


Characterization of Primary Metabolic Pathways and of Cytochrome P450 Isozymes involved
Abstract
Biotransformation of cerivastatin, a new cholesterol-lowering drug, by human liver microsomes was investigated using the14C-labeled drug. Metabolite profiles were established by HPLC and structures of metabolites were elucidated. Two metabolic pathways were equally important, demethylation of the benzylic methyl ether and hydroxylation at one methyl group of the 6-isopropyl substituent. The product of combined hydroxylation and demethylation was observed as a minor metabolite. During sample preparation the lactone forms of both primary metabolites were isolated in small amounts. Detailed structural analysis by NMR and LC-ESI-MS showed that hydroxylation occurred with high regio- and stereoselectivity. The proposed structures were confirmed by chemical synthesis of enantiomerically pure reference compounds. Microsomes from a human lymphoblastoid AHH-1 cell line, stably expressing CYP 3A4, catalyzed the demethylation reaction. Upon incubation of cerivastatin with human liver microsomes in the presence of the specific CYP 3A inhibitor TAO, both hydroxylation and demethylation were considerably reduced. This indicates that CYP 3A enzymes play a major role in cerivastatin metabolism.
Cerivastatin, sodium (E)-(+)-(3R,5S)-7-[4-(4-fluorophenyl)-2, 6-diisopropyl-5-(methoxymethyl)-pyrid-3-yl]-3,5-dihydroxyhept-6-enoate (BAY w 6228), is a highly potent inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA)1 reductase (EC 1.1.1.3.4) and is currently under clinical development for the treatment of hypercholesterolemia (1).
This paper describes detailed structural analysis as well as independent chemical synthesis of cerivastatin metabolites formedin vitro by human liver microsomes. Furthermore, cytochrome P450 3A4 (CYP3A4)1 is identified as a primary enzyme involved in oxidative cerivastatin metabolism.
Materials and Methods
Radiolabeled Compound and Reagents.
For in vitro metabolism studies, sodium (E)-(+)-(3R,5S)-7-[4-(4-fluorophenyl)-2,6-diisopropyl-5-methoxymethyl-pyrid-3-yl]-3,5-dihydroxy-[7-14C]hept-6-enoate ([14C]cerivastatin)2 with a specific activity of 2.2 MBq/mg was used. The radiochemical purity was at least 95% when determined by HPLC.
All reagents and chemicals used for analytical procedures were of analytical or HPLC grade and purchased from E. Merck, Darmstadt, Germany. All reagents and chemicals used for chemical synthesis were of synthetic grade and, with the exception of potassium monomethyl malonate (Fluka), methyl isobutanoylacetate (Wacker Chemie), and (R)-5-carbomethoxy-4-tert.butyldimethylsilyloxy-2-oxopentylphosphonate9 (Ube Industries Ltd.), purchased from Aldrich-Chemie, Steinheim, Germany.
Chemical Syntheses..
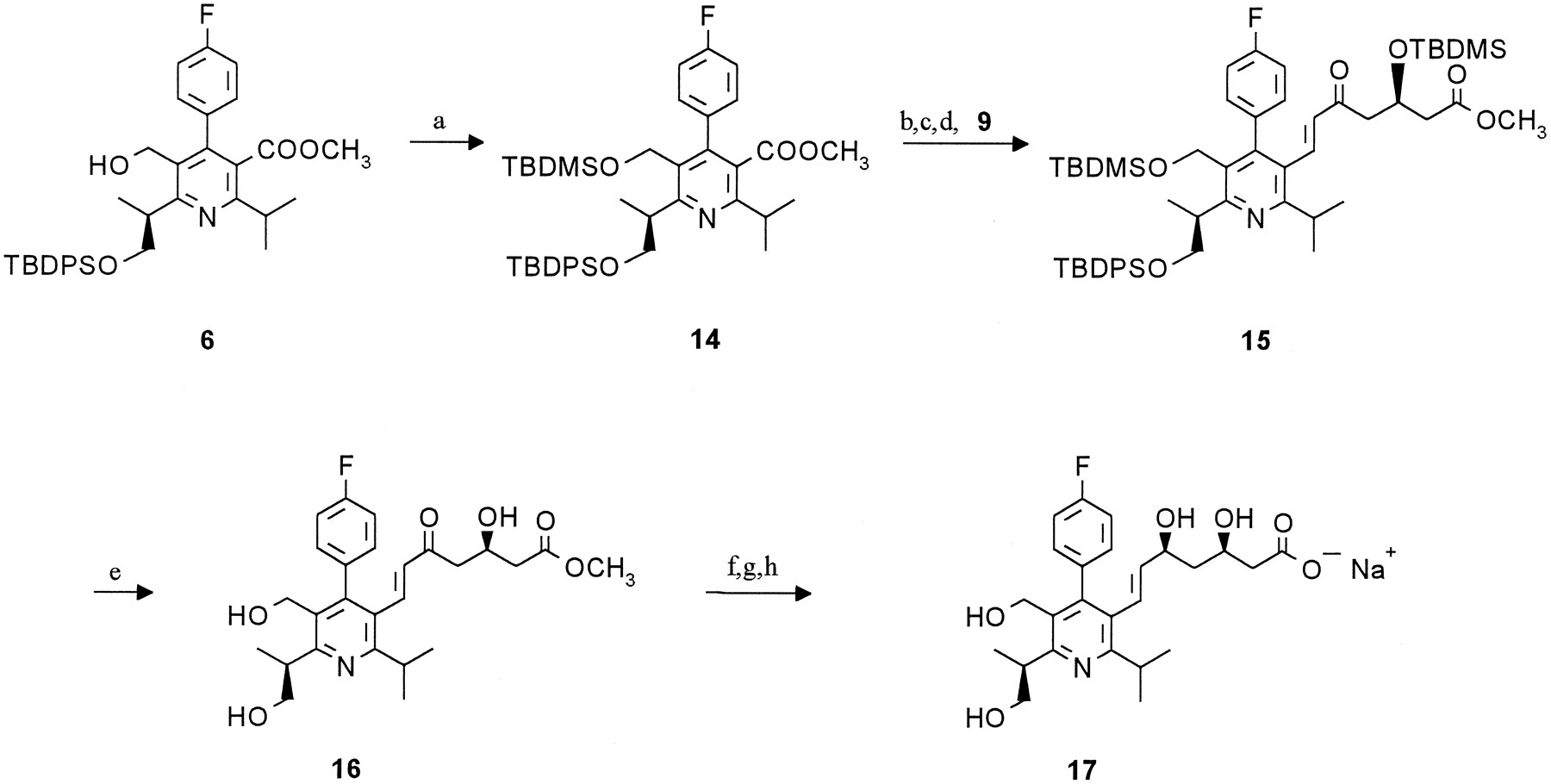
The synthesis of desmethylcerivastatin (metabolite M-1 reference) has been described previously [2]. Metabolite M-23 reference was synthesized as outlined in scheme FS1. Owing to partial racemization, which is supposed to occur during formation of the TBDPS-oxypentanoate precursor 2, all following intermediates contained the corresponding diastereomers (structures not shown). Separation was performed on the last but one step and the enantiomerically pure ester 12 was subsequently hydrolyzed to the final product. The synthesis of M-24 reference followed the same synthetic strategy, the TBDMS-group was used for the protection of the 5-hydroxymethyl function (scheme FS2).

Synthesis of metabolite M-23 reference.
Reagents: (a) TBDPSCl; (b) NaOH;(c) N,N carbonyldiimidazole; (d)CH3OOCCH2COOK, MgCl2, Et3N; (e) NaOCH3;(f) CH3COONH4, CH3COOH; (g) ammonium cerium (IV)nitrate;(h) DIBAL; (i) NaH, CH3J;(k) LiAlH4; (l)Al2O3 PCC; (m)K2CO3; (n) HCl;(o) Et3B, NaBH4;(p) chromatography; (q) NaOH.

Synthesis of metabolite M-24 reference.
Reagents: (a) TBDMSCl; (b)LiAlH4; (c) Al2O3PCC; (d) K2CO3;(e) HCl; (f) Et3B, NaBH4; (g) chromatography;(h) NaOH.

Numbering assignment used.
(R)-3-t-Butyldiphenylsilyloxy-2-methylpropionic acid1. In a 40-liter vessel equipped with a stirrer, 2667.0 g (22.60 mol) methyl (R)-(-)-3-hydroxy-2-methylpropionate (EGA, Steinheim, Germany) was dissolved in 14 liters DMF p.a. After the addition of 6768.8 g (24.65 mol) TBDPSCl, 3376.4 g (49.65 mol) imidazole, and 10 g 4-dimethylaminopyridine, the reaction temperature rose to 45°C. The reaction mixture was stirred with cooling to room temperature for 16 hr until the reaction was complete. The mixture was then poured into 75 liters water and washed with 2 × 20 liters EtOAc and the combined organic phases were washed with 2 × 10 liters water, dried over Na2SO4, and concentrated, yielding 8928 g (110%) of a crude oil of methyl (R)-3-t-butyldiphenylsilyloxy-2-methylpropionate.
1H-NMR (DMSO-d6): δ = 0.98 (s, 9H, t-Bu), 1.10 (d, 3H, CH3), 2.74 (m, 1H, CH), 3.64 (s, 3H, OCH3), 3.78 (d, 2H, OCH2), 7.47 (m, 6H, Ar), 7.61 (m, 4H, Ar) ppm.
A solution of 4464 g (11.3 mol) of this crude product in 27.5 liters THF was heated under reflux (internal temperature 65°C) with 5.65 liters (11.3 mol) 2 N NaOH for 46 hr in a 40-liter vessel equipped with a stirrer. The THF was removed by distillation on a rotary evaporator and the residue was diluted with 5 liters water and 3 liters CH2Cl2 and adjusted to pH 4 with 15% HCl. The phases were separated, the aqueous phase was washed with 3 liters CH2Cl2, and the combined organic phases were dried over Na2SO4 and concentrated to an oil, yielding 3930 g (100%) of crude 1.
1H-NMR (DMSO-d6): δ = 1.00 (s, 9H, t-Bu), 1.08 (d, 3H, CH3), 2.60 (m, 1H, CH), 3.74 (m, 2H, OCH2), 7.43 (m, 6H, Ar), 7.61 (m, 4H, Ar), 12.26 (s, 1H, COOH) ppm.
Methyl (R)-5-t-butyldiphenylsilyloxy-4-methyl-3-oxopentanoate2. To a solution of 1927.5 g (3.77 mol) 1 (67%) in 13 liters THF 744.4 g (4.59 mol) N,N′-carbonyldiimidazole was added at room temperature. The reaction mixture was stirred for 1 hr at room temperature and for 1 hr under reflux. After cooling to room temperature this solution A was used without further purification in the next step.
In a 40-liter vessel equipped with a stirrer 1258.3 g (8.06 mol) of potassium monomethyl malonate was suspended in 12.4 liters CH3CN at 0°C. After the addition of 1124.5 ml (8.06 mol) triethylamine and 847.1 g (8.92 mol) anhydrous MgCl2, the mixture was stirred for 5 hr at room temperature. The reaction solutionA and 112.4 ml (0.81 mol) triethylamine were added over 15 min and the mixture was stirred for 16 hr at room temperature, diluted with 20 liters EtOAc, and adjusted to pH 4 with 15% HCl. The organic phase was separated, washed with 10 liters water, concentrated, and taken up in 20 liters EtOAc. The residual water was separated. The organic phase was neutralized by washing with 2 × 10 liters saturated NaHCO3 solution, dried over Na2SO4, and concentrated on a rotary evaporator, affording 2012 g (85%) of 2 as a crude oil.
1H-NMR (DMSO-d6): δ = 1.00 (s, 9H, t-Bu), 1.00 (d, 3H, CH3), 2.95 (m, 1H, CH), 3.65 (s, 3H, OCH3), 3.72 (m, 4H, CH2, OCH2), 7.40 (m, 6H, Ar), 7.61 (m, 4H, Ar) ppm.
Dimethyl 6-[(S)-(t-butyldiphenylsilyloxymethyl)ethyl]-4(R,S)-(4-fluorophenyl)-2-isopropyl-1,4-dihydropyridine-3,5-dicarboxylate4. After 2094 g (14.54 mol) methyl isobutanoylacetate, 1442 g (11.6 mol) 4-fluorobenzaldehyde, 46 ml glacial acetic acid, and 81 ml piperidine were dissolved in 1.4 liters cyclohexane, the solution was heated under reflux with a Dean-Stark trap. 240 ml water separated within 2.5 hr. The cyclohexane and acetic acid were removed by distillation at 80 mbar, and the starting materials were then distilled off at 2 mbar (bath temperature 120°C, head temperature 70°C). The residue was treated with 3 liters EtOAc at room temperature, washed with NaHCO3, dried over Na2SO4, and concentrated to an oil, yielding 2930 g (61%) of crude (E/Z) methyl 2-(4-fluorophenyl)methylen-4-methyl-3-oxopentanoate 3.
1H-NMR (CDCl3): δ = 1.10 and 1.18 (2d, 6H, CH3), 2.71 and 3.18 (2 sept., 1H, CH), 3.84 (2s, 3H, OCH3), 7.07 (m, 2H, Ar), 7.40 (m, 2H, Ar), 7.58 and 7.75 (2s, 1H, olefin H) ppm.
A solution of 560 g (2.01 mol) of crude 3 and 1.56 kg (2.01 mol) 2 (crude product, 50%) in 3 liters CH3OH was cooled to 10°C and treated in portions with 104 g (1.92 mol) NaOCH3, which lead to slight warming. The mixture was stirred at room temperature for 1.5 hr (HPLC monitoring), treated with 480 g (6.25 mol) ammonium acetate, and 1.65 liters acetic acid, and the volatile constituents were then removed by distillation through a distillation bridge at a bath temperature of 130–140°C (internal temperature 112 °C, head temperature 105°C). After 90 min. 3 liters water was added at room temperature, the mixture was extracted with 3 × 1.5 liters EtOAc, and the combined organic phases were washed with 2 liters water and 2 liters NaHCO3 solution, dried over Na2SO4, and concentrated to an oil, giving 2.015 kg (72%) of 4 as a mixture of 2 diastereomers.
1H-NMR (CDCl3): δ = 0.94–1.35 (several d, 9H, CH3), 1.13 (several s, 9H, t-Bu), 3.59, 3.63, 3.64, 3.67 (4s, 6H, 2 OCH3), 3.73–4.30 (complex region, 4H, CH, OCH2), 4.99 and 5.03 (2s, 1H, H4), 6.80–7.78 (complex region, 14H, Ar) ppm.
Dimethyl 6-[(S)-(t-butyldiphenylsilyloxymethyl)ethyl]-4-(4-fluorophenyl)-2-isopropyl-pyridine-3,5-dicarboxylate5. To a solution of 2.015 kg (about 1.45 mol) 4(45%) in 10 liters CH3CN 1698 g (3.1 mol) (NH4)2Ce(NO3)6 was added in portions over 10 min at room temperature with stirring, the reaction mixture (suspension) warming up to about 34°C. After 30 min the reaction was complete (HPLC monitoring). The reaction mixture was stirred with 12 liters water and the CH3CN was largely removed from the two-phase mixture by distillation on a rotary evaporator. Because of the danger of explosion, water should always be present during the concentration. The aqueous residue was washed with 3 × 3 liters EtOAc and the combined organic phases were washed with 3 liters of 10% KI solution, 3 liters Na2S2O3 solution, and 5 liters water, dried over Na2SO4, and concentrated to 1869 g crude oil. The oil was chromatographed on 12 kg silica with PE/EtOAc 97:3 to yield 577.4 g (36%) of crude oily 5.
1H-NMR (CDCl3): δ = 0.96 (s, 9H, t-Bu), 1.25 (dd, 9H, CH3), 3.07 (sept., 1H, CH(CH3)2), 3.30 (m, 1H, CH-CH2), 3.48 (s, 3H, OCH3), 3.57 (s, 3H, OCH3), 3.70 and 4.05 (m, each 1H, CH2O), 7.00–7.65 (m, 14H, Ar) ppm.
Methyl 6-[(S)-(t-butyldiphenylsilyloxymethyl)ethyl]-4-(4-fluorophenyl)-5-hydroxymethyl-2-isopropylpyridine-3-carboxylate6. A solution of 561 g (0.51 mol) 5 (purity 57%) in 1.7 liters toluene p.a. was cooled to −60°C under argon. To this solution 1367 ml (2.05 mol) of a 1.5 M solution of DIBAL in toluene was added dropwise, so that the internal temperature did not exceed −53°C. At the end of the addition, the solution was stirred for 30 min at −60°C and for 16 hr at −30°C until the starting material had almost completely reacted (TLC check using PE/EtOAc 9:1 on silica). To hydrolyze the aluminium compounds the reaction mixture was added to 8 liters of 10% KOH solution over 20 min with stirring, the stirring was continued for a further 15 min, the aqueous phase was separated, extracted with 2 × 3 liters EtOAc, and passed through kieselguhr under suction to remove flocculations, which hindered the phase separation. The combined organic phases were washed with 2 × 2 liters saturated brine, dried over Na2SO4, and concentrated on a rotary evaporator to 525 g crude oil. The crude oil was chromatographed on silica with PE/EtOAc 9:1 and 8:2, affording 124.9 g (43%) of 6 as an oil.
1H-NMR (CDCl3): δ = 0.95 (s, 9H, t-Bu), 1.17 (d, 3H, CH3), 1.25 (2d, 6H, CH3), 3.05 (sept., 1H, CH), 3.45 (m, 1H, CH-CH2), 3.57 (s, 3H, OCH3), 3.62–3.77 (m, 2H, CH2O), 4.30 and 4.61 (m, each 1H, CH2OSi), 7.02–7.65 (m, 14H, Ar) ppm.
Methyl 6-[(S)-(t-butyldiphenylsilyloxymethyl)ethyl]-4-(4-fluorophenyl)-2-isopropyl-5-methoxymethylpyridine-3-carboxylate7. A suspension of 9.35 g (0.313 mol) NaH (80%) in 500 ml anhydrous THF was heated to boiling and a solution of 125 g (0.209 mol) 6 in 300 ml anhydrous THF was added dropwise under reflux. A solution of 35.5 g (0.25 mol) CH3I in 100 ml anhydrous THF was next added dropwise, also under reflux. The reaction mixture was then refluxed for a further 3 hr. After cooling to room temperature 250 ml water was cautiously added, the solution was extracted with 3 × 300 ml EtOAc, and the combined organic phases were washed with saturated brine and dried over Na2SO4. After removal of the solvent in vacuo, the residue was chromatographed on silica with EtOAc/PE 5:95, giving 117.4 g (92%) of 7.
1H-NMR (CDCl3): δ = 0.95 (s, 9H, t-Bu), 1.22 (2d, 6H, CH3), 1.30 (d, 3H, CH3), 3.05 (sept., 1H, CH), 3.20 (s, 3H, OCH3), 3.51 (s, 3H, OCH3), 3.58 (m, 1H, CH-CH2), 3.8–4.0 (m, 2H, CH2O and 1H CH2OSi), 4.45 (dd, 1H, CH2OSi), 7.0–7.6 (m, 14H, Ar) ppm.
6-[(S)-(t-Butyldiphenylsilyloxymethyl)ethyl]-4-(4-fluorophenyl)-2-isopropyl-5-methoxymethylpyridine-3-carbaldehyde8. A suspension of 9 g (0.0236 mol) LiA1H4in 500 ml anhydrous THF under argon was heated to boiling. A solution of 72.3 g (0.118 mol) 7 in 300 ml anhydrous THF was added dropwise under reflux and the refluxing was continued for 1 hr. After cooling to room temperature, 80 ml water was cautiously added dropwise. 80 ml of 10% KOH solution was then added and the precipitate was removed by filtration under suction. The precipitate was extracted by boiling with 3 × 300 ml ether. The mother liquors were combined, dried over Na2SO4, and concentratedin vacuo. The crude product (69 g, 99%) was used in the next step without further purification.
To a solution of 138 g (0.23 mol) of the above crude product in 3.5 liters CH2Cl2, 48.1 g (0.47 mol) Al2O3 and 101.7 g (0.47 mol) PCC were added. After stirring for 1 hr at room temperature, the reaction mixture was filtered through a frit with silica and washed through with sufficient CH2Cl2. The filtrate was concentrated in vacuo and dried, yielding 95.4 g (69%) of 8 as an oil.
1H-NMR (CDCl3): δ = 0.9 (s, 9H, t-Bu), 1.22 (d, 6H, 2 × CH3), 1.30 (d, 3H, CH3), 3.21 (s, 3H, OCH3), 3.62 (m, 1H, CH), 3.8–4.0 (m, 2H, CH2O; 1H, CH-CH2; 1H, CH2OSi), 4.46 (dd, 1H, CH2OSi), 7.0–7.7 (m 14H, Ar), 9.78 (s, 1H, CHO) ppm.
Methyl (E)-(3R)-7-{6-[(S)-(t-butyldiphenylsilyloxymethyl)ethyl]-4-(4-fluorophenyl)-2-isopropyl-5-methoxymethylpyrid-3-yl}-3-t-butyldimethylsilyloxy-5-oxohept-6-enoate10. A solution of 82.5 g (0.22 mol) dimethyl (R)-5-carbomethoxy-4-t-butyldimethylsilyloxy-2-oxopentylphosphonate9, 29.6 g (0.22 mol) K2CO3, and 2.9 ml water in 635 ml isopropanol was stirred for 1 hr at room temperature. Then 95.4 g (0.16 mol) 8 suspended in 150 ml isopropanol was added. After stirring for 4 days at room temperature (TLC monitoring), 500 ml water was added and the reaction mixture was extracted with 3 × 500 ml EtOAc. The combined EtOAc phases were washed with saturated brine, dried over Na2SO4, and concentrated in vacuo. The residue was chromatographed on silica with EtOAc/PE 5:95, giving 112.1 g (82%) of 10.
1H-NMR (CDCl3): δ = 0.2 (s, 3H, CH3Si), 0.3 (s, 3H, CH3Si), 0.75 (s, 9H, t-Bu), 0.9 (s, 9H, t-Bu), 1.15–1.35 (m, 9H, 3 × CH3), 2.38 (m, 2H, CH2), 2.56 (d, 2H, CH2), 3.12 (s, 3H, OCH3), 3.23 (sept., 1H, CH), 3.52 (m, 1H, CH-CH2), 3.61 (s, 3H, OCH3), 3.75–3.95 (m, 2H, CH2O, and 1H, CH2OSi), 4.38 (dd, 1H, CH2OSi), 4.47 (m, 1H CHOSi), 5.85 (d, 1H, =CH), 6.9–7.6 (m, 14H, Ar, and 1H, =CH) ppm.
Methyl (E)-(3R)-7-{4-(4-(fluorophenyl)-6-[(S)-(hydroxymethyl)ethyl]-2-isopropyl-5-methoxymethylpyrid-3-yl}-3-hydroxy-5oxohept-6-enoate11. A solution of 112 g (0.13 mol) 10 in 1170 ml anhydrous methanol and 130 ml 1 N HCl was stirred for 4 days at room temperature (TLC monitoring). Then 1000 ml CH2Cl2 was added and the solution was washed with 2 × 500 ml saturated NaHCO3 solution. The organic phase was dried over Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica with EtOAc/PE 1:1 to afford 59.2 g (91%) of 11.
1H-NMR (CDCl3): δ = 1.28 (2d, 6H, CH3), 1.42 (d, 3H, CH3), 2.48 (m, 2H, CH2), 2.61 (m, 2H, CH2), 3.20 (s, 3H, OCH3), 3.28 (sept., 1H, CH), 3.32 (m, 2H, CH2OH), 3.71 (s, 3H, OCH3), 3.88 (m, 1H, CH-CH2), 4.0–4.2 (m, 2H, CH2-O), 4.41 (m, 1H, CH-OH), 5.90 (d, 1H, =CH), 7.0–7.2 (m, 4H, Ar), 7.45 (d, 1H, =CH) ppm.
Methyl (E)-(3R,5S)-7-{4-(4-fluorophenyl)-6-[(S)-(hydroxymethyl)ethyl]-2-isopropyl-5-methoxymethylpyrid-3-yl}-3,5-dihydroxyhept-6enoate12. A mixture of 600 ml anhydrous THF, 240 ml anhydrous methanol, and 243.2 ml (0.24 ml) of a 1 M Et3B solution in THF was stirred for 1 hr at room temperature. After cooling to −75°C (internal temperature, acetone/dry ice bath), 59.2 g (0.12 mol)11, dissolved in 150 ml anhydrous THF was added. After 30 min at −75°C, 6.9 g (0.18 mol) NaBH4 was added in portions and stirring was continued for further 3 hr at −75°C. The cooling bath was removed and 100 ml of saturated NH4Cl solution was added dropwise at 0°C. Then 700 ml water and 500 ml EtOAc were added and the aqueous phase was separated and extracted with 2 × 200 ml EtOAc. The combined organic phases were washed with 400 ml saturated brine, dried over Na2SO4, and concentrated in vacuo. The residue was dissolved in 500 ml methanol and concentrated again on the rotary evaporator. This procedure was repeated 5 times and the residue was finally chromatographed on silica with EtOAc/PE 1:1. The fractions containing the product were concentrated to give 50.5 g crude product, which was chromatographed again on silica yielding 33.9 g (58%) of12 as a mixture of diastereomers (de = 59%, HPLC).
1H-NMR (CDCl3): δ = 1.25 (2d, 6H, CH3), 1.40 (m, 2H, CH2), 1.43 (d, 3H, CH3), 2.41 (m, 2H, CH2), 3.18 (s, 3H, OCH3), 3.2–3.4 (m, 2H, CH, and CH-CH2), 3.71 (s, 3H, OCH3), 3.85 (m, 2H, CH2-OH), 4.0–4.2 (m, 3H, CH2-O, and CH-O), 4.32 (m, 1H, CH-O), 5.28 (dd, 1H, =CH), 6.31 (d, 1H, =CH), 7.0–7.2 (m, 4H, Ar) ppm.
For isolation of the pure diastereomer by preparative chiral HPLC, 30 g of this mixture was dissolved in 160 ml ethanol and diluted with 640 ml n-heptane. A total of 940 portions of 0.8 ml (30 mg) each were injected onto the HPLC column every 15 min with an autoinjector, and 13 fractions were collected with the aid of a fraction collector with peak/time control. After a purity check of these fractions by HPLC, fractions 1–6 (pure diastereomer 12) and 7+8 (mixture of diastereomers) were combined accordingly. The solvent was removed by distillation in vacuo. The mixed fractions were again separated analogously.
Finally 16.9 g pure 12 (de = 99.2%, 76% yield as referred to crude 12) were obtained.
To remove colored impurities, 16.9 g separated 12was chromatographed on silica with EtOAc/PE 1:1, yielding 15.9 g12 as colorless oil.
Sodium (E)-7-(3R,5S)-{4-(4-fluorophenyl)-6-[(S)-(hydroxymethyl)ethyl]-2-isopropyl-5-methoxymethylpyrid-3-yl}-3,5-dihydroxyhept-6enoate13. To a solution of 10.6 g (21.7 mmol) 12in 150 ml THF, 238.5 ml of a 0.1 N NaOH solution was added at room temperature. After 1 hr at room temperature, the THF was removed on a rotary evaporator and the aqueous residue was freeze dried, affording 10.7 g (99%) of 13 (metabolite M-23 reference sodium salt). NMR data are depicted in table 1.
1H-NMR of cerivastatin and metabolites 1-a
Methyl 6-[(S)-(t-butyldiphenylsilyloxymethyl)ethyl]-5-(t-butyldimethylsilyloxymethyl)-4-(4-fluorophenyl)-2-isopropylpyridine-3-carboxylate14. A solution of 30 g (0.05 mol) 6, 16.6 g (0.11 mol) t-butyldimethylsilylchloride, 15 g (0.22 mol) imidazole, and 1.0 g (8 mmol) 4-dimethylaminopyridine in 400 ml DMF was stirred at 50°C for 16 hr. After cooling to room temperature 100 ml ether and 200 ml brine were added. The organic phase was separated and dried over Na2SO4. After removal of the solvent in vacuo the residue was chromatographed on silica with EtOAc/PE 5:95, giving 35.6 g (99.8%) of 14.
1H-NMR (CDCl3): δ = -0.3 (s, 3H, CH3Si), -0.1 (s, 3H, CH3Si), 0.87 and 0.92 (2s, 18H, t-Bu), 1.2–1.35 (3d, 9H, CH3), 3.0 (sept, 1H, CH), 3.51 (s, 3H, OCH3), 3.6–3.75 (m, 1H, CH-CH2), 3.8–4.25 (m, 2H, CH2O and 1H CH2OSi), 4.78 (dd, 1H, CH2OSi), 6.95–7.65 (m, 14H, Ar) ppm.
6-[(S)-(t-Butyldiphenylsilyloxymethyl)ethyl]-5-(t-butyldimethylsilyloxymethyl-4-(4-fluorophenyl)-2-isopropylpyridine-3-carbaldehyde. A suspension of 3.79 g (0.1 mol) LiA1H4 in 250 ml anhydrous THF was heated under argon to boiling. A solution of 35.57 g (0.05 mol) 14 in 150 ml anhydrous THF was then added dropwise under reflux and the refluxing was continued for 1 hr. After cooling to room temperature 30 ml water was cautiously added dropwise. 30 ml of 10% KOH solution was added and the precipitate was removed by filtration under suction. The precipitate was extracted by boiling with 3 × 100 ml ether. The mother liquors were combined, dried over Na2SO4, and concentrated in vacuo. The crude product (33.5 g, 98%) was used in the next step without further purification. It was dissolved in 600 ml CH2Cl2 and 9.97 g (0.1 mol) Al2O3 and 21.08 g (0.1 mol) PCC were added. After stirring for 1 hr at room temperature, the reaction mixture was filtered through a frit with silica and washed through with sufficient CH2Cl2. The filtrate was concentrated in vacuo and dried, yielding 26.56 g (79.5%) of the title compound as an oil.
1H-NMR (CDCl3): δ = −0.5 (s, 3H, CH3Si), −0.1 (s, 3H, CH3Si), 0.87 and 0.89 (2s, 18H, t-Bu), 1.2 - 1.3 (3d, 6H, 3 × CH3), 3.65–4.2 (m, 2H, CH2O; 1H, CH-CH2; 1H, CH2OSi), 4.78 (dd, 1H, CH2OSi), 7.05–7.65 (m 14H, Ar), 9.78 (s, 1H, CHO) ppm.
Methyl (E)-(3R)-7-{6-[(S)-(t-butyldiphenylsilyloxymethyl)ethyl]-5-(t-butyldimethylsilyloxymethyl)-4-(4-fluorophenyl)-2-isopropyl-pyrid-3-yl}-3-t-butyldimethylsilyloxy-5-oxohept-6-enoate15. A solution of 2.4 g (6.38 mmol) dimethyl (R)-5-carbomethoxy-4-t-butyldimethylsilyloxy-2-oxopentylphosphonate9, 0.875 g (6.33 mmol) K2CO3 and 0.09 ml water in 18.8 ml isopropanol was stirred for 1 hr at room temperature. Then 3.3 g (4.83 mmol) aldehyde (previous step) suspended in 18.8 ml isopropanol was added. After stirring for 5 days at room temperature (TLC monitoring), 100 ml water was added and the reaction mixture was extracted with 3 × 100 ml EtOAc. The combined EtOAc phases were washed with saturated brine, dried over Na2SO4, and concentrated in vacuo. The residue was chromatographed on silica with EtOAc/PE 5:95, giving 3.6 g (79.4%) of 15.
1H-NMR (CDCl3): δ = −0.5, −0.1, and 0.2 (3s, 12H, CH3Si), 0.8, 0.87, and 0.9 (3s, 27H, t-Bu), 1.15–1.35 (m, 9H, 3 × CH3), 2.42 (m, 2H, CH2), 2.6 (d, 2H, CH2), 3.28 (sept., 1H, CH), 3.65 (s, 3H, OCH3, m, 1H, CHCH2), 3.8–4.2 (m, 2H, CH2O, and 1H, CH2OSi), 4.52 (dd, 1H, CH2OSi), 4.73 (d, 1H, CHOSi), 5.90 (d, 1H, =CH), 6.9–7.65 (m, 14H, Ar, and 1H, =CH) ppm.
Methyl (E)-(3R)-7-{4-(4-(fluorophenyl)-6-[(S)-(hydroxymethyl)ethyl]-5-hydroxymethyl-2-isopropyl-pyrid-3-yl}-3-hydroxy-5-oxohept6-enoate16. A solution of 3.6 g (3.8 mmol) 15 in 63 ml anhydrous methanol and 7 ml 1 N HCl was stirred for 5 days at room temperature (TLC monitoring). Then 100 ml CH2Cl2 was added and the solution was washed with 2 × 100 ml saturated NaHCO3 solution. The organic phase was dried over Na2SO4 and concentrated in vacuo. The residue was chromatographed on silica with EtOAc/PE 6:4 to afford 1.4 g (77.9%) of16.
1H-NMR (CDCl3): δ = 1.2–1.45 (3d, 9H, CH3), 2.48 (m, 2H, CH2), 2.6 (m, 2H, CH2), 3.2–3.6 (m, 1H, CH, and 2H, CH2OH), 3.7 (s, 3H, OCH3), 3.8–4.6 (m, 1H, CH-CH2, m, 2H, CH2OH, m, 1H, CH-OH), 5.92 (d, 1H, =CH), 7.0–7.4 (m, 4H, Ar, and 1H =CH) ppm.
Methyl (E)-(3R,5S)-7-{4-(4-fluorophenyl)-6-[(S)-(hydroxymethyl)ethyl]-5-hydroxymethyl-2-isopropylpyrid-3-yl}-3,5-dihydroxyhept-6enoate. A mixture of 24 ml anhydrous THF, 6 ml anhydrous methanol, and 5.9 ml (5.9 mmol) of a 1 M Et3B solution in THF was stirred for 1 hr at room temperature. After cooling to −75°C (internal temperature, acetone/dry ice bath), 1.4 g (2.96 mmol)16, dissolved in 20 ml anhydrous THF was added. After 30 min at −75°C, 168 mg (4.44 mmol) NaBH4 was added in portions and stirring was continued for a further 3 hr at −75°C. The cooling bath was removed and 100 ml of saturated NH4Cl solution was added dropwise at 0°C. Then 100 ml water and 100 ml EtOAc were added and the aqueous phase was separated and extracted with 2 × 100 ml EtOAc. The combined organic phases were washed with 50 ml saturated brine, dried over Na2SO4, and concentratedin vacuo. The residue was dissolved in 100 ml methanol and concentrated again on the rotary evaporator. This procedure was repeated 5 times and the residue was finally chromatographed on silica with EtOAc/PE 6:4 yielding 1.16 g (82.5%) of the crude title compound as a mixture of diastereomers (de = 59%, HPLC).
1H-NMR (CDCl3): δ = 1.15–1.3 and 1.4 (3d, 9H, CH3), 2.42 (m, 2H, CH2), 3.1 (m, 1H, CH), 3.2–3.65 (m, 2H, CH2, and m, 1H, CH-CH2), 3.71 (s, 3H, OCH3), 3.8 - 4.55 (m, 4H, CH2-OH, and m, 2H, CH-O), 5.78 (dd, 1H, =CH), 6.3 (d, 1H, =CH), 7.0–7.25 (m, 4H, Ar) ppm.
For isolation of the pure diastereomer by preparative chiral HPLC, 6.7 g of this mixture (synthesized accordingly in several batches) was dissolved in 34.4 ml ethanol and diluted with 137.6 ml n-heptane. A total of 344 portions of 0.5 ml (19.8 mg) each were injected onto the HPLC column every 19 min with an autoinjector, and 10 fractions were collected with the aid of a fraction collector with peak/time control. After a purity check of these fractions by HPLC, fractions 1–5 (pure diastereomer) and 6+7 (mixture of diastereomers) were combined accordingly. The solvent was removed by distillation in vacuo. The mixed fractions were again separated analogously.
Finally 4.43 g pure title compound (de > 99%, 87% yield as referred to crude product) were obtained.
Sodium (E)-7-(3R,5S)-{4-(4-fluorophenyl)-6-[(S)-(hydroxymethyl)ethyl]-5-hydroxymethyl-2-isopropylpyrid-3-yl}-3,5-dihydroxyhept-6enoate17. To a solution of 504 mg (1.06 mmol) pure diastereomer (previous step) dissolved in 10 ml THF 10.6 ml of 0.1 N NaOH solution was added at room temperature. After 1 hr at room temperature, the THF was removed on a rotary evaporator and the aqueous residue was freeze dried, affording 511 mg (99.7%) of 17 (metabolite M-24 reference sodium salt). NMR-data are depicted in table 1.
Incubations.
Human liver microsomes were purchased from Human Biologics, Inc. (Phoenix, AZ) and IIAM Keystone Skin Bank (Exton, PA). The standard incubation mixture contained 250 μmol HEPES buffer (pH 7.4), 50 μmol MgCl2, 100 μmol glucose-6-phosphate, 4 units of glucose-6-phosphate dehydrogenase, 5 μmol NADPH, and 5 μmol NADH (all reagents from Boehringer Mannheim, Mannheim, Germany) in a final volume of 5.25 ml. Microsomes were added so that ratios of 1:5 to 1:18 (CYP: substrate) on a molar basis were achieved. After 3 min of preincubation at 37°C, the reaction was started by addition of 1 μl of a solution of 10 mg [14C]cerivastatin in 1 ml methanol. The reaction mixture was incubated at 37°C in a 20 × 125 mm flint glass tube under an air atmosphere using a JULABO U3 (JULABO Labortechnik, Seelbach, Germany) shaker at 100 rpm. Samples were taken 0.5, 5, 10, 20 (1 ml per sample), and 60 min (1.25 ml) after starting the incubation. The reaction was stopped by addition of 1 ml of ice-cold acetonitrile. The reaction mixtures were centrifuged, the supernatants were concentrated to 200 μl by vacuum evaporation in a Speed Vac® evaporator (Savant Instruments Inc., Farmingdale, NJ) and stored at −20°C until HPLC analysis. All incubations were performed in duplicate.
To inhibit CYP3A mediated metabolism, 25 nmol of triacetyloleandomycin were preincubated for 20 min or added to the mixture at the start of the enzymatic reaction.
For isolation and structure elucidation of single metabolites larger scale incubations were performed using the same procedure, but different CYP: substrate ratios and longer incubation times. Typically, 315 μg (601 nmol) of [14C]cerivastatin was incubated with 2 ml human liver microsomal protein at a ratio of CYP: substrate of 1:55.7 on a 20 ml scale for 5 hr.
For the cytochrome P450 (CYP) assays, microsomes prepared from the following AHH-1 cell lines, expressing single CYP isozymes were used: M102b (CYP1A1), M111b (CYP1A1), M103c (CYP1A2), M104a (CYP2A6), M107r (CYP3A4), M110a (CYP2B6), M112a (CYP2C8), M109r (CYP2C9), M105b (CYP2D6Met), M117r (CYP2D6Val), M106k (CYP2E1), M101a (control) (Gentest Corporation, Woburn, MA).
Ten μg [14C]cerivastatin was incubated for 3 hr at 37°C with 0.5 mg microsomal protein in 1 ml HEPES buffer, supplemented with a NADH and NADPH generating system as described above. An incubation with control microsomes (native AHH-1 cells) was performed in parallel. The reaction was initiated by adding ice cold microsomes to prewarmed buffer, containing substrate and the cofactors. After initial mixing of the microsomes, samples were taken at 0, 0.5, 1, 2, and 3 hr (1 ml per sample) and further processed as described above.
Chromatographic Methods.
Analytical HPLC was performed on a HP 1090 M liquid chromatograph with diode array detection (Hewlett-Packard, Waldbronn, Germany) and online radioactivity monitor Ramona®5 (Raytest, Straubenhardt, Germany), connected via A/D converter that transformed DPM values into mV units.
For metabolite profiling of microsomal incubates a 5 μm LiChrospher® RP8 column (250 × 4 mm) was eluted with a step gradient from 70% solvent A (0.1% aqueous TBAH, adjusted to pH 5 with 0.1% aqueous sodium hydroxide) to 100% solvent B (20% solvent A, 80% acetonitrile) within 130 min. The flow rate was 1.3 ml/min and the oven temperature was 40°C.
The diastereomeric purity of metabolites M-23 and M-24 was determined by chiral HPLC on a 5 μm-Chiral AGP® column (150 × 4 mm, ChromTec, Hägersten, Sweden). Isocratic elution employed a 66 mM phosphate buffer pH 5 (KH2PO4/Na2HPO4), containing 0.5% 2-propanol and a flow of 0.9 ml/min at room temperature. For this chiral HPLC assay M-23 and M-24 were separated from other metabolites by preceding achiral HPLC.
Preparative separation of the diastereomeric esters of metabolite M-23 and M-24 reference, respectively, was performed using two HPLC pumps 305 and 306, a fraction collector 201, an autoinjector model 231 × 6 (Gilson, Beltline-Middleton, WI), a UV detector SP100 (Spectra Physics, San Jose, CA) and a recorder 320D Servogor (Metrawatt, Nuernberg, Germany). A Chiralpak® AS column (250 × 20 mm, Daicel Chemical Ind., Tokyo, Japan) was eluted isocratically with n-heptane/ethanol 95:5 (M-23) or 88:12 (M-24) at a flow rate of 10 ml/min. UV detection was used at 230 nm, the oven temperature was 40°C.
Measurement of radioactivity.
Radioactivity of liquid samples was measured at 13°C in a Canberra Packard TriCarb® 2500 TR liquid scintillation spectrometer with automatic quench correction by the external standard channel ratio method using Ultima Gold® (Canberra Packard) as scintillation cocktail.
Spectroscopic Methods..
Positive mode ESI mass spectra were recorded by LC-MS using an ABI 140B HPLC system (Applied Biosystems, Inc., CA) coupled with a PE/Sciex/API III mass spectrometer (Perkin Elmer Sciex Instruments, Ontario, Canada). A 5 μm-Supelcosil® LC-18 (250 × 2.1 mm) column was eluted with a step gradient from 50% 10 mM ammoniumacetate to 90% acetonitrile over a period of 30 min. The column effluent (250 μl/min) was connected to the Ionspray® interface, with a splitting ratio of 1:10. 1H-NMR spectra of isolated metabolites and reference compounds were recorded at 500 MHz on a Bruker AMX 500 (Bruker, Rheinstetten, Germany) using methanol-d4 (99.96% deuterium incorporation) as solvent. The two-dimensional NOESY spectrum (Nuclear Overhauser Effect Spectroscopy) was performed with a mixing time of 600 ms.
Isolation and Purification of Metabolites.
Approximately 20 ml incubation mixture with human liver microsomes were separated by solid phase extraction on a 20 ml Isolute®E18 cartridge into 3 fractions (recovery 80%) using 0.1% phosphoric acid/acetonitrile gradient elution. From fractions 1 and 2 single metabolites were isolated by HPLC on a 7 μm-Nucleosil®C18 column (250 × 8 mm) using 0.1% aqueous trifluoroacetic acid/acetonitrile gradient elution. Fractions containing metabolites M-1, M-22, M-23, M-24, and M-31 were combined and submitted to spectroscopic analysis.
Results
Metabolite Profiles.
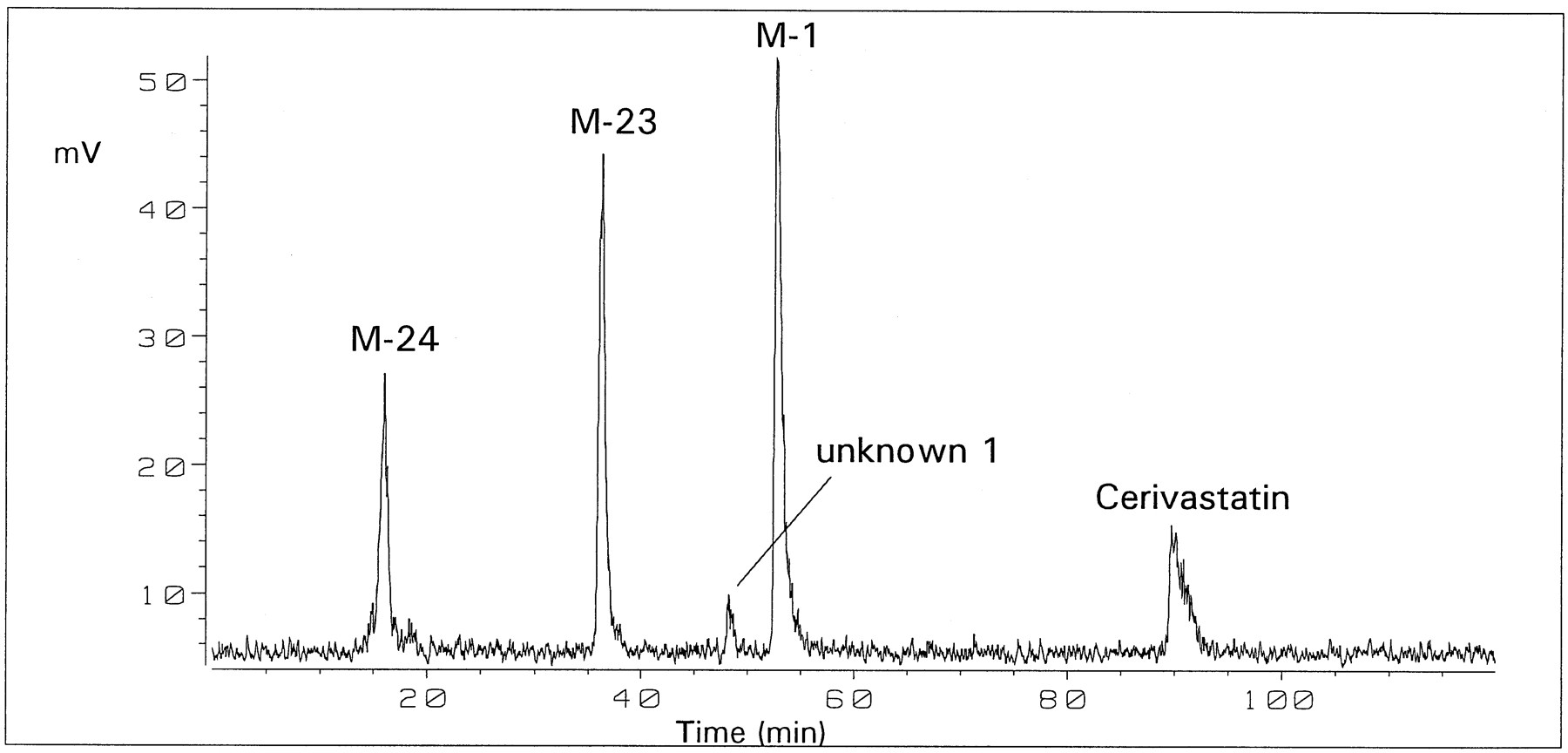
Incubation of [14C]cerivastatin with four different batches of human liver microsomes for 60 min resulted in qualitatively similar, but quantitatively different metabolite profiles (table2 ). Two major metabolites were equally important, the demethylated drug M-1 and metabolite M-23, hydroxylated at one methyl group of the 6-isopropyl substituent. In microsomal batches 1–3 total drug turnover was similar (17–28% after 60 min), whereas in batch no. 4 metabolism was more extensive (66%). Besides M-1 and M-23, considerable amounts of metabolite M-24, comprising both biotransformations, were formed by batch no. 4, in particular when incubated on a larger scale for 5 hr (fig 1).
Metabolism of [14C] cerivastatin by human liver microsomes 2-a
Structure Elucidation of Metabolites.
The ESI mass spectra as well as the 1H-NMR spectra of isolated and purified metabolites M-1, M-23, and M-24 were identical with those of the synthetic reference compounds (tables 1 and3). Identity was also proven by chromatographic comparison using achiral, and - for M-23 and M-24 - also using chiral HPLC. Minor metabolites M-22 and M-31 were identified by their ESI mass spectra (table 3).
ESI mass spectra of cerivastatin and metabolites
All mass spectra comprise [M+H]+ ions, which are in accordance with the proposed structures.
The 1H-NMR spectrum of metabolite M-1 was highly similar to the spectrum of cerivastatin (table 1). Essential differences were the lack of the methylether group and the downfield shift of the -CH2O- AB spin system to δ = 4.32 ppm. By means of a NOESY (Nuclear Overhauser Effect Spectroscopy) experiment (scheme FS4, the short distances found in the NOESY spectrum are indicated by arrows) it was shown that the upfield absorbing 6H doublet at δ = 1.23 ppm represented the isopropyl methyl groups in C2′ position, whereas the doublet at δ = 1.31 ppm represented the C6′ isopropyl moiety.

Observed NOESY signals of metabolite M-1 (desmethyl cerivastatin).
The 1H-NMR spectrum of metabolite M-23 comprised two important differences as compared with the cerivastatin spectrum: one downfield absorbing methyl group had disappeared and a new 2H doublet at δ = 3.88 ppm, typical for a CH2OH group indicated hydroxylation at this methyl group. Based on the NOESY results for metabolite M-1, it was concluded that the 6′-isopropyl moiety was functionalized. This structure proposal was in agreement with the molecular weight (475 dalton) determined by mass spectrometry and was confirmed by the independent synthesis.
The 1H-NMR spectrum of metabolite M-24 was very similar to that of metabolite M-23 (table 1). In the mass spectrum the molecular weight difference of 14 dalton, as compared with M-23 (table 3), indicated metabolic demethylation. Accordingly, the -CH2O- AB spin system was shifted to δ = 4.33 ppm. The new CH2OH group resulting from the hydroxylation of the 6′-isopropyl moiety appeared as an ABX spin system centered at δ = 3.87 ppm.
The hydroxylation leading to metabolite M-23 created a new chiral center in the cerivastatin molecule. As the stereochemistry of the dihydroxyheptenoic acid side chain is fixed, two diastereomeric metabolite structures were theoretically conceivable. In the NMR spectrum of the isolated M-23 sample, no additional signal splitting provided evidence for the presence of diastereomers. Using a chiral HPLC column, a diastereomeric excess (de) of 93% was determined for the isolated M-23 (fig. 2). The reference sample used for this assay was a synthetic mixture of both diastereomers.

Profile of metabolites formed from [14C]cerivastatin by human liver microsomes on a 20 ml scale after 5 hr.
HPLC was performed as described under Materials and Methods with radioactivity detection.

Determination of diastereomeric excess (de) of (A) metabolite M-23 formed from [14C]cerivastatin by human liver microsomes. Comparison with (B) reference mixture of M-23 and its diastereomer.
HPLC was performed on a chiral column as described in Materials and Methods with UV detection at 230 nm.
Based on the configuration of the starting materials used for reference synthesis, the absolute configuration at the new chiral center of M-23 was assigned to be (S)-(hydroxymethyl)ethyl.
The 1H-NMR spectrum of metabolite M-24 resembled the characteristic features of both, M-1 and M-23 (table 1). Like M-23, M-24 was formed with high stereoselectivity, for the isolated sample a de of 78% was determined by chiral HPLC. The new chiral center exhibits the (S)-configuration, as inferred from the stereochemistry of the synthetic starting materials.
The lactones of main metabolites M-1 and M-23, M-22 and M-31, respectively, were not found in the original microsomal incubation mixtures. They were most likely formed in small amounts from their open hydroxy acid precursors during the workup process. Lactonization of cerivastatin and metabolites occurs preferentially under acidic conditions, as used for preparative HPLC.
Cytochrome P450 specific in vitro metabolism.
[14C]Cerivastatin was incubated with microsomes from 11 human B lymphoblastoid AHH-1 cell lines, which stably express single human CYP isozymes (3). CYP 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2D6Met, 2D6Val, and 2E1 did not metabolize the drug to a measurable extent. With microsomes from cell line no. M107r, expressing human CYP 3A4, a 2% turnover was achieved within 2 hr. The only product detected was the demethylated metabolite M-1.
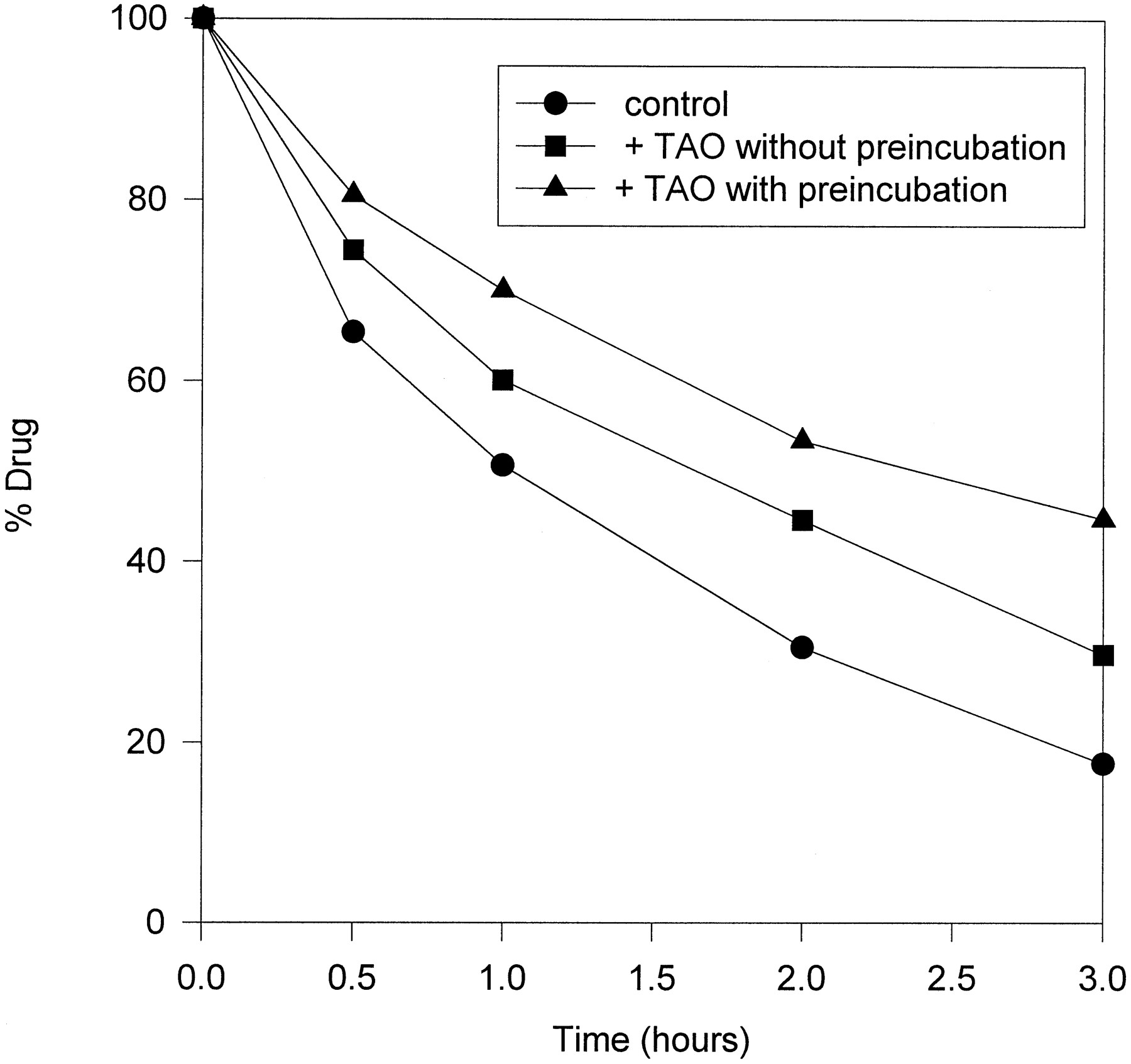
To further substantiate the extent to which CYP 3A enzymes are involved in the metabolism of cerivastatin, specific inhibition experiments were performed. Human liver microsomes from batch 4, which exhibited the highest cerivastatin turnover, were used. Incubations were performed with and without the addition of TAO, a highly selective suicide inhibitor of CYP 3A enzymes (4, 5). Following a 20 min preincubation with TAO a higher inhibitory effect was achieved than without preincubation (fig. 3). After 3 hr 45% of [14C]cerivastatin remained unmetabolized whereas in the control experiment with-out TAO only 18% was left. The amounts of metabolites M-1, M-23, and M-24 formed decreased to a similar extent under the influence of the inhibitor (table 4). After preincubation with TAO 73% of M-1, 82% of M-23, and 31% of M-24 were formed after 3 hr as compared with the control experiments.

Metabolic degradation of cerivastatin by human liver microsomes. Effect of the specific cytochrome P450 3A inhibitor TAO.
Inhibition of cerivastatin metabolism in human liver microsomes by coincubation with TAO
Discussion
In human liver microsomes cerivastatin is subject to two primary biotransformation reactions. Demethylation of the benzylic methylether leads to metabolite M-1, whereas hydroxylation of one methyl group in the 6′-isopropyl moiety furnishes metabolite M-23. Metabolite M-24 comprises the combination of both primary reactions (scheme FS5). Lactonization of M-1 and M-23 to M-22, and M-31, respectively, most likely occurred during sample workup. The hydroxylation reaction was found to be highly stereoselective, producing the 3R,5S,1“S-enantiomers of the primary metabolite M-23 as well as of its demethylated analogue M-24.
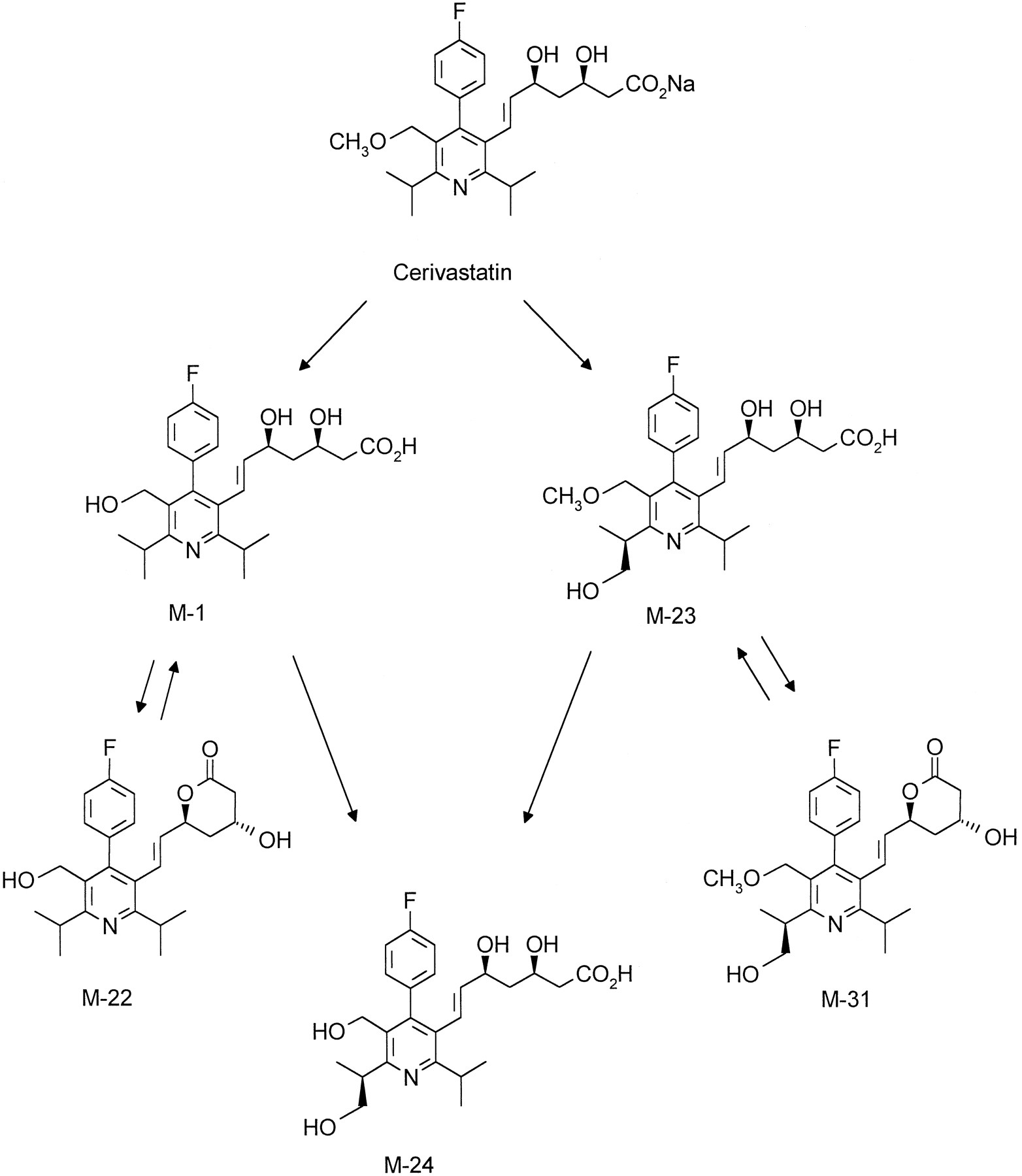
Pathways of cerivastatin metabolism by human liver microsomes.
The absolute configuration of these single enantiomers was determined by comparison with the synthetic reference compounds. Although each synthesis started from an enantiomerically pure precursor, racemization on one of the following steps afforded both diastereomeric products. Racemization was not complete, thus the configuration of the major diastereomer had to be identical to the starting material. This conclusion was confirmed by the finding that the opposite diastereomeric ratio was obtained for the final products when the starting material with opposite configuration was used (data not shown). Chromatographic comparison unequivocally showed that the isolated metabolites M-23 and M-24 were identical with the main component in the respective reference mixture.
The incubations of cerivastatin with microsomes from single CYP isozyme expressing cell lines suggest that only the demethylation pathway, but not the hydroxylation pathway is mediated by CYP 3A4.
In the inhibition experiments, the selective CYP3A4 inhibitor TAO was used in a concentration (approximately 5 μM) which has been shown to exert the maximum inhibitory effect on CYP 3A4 in human liver microsomes (5). Using testosterone 6β-hydroxylation as a specific probe reaction, CYP 3A4 activity was reduced to less than 30% of control. Virtually no inhibitory effect was observed for reactions mediated by CYP 1A2, 2C9, 2D6, or 2E1 (5).
Cerivastatin metabolism was reduced by TAO to 56% of control after 30 min of incubation. After 3 hr the relative turnover had increased to 67% of the control value, indicating that despite the suicide destruction of CYP 3A4 by TAO other enzymes are able to metabolize the drug. Moreover, contrary to the CYP 3A4 expressing cell line, TAO inhibition equally affects both metabolic pathways, hydroxylation and demethylation. The formation of metabolites M-1 and M-23 was inhibited to similar extents over a 3-hr reaction peroid. A possible explanation for these findings is that an enzyme other than CYP 3A4 is responsible for the hydroxylation pathway to M-23 and that this enzyme is inhibited by TAO and also demethylates cerivastatin to M-1. The characterization of this enzyme, as well as its clinical importance, e.g. for drug interaction studies, will be the subject of future investigations.
Footnotes
-
Send reprint requests to: Dr. M. Boberg, Bayer AG, PH-PD P Drug Metabolism and Isotope Chemistry, D-42096 Wuppertal, Germany.
-
↵2 M. Radtke and R. Angerbauer, manuscript in preparation
- Abbreviations used are::
- HMG-CoA
- 3-hydroxy-3-methylglutaryl-coenzyme A
- CYP
- cytochrome P450
- TBAII
- tetrabutylammonium hydrogensulfate
- TAO
- triacetylolcandomycin
- TBDMS
- tert. butyldimethylsilyl
- TBDS
- tert. butyldiphenylsilyl
- BuLi
- butylithium
- DMF
- dimethylformamide
- EtOAc
- ethyl acetate
- THF
- tetrahydrofuran
- Et3N
- triethylamine
- PE
- petroleum ether
- DIBAL
- diisobutylaluminiumhydride
- PCC
- pyridinium chlorochromate
- Et3B
- triethylborane
- ESI
- electrospray ionization
- HEPES
- N-2-hydroxyethpiperazine-N′-2-ethanesulfonic acid
- Received July 24, 1996.
- Accepted October 29, 1996.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}