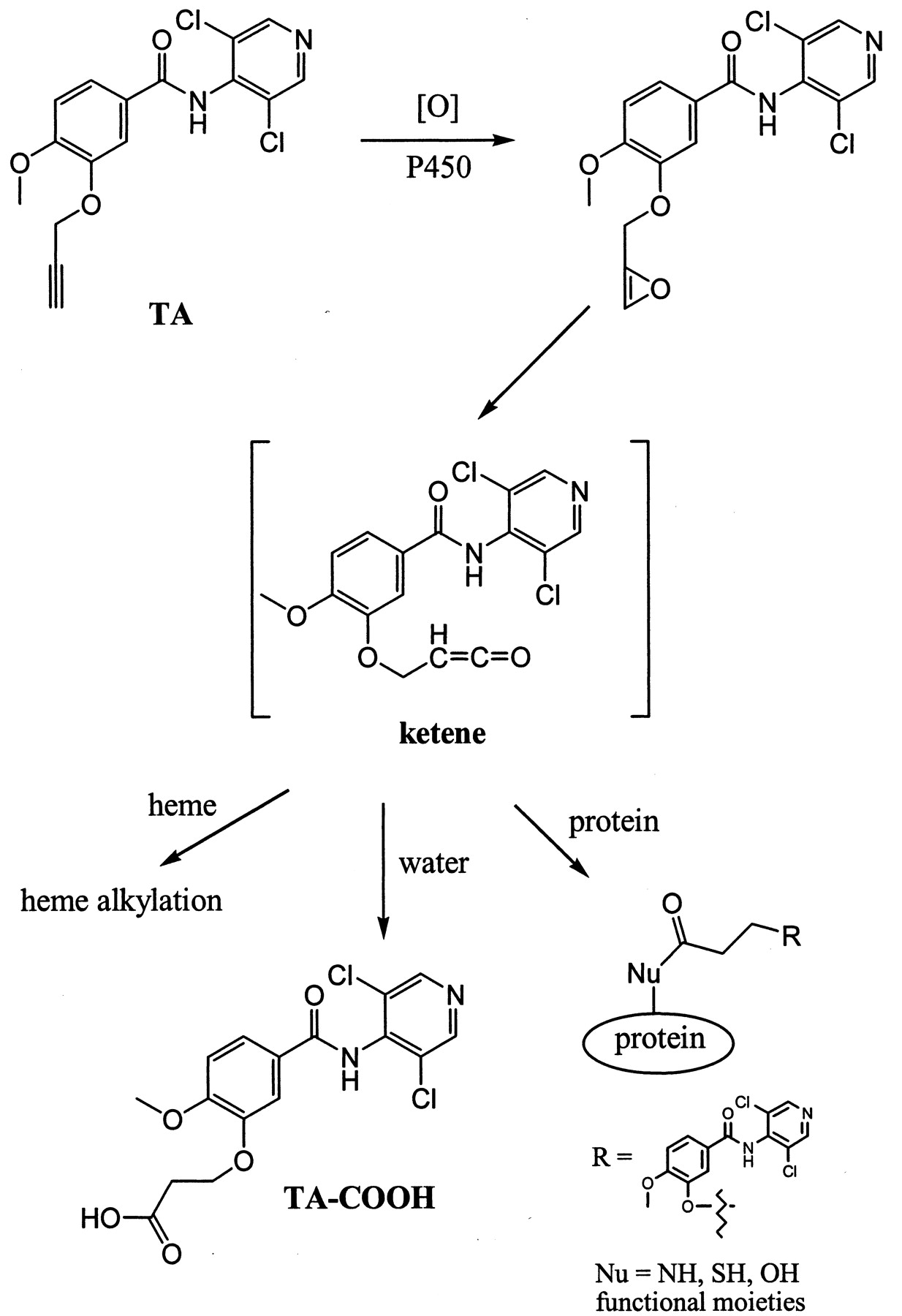
Proposed bioactivation mechanism of TA by cytochrome P450 and the fate of the reactive ketene intermediate.
The ketene could proceed to form heme or protein adducts or the carboxylic acid metabolite TA-COOH.
Typically, the stoichiometry of enzyme binding has been calculated based on nanomoles of inhibitor bound per nanomoles of P450 inactivated using radiolabeled mechanism-based inhibitors (Ortiz de Montellano, 1991; Chan et al., 1993). Numerous examples in the literature have also demonstrated 1:1 binding stoichiometry with most terminal acetylene inhibitors, in which 1 nmol of inhibitor inactivated 1 nmol of P450 (Ortiz de Montellano, 1991; Chan et al., 1993; Lin et al., 2002). Due to the lack of radiolabeled TA and purified 2B6 enzyme, the stoichiometry of binding was not measured. However, our studies did provide a measure of the efficiency of enzyme inactivation as defined by the partition ratio, with ketene formation as the rate-limiting step. Specifically, a partition ratio of 1.5 was determined by the amount of TA-COOH produced per inactivation event. This value is comparable with the ratio of 1 nmol of acid formed from 10-undecynoic acid per nanomole of lauric acid hydroxylase inactivated (CaJacob et al., 1988).
Despite the selectivity of P450 2B6 in the hydroxylation of the cyclopentyl derivative DCMB, the TA derivative was found to inactivate members of the P450 2C family in addition to 2B6. In contrast, human liver P450 2D6, 2E1, and 3A4 residual activities were not decreased to an appreciable extent. These results suggest that subtle changes in theO-alkyl chain of the parent, DCMB, are critical for the selectivity of enzyme inactivation. Several reports in the literature have shown that minor modifications of substrates can result in a dramatic change in inhibition potency as well as inactivation mechanism. Examples to date include the inactivation of rat liver P450 3A1 and 3A2 byN-(2-p-nitrophenethyl)dichloroacetamide but not by N-(2-phenethyl)dichloroacetamide (Stevens and Halpert, 1988). In addition, Foroozesh et al. (1997) have shown that by replacing the terminal hydrogen of aryl acetylenes with a methyl group, the resulting propynes from ethynes enhance the inhibition of P450 1A enzymes but decrease the inhibition of P450 2B-dependent dealkylations. In some instances, such a modification converted a reversible inhibitor of P450s into a suicide inhibitor.
Preliminary quantum chemical calculations of the energy of hydrogen abstraction at various sites on the DCMB molecule have been performed by our laboratory and indicate that trans-hydroxylation of the cyclopentyl group seems to be driven largely by steric rather than electronic factors. Thus the selectivity of 2B6 for DCMB likely requires orientation of the cyclopentyl group within the active site of 2B6 to allow access to the prosthetic heme (Domanski et al., 1999). We have demonstrated here that the terminal acetylene group in TA is able to reach the heme of the P450 by ultimately irreversibly reducing the P450 content in expressed 2B6. However, selectivity of inactivation is lost, possibly due to the free rotation about the methylene carbon adjacent to the acetylene functional group of TA. Consequently, the terminal acetylene moiety is able to affect 2C9 and 2C19 as well. TA does not, however, affect enzyme activities for P450 2D6, 2E1, or interestingly 3A4, a P450 often distinguished by broad substrate specificity. Recently, efforts have been made to generate quantitative structure-activity models to understand the substrate specificity for P450 2B6 in the absence of a crystal structure (Domanski et al., 1999;Ekins et al., 1999; Wang and Halpert, 2002). Domanski et al. (1999)have demonstrated that within the active site of 2B6, two amino acid residues, F107 and L363, have been identified as critical to the substrate specificity of 2B6, including DCMB hydroxylation. Such models may help to deduce the orientations and positions of substrate pharmacophores in the active site. Therefore future studies would involve a comprehensive structure-activity analysis by designing a set of more sterically hindered acetylene analogs to increase the selectivity of human P450 form inhibition.
Footnotes
- Abbreviations used are::
- P450
- cytochrome P450
- DCMB
- N-(3,5-dichloro-4-pyridyl)-3-(cyclopentyloxy)-4-methoxybenzamide
- TA
- N-(3,5-dichloro-4-pyridyl)-4-methoxy-3-(prop-2-ynyloxy)benzamide (terminal acetylene)
- DCMB-OH
- N-(3,5-dichloro-4-pyridyl)-3-(3-hydroxycyclopentyloxy)-4-methoxybenzamide (hydroxy metabolite of DCMB)
- TA-COOH
- N-(3,5-Dichloro-4-pyridyl)-3-(2-carboxyethoxy)-4-methoxybenzamide
- LC
- liquid chromatography
- MS/MS
- tandem mass spectrometry
- MS
- mass spectrometry
- kinactivation
- maximal rate of inactivation at saturation
- KI
- half-maximal inactivation
- SRM
- selected reaction monitoring
- Received June 13, 2002.
- Accepted September 25, 2002.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}