


Abstract
Ticlopidine is an agent that inhibits adenosine diphosphate-induced platelet aggregation. Metabolic studies with ticlopidine have indicated that the principal routes of metabolism are N-dealkylation, N-oxidation, and oxidation of the thiophene ring. However, ticlopidine shares some structural features that are similar to those of cyclic tertiary amines such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and tetrahydroisoquinolines, which are converted to neurotoxic pyridinium metabolites, via the iminium (dihydropyridinium) species. The current in vitro studies examined the potential of ticlopidine to undergo a similar conversion by cytochrome P450 (P450), peroxidases, and monoamine oxidase (MAO). The results from these studies have suggested that ticlopidine undergoes an overall 4-electron oxidation to the novel thienopyridinium metabolite (M6) via the intermediate 2-electron oxidation product, the thienodihydropyridinium metabolite (M5) by P450, horseradish peroxidase, and myeloperoxidase and, to a lesser extent, by MAO. The structures of these metabolites were characterized by liquid chromatography (LC)-tandem mass spectrometry and LC-NMR. Qualitative studies with baculovirus-expressed P450s revealed the involvement of P450 3A4 in this conversion. Interestingly, M5 was the primary metabolite in the peroxidase-mediated reactions and was quite stable to air oxidation or disproportionation. It was less electrophilic and did not form cyanide, glutathione, or N-acetylcysteine adducts. On the other hand, M6 was the major metabolite in P450-catalyzed oxidation of ticlopidine. The results from this study have revealed that in addition to metabolism of the thiophene ring of ticlopidine, the tetrahydropyridine moiety of the compound is susceptible to a 2-electron and a 4-electron oxidation like other cyclic tertiary amines.
Ticlopidine [5-(2-chlorophenyl)methyl-4,5,6,7-tetrahydrothieno[3,2-c] pyridine (1); Fig. 1] is a potent and long-acting inhibitor of platelet aggregation (Noble and Goa, 1996; Kam and Nethery, 2003). It inhibits ADP-induced platelet aggregation by a mechanism that appears to involve effects on the 2-methylthio-ADP-binding receptor. Although effective in preventing atherothrombotic events in cardiovascular, cerebrovascular, and peripheral vascular disease, administration of ticlopidine results in a relatively high incidence of hematological toxicities (Lesesve et al., 1994; Love et al., 1998) such as agranulocytosis (Ono et al., 1991), thrombotic thrombocytopenic purpura (Muzkat et al., 1998; Steinhubl et al., 1999), and aplastic anemia (Mataix et al., 1992; Ferrer et al., 1998). Recently, reports of ticlopidine-induced hepatotoxicity have also appeared (Grieco et al., 1998; Perez-Balsa et al., 1998; Zeolla and Carson, 1999). Ticlopidine is also a broad-spectrum inhibitor of P450,1 but at clinically relevant concentrations, it inhibits human P450 2C19 and 2D6 with a Ki of 1.2 and 3.4 μM, respectively (Ko et al., 2000). Recent studies have shown that ticlopidine is a selective mechanism-based inhibitor of P450 2C19 and, like tienilic acid, an electrophilic thiophene-S-oxide, is postulated as the reactive intermediate causing inactivation (Ha-Duong et al., 2001a,b). The mechanisms of hematological toxicity and hepatotoxicities caused by ticlopidine are unclear. However, it has been suggested that ticlopidine-induced agranulocytosis is caused either by direct cytotoxicity or via an immune-mediated reaction (Ono et al., 1991). Liu and Uetrecht (2000) have recently shown the involvement of a myeloperoxidase/H2O2/Cl– system and activated neutrophils in the metabolism of ticlopidine to a reactive intermediate, thiophene-S-chloride.
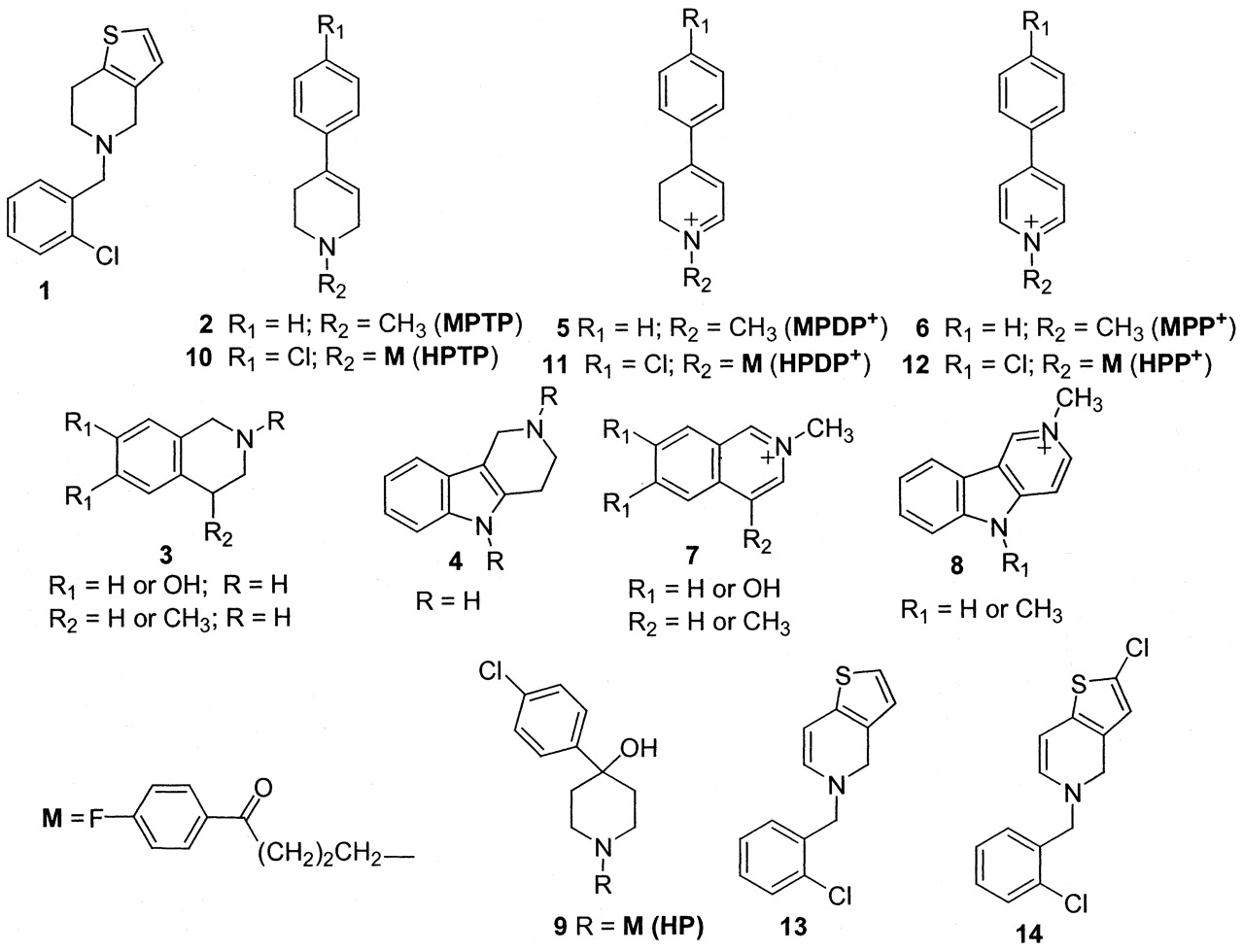
Structures of ticlopidine, MPTP, tetrahydroquinoline, and tetrahydro-β-carboline derivatives, haloperidol (HP), and their metabolites.
Ticlopidine shares some structural features that are similar to those of the parkinsonian-inducing agent 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine [MPTP (2), Fig. 1], and several naturally occurring or endogenously formed tetrahydroisoquinoline (3) and tetrahydro-β-carboline (4) derivatives. MPTP is activated by cytochrome P450 or monoamine oxidase B (MAO B) to an unstable dihydropyridinium intermediate, MPDP+ (5), which is further oxidized to the N-methyl pyridinium metabolite 1-methyl-4-phenylpyridinium (6) (Castagnoli et al., 1997). Tetrahydroisoquinoline and tetrahydro-β-carboline derivatives are also activated to the respective N-methyl-isoquinolinium (7) or N-methyl-β-carbolinium (8) metabolites (Castagnoli et al., 1997) by two metabolic steps, which involve N-methylation by a nonspecific N-methyltransferase and subsequent oxidation by MAO (Naoi et al., 1994). Besides P450, recent reports have also demonstrated the role of peroxidases in the conversion of cyclic tertiary amines like the neuroleptic agent, haloperidol (HP) (9;Fig. 1) and its tetrahydropyridine metabolite HPTP (10) to the pyridinium metabolite HPP+ (12) via the dihydropyridinium intermediate HPDP+ (11) (Igarashi et al., 1995, 2000; Usuki et al., 1996).
In vivo metabolic studies with ticlopidine have indicated that the principal routes of metabolism are N-dealkylation, N-oxidation, and oxidation of the thiophene ring (Tuong et al., 1981; Fraire, 1984; Desager, 1994; Noble and Goa, 1996). In vitro metabolism of ticlopidine by recombinant human P450 2C19 and 2D6 has recently led to the identification of thiophene-S-oxide dimer TSOD (M2) and 2-oxoticlopidine (M4) (see Fig. 2) (Ha-Duong et al., 2001a,b). Previous reports have also suggested that the myeloperoxidase (MPO)-catalyzed oxidation of ticlopidine yields dehydrogenated ticlopidine (13) and 2-chloroticlopidine (14) metabolites (Fig. 1) (Liu and Uetrecht, 2000). In view of the structural similarity of ticlopidine with other tertiary cyclic amines, we postulated that ticlopidine could also be oxidized to the corresponding dihydrothienopyridinium (M5) and thienopyridinium (M6) metabolites (Fig. 2). To our knowledge, the oxidation of ticlopidine to M5 or M6 has not been shown. These considerations prompted us to investigate the in vitro biotransformation of ticlopidine to M5 and M6 using the three common oxidative enzyme systems, P450, peroxidases, and MAO.

Proposed schematic representation for the metabolism of ticlopidine.
Materials and Methods
Materials. Ticlopidine hydrochloride, horseradish peroxidase (HRP; type VI-A, 1000 units/mg of protein), MPO (from human leukocytes, 50 units/mg), catalase (2950 units/mg), desferoxamine mesylate, glutathione, N-acetylcysteine, potassium cyanide (KCN), and hydrogen peroxide (30%) were obtained from Sigma-Aldrich (St. Louis, MO). NADPH was purchased from ICN Biomedicals, Inc. (Costa Mesa, CA). Human liver microsomes (pooled from 53 livers) were prepared at Pfizer. The protein concentration was 17.6 mg/ml and was determined using the BCA assay (Pierce Chemical, Rockford, IL). The P450 content in the pooled microsomes was 0.32 nmol/mg and was determined by the procedure described by Omura and Sato (1964). Individual recombinant P450s 1A2, 2C9, 2C19, 2D6, 3A4, and 3A5 from baculovirus-infected insect cells were obtained from PanVera Corp. (Madison, WI). Recombinant human MAO A and B expressed in yeast were acquired from Prof. Dale Edmondson (Emory University). The protein concentration was 14.2 mg/ml for MAO A and 33.2 mg/ml for MAO B. The specific activity of each isozyme was determined using kynuramine as a substrate and was 2.5 and 4.5 nmol/min/mg of protein for MAO A and B, respectively. Stock solutions of ticlopidine were prepared in a mixture of water and acetonitrile (60:40). All deuterated solvents were obtained from Cambridge Isotope Laboratories (Andover, Massachusetts). Other chemicals and reagents were of the highest quality available.
LC-MS/MS Analysis. LC-MS/MS analyses were performed on a chromatographic system consisting of a HP-1100 solvent delivery pump, a HP-1100 membrane degasser (Agilent Technologies, Palo Alto, CA.) connected to a PerkinElmer Sciex 3000 triple quadrupole mass spectrometer (PerkinElmer-Sciex Instruments, Boston, MA). The samples were injected via a LEAP autosampler (LEAP Technologies, Inc., Carrboro, NC). The separation of metabolites was achieved at ambient temperature on a kromasil C4 column (5 μm, 4.6 × 250 mm, MetaChem Technologies, Lake Forest, CA) by reversed phase chromatography. The mobile phase consisted of 5 mM ammonium formate (adjusted to pH 3 with formic acid; solvent A) and acetonitrile (solvent B) and was delivered at 0.9 ml/min. A gradient was used to separate the metabolites and ticlopidine. The initial composition of solvent B was maintained at 5% for 5 min and then increased in a linear manner as follows: 30% at 30 min; 90% at 33 min. It was then maintained at 90% for up to 38 min and then decreased to 5% in the next 5 min. The column was allowed to equilibrate at 5% solvent B for 5 min before the next injection. The postcolumn eluate was split such that 5% of the flow was introduced to the PerkinElmerSciex API 3000 triple quadrupole mass spectrometer. The HPLC effluent going to the mass spectrometer was directed to waste through a divert valve for the initial 5 min after sample injection. The mass spectrometer was equipped with a turbo ionspray source, maintained at 450°C, and operated at a 4.5-kV potential in the positive ionization mode, and the orifice voltage was 25 V. MS/MS studies were performed using nitrogen gas at collision energy of 40 V and collision gas thickness of 6 × 1014 molecules/cm2. The instrument parameters were determined to be optimum for ticlopidine. A full scan and precursor ion scan (m/z 125) were initially performed on all samples to identify drug-related peaks. Product ion spectra were then obtained by collision-induced dissociation of the precursor ions that were detected in the full scan or precursor ion scan. Subsequent experiments were performed using multiple-reaction monitoring to detect the specific m/z signals for ticlopidine and its metabolites. The transitions scanned were m/z 264 to 125, 262 to 125, 260 to 125, and 278 to 125 for ticlopidine, M5, M6, and M8, respectively. The hydrogen-deuterium exchange studies were performed using deuterated 10 mM ammonium formate in D2O (pH 4.0) and acetonitrile-d3 on a Bruker Daltonics Esquire 3000 ion trap mass spectrometer (Bruker Daltonics Inc., Billerica, MA) equipped with an electrospray ionization source in the positive ion mode. The HPLC system consisted of an HP-1100 binary pump (Agilent Technologies), a Bruker BSFU-0 injector and column oven (Bruker BioSpin GmbH, Rheinstetten, Germany). The metabolites were separated using similar gradient conditions, as described, on a kromasil C4 (150 × 4.6 mm, 5-μm; MetaChem Technologies Inc.). The retention times of M5, M6, ticlopidine, and M8 using this column were 29.0, 29.5, 31.0, and 40.6 min. The mass spectrometer was operated using optimized parameters. Data were then collected with total ion scanning, and the selected ions were extracted from the scan.
LC-NMR Analysis. The HPLC system was similar to that used in the hydrogen-deuterium exchange experiments (see above) and was interfaced with a Bruker Daltonics Esquire 3000 ion trap MS equipped with an electrospray source, a Bruker BPSU-36 peak storage unit, and a Bruker 500 MHz Avance DRX spectrometer equipped with a 4-mm 1H-13C inverse z-gradient LC flow probe. The NMR to MS split ratio was 20:1. Chromatographic separation was achieved on a Luna Hexyl-Phenyl column (3 × 150 mm, 5 μm; Phenomenex) at ambient temperature. A gradient mobile phase of (0.1% trifluoroacetic acid-d in D2O)-(acetonitrile-d3) 85:15 to 70:30 over 30 min at a flow rate of 0.5 ml/min was used to separate the metabolites. Five percent of the effluent was split off postcolumn and diluted with 9:1 acetonitrile-d3:D2O containing 0.2% acetic acid-d4 at a flow rate of 125 μl/min before entering the MS. The remaining 95% passed through the diode array detector, and the peaks of interest were stored in the loop storage unit using the loop storage technique and subsequently introduced into the NMR spectrometer. 1H NMR spectra were obtained at 298 K on the LC peaks using one-dimensional nuclear Overhauser effect spectroscopy -type double presaturation of solvent NMR resonances. COSY spectra were obtained using double presaturation of solvent NMR resonances. All chemical shifts are reported in ppm downfield from tetramethylsilane as referenced to acetonitrile-d3 at 1.94 ppm.
Incubations with Human Liver Microsomes. Incubations (total volume 500 μl) were carried out at 37°C for 30 min and contained 10 mM magnesium chloride, desferoxamine mesylate (10 μM), microsomal protein (0.53 mg/ml), ticlopidine (5 or 50 μM), and 2.4 mM NADPH (prepared daily) in 100 mM potassium phosphate buffer (pH 7.4). Reactions were terminated with 500 μl of acetonitrile. The mixture was vortexed and centrifuged to remove precipitated proteins. The resulting supernatant was used for all subsequent mass spectral analyses. Incubations that lacked NADPH and microsomes served as negative controls.
Incubations with Microsomes Containing cDNA-Expressed Enzymes. Incubations with P450 3A4, 2C9, 2C19, 1A2, and 2D6 were performed using baculovirus-expressed enzymes. The incubations were prepared and conducted as described above for human liver microsomes, except that in each case ∼50 pmol/ml of enzyme was used.
Incubations with HRP. HRP incubations (total volume 500 μl) were carried out at 37°C and contained ticlopidine (5 or 50 μM), desferoxamine (10 μM), and HRP (3 units/ml) in 100 mM phosphate buffer (pH 7.0). The reaction was initiated by addition of H2O2 (500 μM), and after 45 min, the reactions were treated with catalase (300 units). The mixture was incubated for an additional 3 min and terminated by addition of acetonitrile (500 μl). The precipitated proteins were vortexed and centrifuged, and the resulting supernatant was used for all subsequent mass spectral analyses. Incubations that lacked HRP or H2O2 served as negative controls.
The effect of peroxidase inhibitors on the oxidation of ticlopidine was investigated by preincubating the reaction with ascorbic acid and sodium azide (final concentration, 1 mM) before addition of ticlopidine and H2O2. The concentrations of reagents and the conditions for the incubation were similar to those described previously. The importance of H2O2 in the oxidation of ticlopidine by HRP was investigated by adding 300 units of catalase to the incubation mixture before addition of ticlopidine and initiation of the reaction by hydrogen peroxide. All incubations were worked up as described above.
Incubations with MPO. MPO-mediated incubations (total volume 500 μl) were carried out using 0.5 unit/ml MPO in 100 mM phosphate buffer (pH 7.0) in the presence or absence of 100 mM KCl. All other conditions and reagents were similar to those described for HRP-mediated oxidations. The effect of inhibitors (ascorbic acid and sodium azide) on MPO-mediated oxidation of ticlopidine was investigated using the conditions described above.
Incubations Of Ticlopidine with Human Liver Microsomes, HRP, or MPO in the Presence Of Glutathione,N-Acetylcysteine, or KCN. Incubations containing ticlopidine (50 μM) were supplemented with glutathione (5 mM), N-acetylcysteine (5 mM), or KCN (0.5 mM) in 100 mM phosphate buffer at 37°C for 45 min. The remaining conditions and reagents were similar to those described in human liver microsomes, and HRP- and MPO-mediated oxidations. The reactions were terminated with acetonitrile. Some incubations were treated with KCN (1 mM) after the reaction was terminated with acetonitrile. All samples were vortexed and centrifuged, and the supernatants were used for subsequent mass spectral analyses.
Incubations with Recombinant Human MAO A and B. Ticlopidine (50 μM) was incubated with recombinant human MAO A (0.36 mg/ml) and B (0.83 mg/ml) in 100 mM potassium phosphate buffer (pH = 7.4) for 45 min at 37°C (final volume 500 μl). The incubation was then terminated with acetonitrile (500 μl), and the reaction was vortexed and centrifuged. The resulting supernatant was used for all subsequent mass spectral analyses.
Results
Metabolism of Ticlopidine. All incubations were conducted at ticlopidine concentrations of 5 and 50 μM. The 5 μM concentration of ticlopidine was selected to mimic the clinically relevant concentrations of ticlopidine following a multiple dose administration (Cmax values for ticlopidine following a multiple dose administration for 21 days are 890-1420 ng/ml, 3.4–5.4 μM) (Noble and Goa, 1996), whereas the 50 μM concentration was used to identify all possible metabolites produced in vitro. Metabolic profiles at 5 and 50 μM concentrations were similar to each other, although the relative proportions of each metabolite differed (data not shown). Total ion mass chromatograms (LC-MS/MS) of the products obtained following incubation of 5 μM ticlopidine after incubations with various enzyme systems are shown in Fig. 3.

Total ion chromatograms of the products obtained after incubation of 5 μM ticlopidine with human liver microsomes (protein concentration 0.5 mg/ml) (A), recombinant P450 3A4 (50 pmol) (B), recombinant P450 2C19 (50 pmol) (C), MPO (0.5 unit/ml) (D), HRP (3 units/ml) (E), and MAO B (0.83 mg/ml; the results with MAO A were similar) (F).
M2, TSOD; M3, hydroxyticlopidine; M4, 2-oxoticlopidine; M5, dihydrothienopyridinium metabolite; M6, thienopyridinium metabolite; M7, ticlopidine N-oxide; M8, lactam metabolite; M9, dihydroxylated ticlopidine. Samples were incubated at 37°C for 30 min (for human liver microsomes and recombinant P450s) and 45 min for (MPO and MAO) and analyzed by LC-MS and LC-MS/MS as described under Materials and Methods.
Incubation of ticlopidine with human liver microsomes resulted in several products. LC-MS/MS analysis showed four main peaks, M2, M4, M6, and M5, at 20.5, 31.5, 32.8, and 33.6 min, respectively, that gave protonated molecular ions at m/z 559, 280, 260, and 262 (Fig. 3A). The remaining three metabolites, M3, M7, and M8, exhibited protonated molecular ions at m/z 280 (for M3 and M7) and 278 (for M8), respectively. A proposed scheme for the metabolism of ticlopidine is presented in Fig. 2. The molecular ions and the product ion mass spectra (not shown) of M2 and M4 were consistent with TSOD and 2-oxoticlopidine metabolites that were previously reported (Ha-Duong et al., 2001a,b). The dimer TSOD was presumably formed from the thiophene-S-oxide as described previously (Ha-Duong et al., 2001a,b). Metabolites M3 and M7 were tentatively identified as the hydroxyticlopidine and ticlopidine N-oxide. Treatment of the incubation mixture with titanium trichloride (TiCl3) resulted in reduction of both TSOD and ticlopidine N-oxide, further confirming their identity (data not shown). Previous reports have shown that N-oxides can be reduced by TiCl3 to their corresponding amines and thiols (Offen et al., 1985). The characterization of the three new metabolites (M5, M6, and M8) was done with additional LC-MS/MS and LC-NMR analysis as described below.
Five major cDNA-expressed human P450 enzymes (P450 3A4, 2C9, 2C19, 2D6, and 1A2) were examined for their ability to metabolize ticlopidine to M5 and M6. Of the isozymes studied, P450 3A4 and 2C19 converted ticlopidine to M6 (Fig. 3, B and C), whereas M5 was a minor component in these incubations. All other isozymes also generated M5 and M6, but to a lesser extent. Metabolite M8 was observed, as well, but only in P450 3A4 incubations. P450 2C19 also formed TSOD in significant amounts, and this was consistent with the previous reports (Ha-Duong et al., 2001a,b).
Metabolism of ticlopidine was HRP-, MPO-, and H2O2-dependent. Addition of ascorbic acid and sodium azide as inhibitors of peroxidases (1 mM final concentrations) or catalase, which catalyzes the breakdown of hydrogen peroxide, also resulted in inhibition of metabolism (data not shown). The addition of desferoxamine mesylate, an iron chelator, to the incubation mixtures did not inhibit ticlopidine metabolism, suggesting that the reaction was enzyme-mediated. Incubations with MPO were conducted in the presence and absence of potassium chloride (100 mM). The lack of chloride ions did not prevent the formation of the metabolites. Total ion chromatograms of the incubation mixtures revealed that MPO and HRP oxidized ticlopidine primarily to M5, and M6 was detected as a minor component (Figs. 3D and 2E). The relative proportions of M5 and M6 in all three systems (MPO with or without Cl– ions/H2O2 and HRP/H2O2) were similar. Metabolite M7 (m/z 280), detected in these incubation mixtures, was also present in the controls containing H2O2 but lacking the enzyme. This was attributed to chemical oxidation of ticlopidine by H2O2 to the N-oxide. The absence of M5 or M6 in the incubation mixtures lacking the enzyme suggested that these metabolites were not derived from M7.
Incubations with recombinant human MAO A and B expressed in yeast were carried out to investigate the ability of these enzymes to oxidize ticlopidine to M5 and M6. Although these enzymes were able to catalyze the formation of M5 (Fig. 3F), there appeared to be substantially less activity, suggesting that these enzymes do not play a significant role in ticlopidine metabolism in vivo.
Characterization of M5, M6, and M8 by LC-MS/MS and LC-NMR. The signals of M5 and M6 at m/z 262 and 260, respectively, were 2 and 4 amu less than m/z 264 (the protonated molecular ion of ticlopidine). The product ion mass spectra of m/z 260 and 262 gave one common fragment ion at m/z 125 corresponding to the chlorotropyllium ion, which was similar to the product ion in the mass spectrum of ticlopidine (Fig. 4). Treatment of the incubation mixture with sodium borohydride also resulted in the disappearance of these signals (data not shown). Based on these results, M5 and M6 were tentatively identified as the thienodihydropyridinium and thienopyridinium metabolites, respectively. The signal of M8 at m/z 278 was 14 amu greater than at m/z 264. The product ion mass spectrum of m/z 278 also gave one fragment ion at m/z 125 (not shown). However, sodium borohydride treatment did not alter the retention time or the mass of this peak, suggesting that M8 was a lactam analog of ticlopidine. Additional confirmation of the structures of M5, M6, and M8 was achieved using deuterium-hydrogen exchange and LC-MS analysis (see “LC-MS/MS Analysis” under Materials and Methods). With deuterated solvents, ticlopidine gave a deuterated molecular ion (M + D+) at m/z 265. Mass spectral analysis of M5, M6, and M8 (generated using 50 μM ticlopidine and P450 3A4, HRP, and MPO) using the same conditions revealed deuterated molecular ions (M + D+) at m/z 262, 260, and 279 for M5, M6, and M8, respectively (Fig. 5). The observation that there was no deuterium exchange in M5 and M6 was consistent with the thienodihydropyridinium and thienopyridinium structure assignments for these two metabolites.

Product ion mass spectrum of ticlopidine at m/z 264 (M + H+).
The product ion mass spectra of M5, M6, and M8 were similar to the product ion mass spectrum of ticlopidine and showed one fragment ion at m/z 125 that corresponded to chlorotropyllium ion.

Extracted ion chromatograms of M6 = 260 (29 min); M5 = 262 (29.5 min); ticlopidine = 265 [M + D+] (31 min) and M8 = 279 [M + D+] (40.8 min) after ticlopidine incubation with P450 3A4, MPO, or HRP following the hydrogen-deuterium exchange experiment.
HPLC analysis was performed using 10 mM deuterated ammonium formate, pH 4.0, and acetonitrile-d3 using a kromasil C4 column (150 × 4.6 mm, 5 μm); the gradient conditions were described in “LC/MS Analysis” under Materials and Methods.
The structures of the three metabolites were further verified by LC-NMR analysis. The metabolites were separated using HPLC and the gradient conditions described under Materials and Methods (“LC-NMR Analysis”). Under these conditions M6, M5, ticlopidine, and M8 eluted at 18.7, 20.3, 22.0, and 53 min, respectively. The 1H NMR spectrum of ticlopidine indicated resonances between 3.15 and 4.59 ppm corresponding to the methylene protons at positions 1, 2, 3, and 4 (see Table 1). The resonances between 6.76 and 7.56 ppm in the aromatic region were assigned to the protons on the chlorophenyl and thiophene rings. The 1H NMR spectrum of M5 showed the disappearance of signals between 4.25 and 4.59 ppm corresponding to the protons at positions 3 and 4 (for ticlopidine) and the appearance of a singlet at 8.8 ppm (1H) and 5.15 ppm (2H). This was consistent with the iminium proton (position 3) and the two benzylic protons (position 4). The two triplets at 3.31 and 3.96 ppm (2H each) were assigned to the methylene protons on positions 1 and 2, respectively (Table 1). These data were consistent with the dihydrothienopyridinium structure for M5. The correlations among the protons in the COSY spectrum (Fig. 6) were also in agreement with the proton assignments for the dihydrothienopyridinium structure. The 1H NMR spectrum of M6 showed the disappearance of all methylene resonances between 3.15 and 4.59 ppm and the appearance of doublets at 8.49 and 8.51 ppm (1H each), corresponding to proton resonances at positions 1 and 2, and singlets at 9.37 ppm (1H) and 5.91 ppm (2H), corresponding to the protons at positions 3 and 4, respectively, which were consistent with the thienopyridinium structure. These chemical shift assignments also matched those previously observed in the 1H NMR spectrum of the synthetic thienopyridinium derivative (Maffrand and Biogegrain, 1979). The 1H NMR spectrum of M8 showed triplets at 3.03 and 3.58 ppm (2H) corresponding to the methylene protons on positions 1 and 2 and a singlet at 4.73 ppm (2H) corresponding to the benzylic protons on position 4 (Table 1). The disappearance of resonances of the protons at position 3 in the ticlopidine spectrum was consistent with the structure of M8 as a lactam. The resonances of the aromatic protons (the protons on the chlorophenyl ring and the thiophene) in the spectra of the three metabolites were similar to those in the spectrum of ticlopidine and appeared in the aromatic region (∼7.28–7.58), as shown in Table 1.
1H NMR chemical shift (δ) assignments for ticlopidine, M5, M6, and M8 after an HPLC-NMR analysis

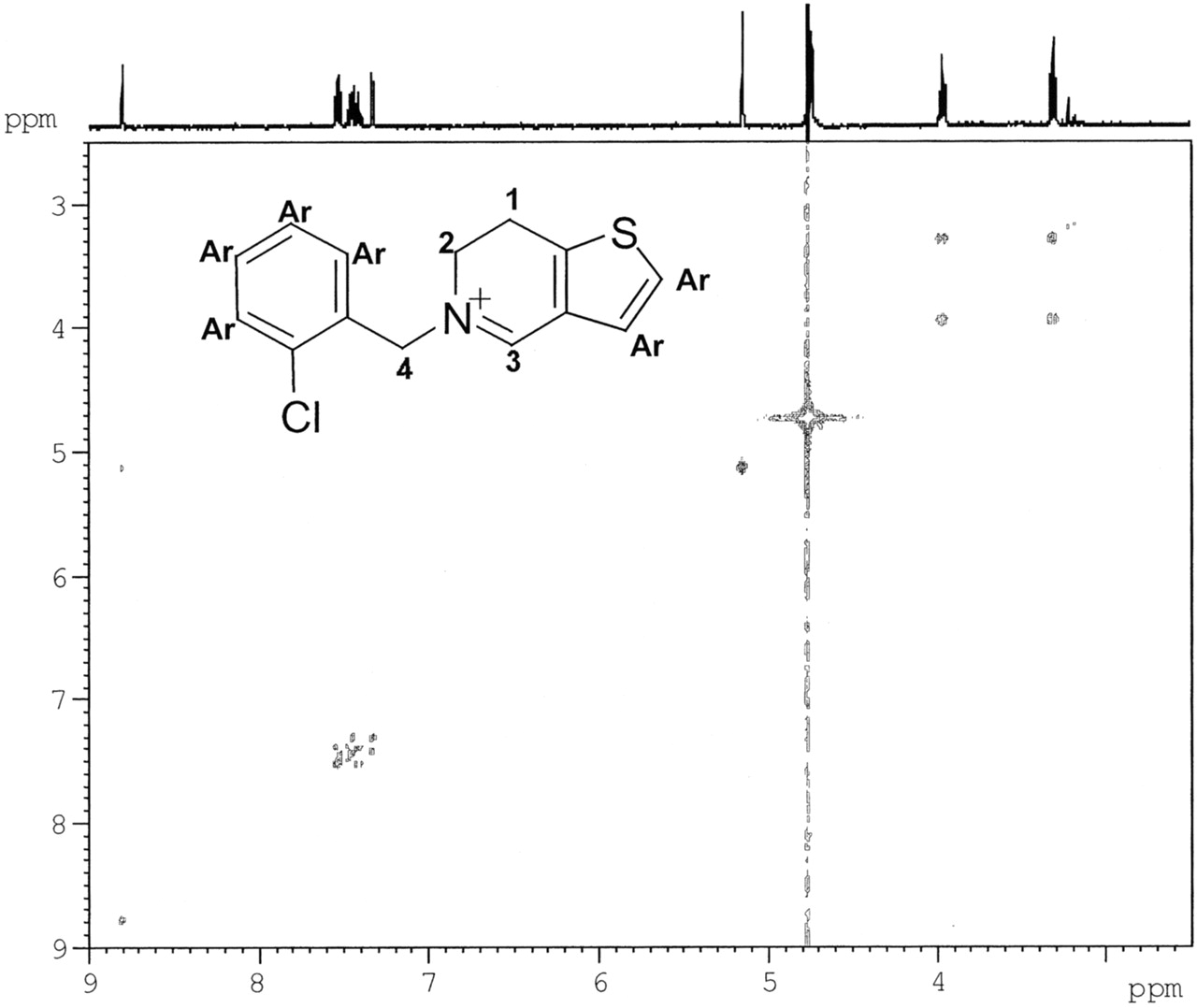
Two-dimensional COSY spectrum of the dihydropyridinium metabolite (M5).
Discussion
The evidence provided here indicates that ticlopidine is metabolized in vitro to dihydrothienopyridinum (M5) and thienopyridinium (M6) metabolites, and this is catalyzed by P450 3A4 as well as by peroxidases including MPO and HRP. Interestingly, our results showed that P450 2C19 is also involved in the oxidation of the tetrahydropyridine moiety of ticlopidine, in addition to the thiophene ring. In contrast, ticlopidine is a weak substrate of MAO A and B. This result was not surprising since it is well documented that compounds with large N-substituents are weak MAO substrates (Youngster et al., 1987). Also, N-methyl tetrahydroisoquinoline and tetrahydro-β-carboline derivatives exhibit poor MAO substrate properties (Naoi et al., 1989; Maret et al., 1990).
Previous studies on MPO-mediated ticlopidine oxidation by Liu and Uetrecht (2000) have also shown that its metabolism is extensive in the presence of MPO, but the extent of oxidation is decreased in the absence of the chloride ions. As mentioned in the introduction, dehydrogenated ticlopidine and 2-chloroticlopidine have been detected in these studies, in addition to the putative reactive metabolite ticlopidine-S-chloride. In our studies we found that the metabolism of ticlopidine was independent of the chloride ions and that the product profiles obtained with and without chloride ions was similar, resulting in formation of M5 as the primary metabolite. Although the reason for this difference is unknown, one possibility could be the differences in the pH of the incubation medium in the two studies (incubations were carried out at a pH of 6 in the previous studies). The effect of pH on the metabolism of compounds by MPO in the presence of chloride ions has been previously reported (Masuda et al., 2001). Also, the metabolism of compounds by the MPO/H2O2 system in the absence of the chloride ions is known (Iverson et al., 2002).
The difference between the peroxidases and the P450 incubations is that ticlopidine undergoes an overall 4-electron oxidation, resulting in the formation of M6 by P450, whereas M5 (an intermediate 2-electron oxidation product) is the primary metabolite in the peroxidase-mediated incubations. The catalytic pathway proposed for the oxidation of ticlopidine to M5 is shown in Fig. 7A. In P450-catalyzed reactions, this may proceed either by initial single-electron transfer (SET) chemistry, via the aminium radical cation (15), or by a direct hydrogen atom transfer (HAT) step, ultimately resulting in the carbinolamine (17), which is in equilibrium with iminium species, M5 (Sono et al., 1996). In the case of ticlopidine, this equilibrium is shifted toward M5 due to stabilization (as shown in Fig. 7A). Since peroxidases oxidize amine substrates by a SET process (Guengerich et al., 1995; Shaffer et al., 2001), the generation of M5 possibly derives directly by spontaneous dismutation of the cation radical 15 (Guengerich et al., 1995).

Proposed mechanism for oxidation of ticlopidine to M5 and M6 (A) and proposed mechanism for disproportionation of dihydrothienopyridinium metabolite M5 to the thienopyridinium M6.
The formation of M6 from M5 by P450 presumably involves a HAT process and may proceed via formation of intermediates 18, 19, and 20 as shown in Fig. 7A. Alternatively, it may be argued that 20 can be formed via the neutral dihydropyridine 21, which is in equilibrium with M5, through the HAT or SET process as described in the formation of M5. The formation of relatively minor amounts of M6 in the peroxidase-mediated reactions compared with P450-mediated reactions is probably attributed to the inability of peroxidases to oxidize substrates via the HAT process. The lactam (M8) detected in the human liver microsomes and recombinant P450 3A4-mediated oxidations is probably the oxidation product of carbinolamine intermediate (17). Previous reports have shown the involvement of aldehyde oxidase, a cytosolic enzyme, in the conversion of a dihydropyridinium metabolite to a lactam (Yoshihara and Ohta, 1998). However, our studies were conducted using recombinant P450s and isolated peroxidases. Hence, the involvement of aldehyde oxidase in the formation of M8 was ruled out.
An alternative possibility for the formation of M6 is by auto-oxidation or chemical disproportionation. Studies with other dihydropyridinium compounds have shown that these molecules undergo spontaneous (concentration-dependent) disproportionation to stoichiometric amounts of the pyridinium and tetrahydropyridine species at pH 7.4 (Peterson et al., 1985). Ticlopidine can undergo a similar process, presumably via a mechanism that appears to involve the conversion of M5 to a free base (21), followed by a hydride transfer to another molecule of M5, resulting in equimolar amounts of ticlopidine and M6 (Fig. 7B). However, since the peroxidase-mediated oxidations yielded very little amounts of M6, this possibility seemed highly unlikely.
The stability of the dihydrothienopyridinium metabolite M5 is quite intriguing and is attributed to the possible delocalization of the positive charge on the nitrogen (Fig. 7A). This stabilization could possibly decrease the electrophilicity of M5, thus making it less reactive toward nucleophilic attack. Alternatively, nucleophiles could add only reversibly to M5. Attempts to trap M5 with cyanide or other nucleophiles such as glutathione or N-acetylcysteine proved unsuccessful, even though cyanide additions to MPDP+ or 3,3-MPDP+ to form the respective cyanide adducts have been reported (Gessner et al., 1985; Peterson et al., 1985; Sayre et al., 1986). Furthermore, stability studies with M5 (generated in large amounts by incubating ticlopidine with the HRP/H2O2 system) at a pH of 7.4 showed that the metabolite was stable to auto-oxidation or disproportionation at this pH (data not shown). One possible interpretation for this enhanced stability toward disproportionation could be the formation of an energetically unfavorable 1,2-dihydropyridine intermediate (21), which may result from the loss of aromaticity of the thiophene ring.
Although the results presented here have shown that M5 is less reactive to various nucleophilic agents, it could induce oxygen radical formation, as shown in the case of MPDP+ (Adams et al., 1993). Also, extensive evidence documents that the neurotoxic effects of the above-mentioned tetrahydropyridines and tetrahydroquinolines are mediated by their pyridinium and isoquinolinium metabolites (Castagnoli et al., 1997). With this in mind, one could speculate that ticlopidine-induced toxicity could be a result of the conversion of ticlopidine to M5 and M6.
In summary, the results of this study have demonstrated that ticlopidine is metabolized to the dihydrothienopyridinium and thienopyridinium species by P450s and MPO, in addition to metabolism of the thiophene ring of ticlopidine. Furthermore, the LC-MS and LC-MS/MS analysis provides evidence that the dihydrothienopyridinium metabolite is the predominant species in the MPO/H2O2 system, whereas it is oxidized further to the thienopyridinium metabolite in P450-mediated reactions. Although the toxic properties of dihydropyridinium and pyridinium metabolites of various tetrahydropyridines are well precedented, the role of M5 and M6 in the ticlopidine-induced toxicities remains to be investigated.
Acknowledgments
We thank Drs. Alfin Vaz, R. Scott Obach, and Amit Kalgutkar for their helpful suggestions during the preparation of the manuscript.
Footnotes
-
↵1 Abbreviations used are: P450, cytochrome P450; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MAO, monoamine oxidase; MPDP+, 1-methyl-4-phenyl-2,3-dihydropyridinium; TSOD, ticlopidine-S-oxide dimer; MPO, myeloperoxidase; HRP, horseradish peroxidase; KCN, potassium cyanide; LC-MS/MS, liquid chromatography-tandem mass spectrometry; HPLC, high-performance liquid chromatography; COSY, correlation spectroscopy; SET, single-electron transfer; HAT, hydrogen atom transfer.
- Received August 1, 2003.
- Accepted September 2, 2003.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}