


Abstract
The metabolism and disposition of [14C]ritonavir (ABT-538, NORVIR), a potent, orally active HIV-1 protease inhibitor, were investigated in male and female Sprague-Dawley rats, beagle dogs, and HIV-negative male human volunteers. Rats and dogs received a 5 mg/kg iv, 20 mg/kg oral or 20 mg/kg intraduodenal dose, whereas humans received a single 600-mg liquid oral dose. Ritonavir was cleared primarily viahepatobiliary elimination in all three species. After iv or oral dosing in either rats or dogs, >92% of the dose was recovered in rat and dog feces and ≤4% was recovered in the urine. Humans excreted 86.3% of the oral dose in feces and 11.3% in urine over 6 days. Bile-exteriorized rats and dogs excreted 85.5% and 39.8%, respectively, of the iv dose in bile, with <3% recovered in urine. Radio-HPLC analysis of bile, feces, and urine from all three species indicated extensive metabolism of ritonavir to a number of oxidative metabolites, some being species-specific, and all involving metabolism at the terminal functional groups of the molecule. Glucuronide metabolites were observed in dog only. Plasma radioactivity consisted predominantly of unchanged parent drug in all three species. M-2, the product of hydroxylation at the methine carbon of the terminal isopropyl moiety of ritonavir, was the only metabolite present in human plasma and made up 30.4% of the total dose recovered in human excreta over 6 days. Tissue distribution of ritonavir in rat was widespread, with good distribution into lymphatic tissue but low CNS penetration. Plasma protein binding of ritonavir was high (96–99.5%) in all species and was nonsaturable in humans at concentrations up to 30 μg/ml. Partitioning into the formed elements of whole blood was minimal.
Ritonavir1 (ABT-538, NORVIR, fig. 1) is a rationally-designed peptidomimetic inhibitor of the HIV-1 protease, the virus-encoded enzyme responsible for the proteolytic processing of the viral gag and gag-pol polyproteins (1). Inhibition of the HIV-1 protease prevents assembly and maturation of infectious virions, resulting in the formation of noninfectious viral progeny and effectively blocking HIV replication. This therapeutic target has made the discovery of potent, orally-bioavailable HIV protease inhibitors for the treatment of AIDS the subject of intense scientific effort (2). Ritonavir has been shown to possess excellent in vitroantiviral potency (EC50 = 0.022 μM against HIV-1IIIB in MT4 cells) and oral pharmacokinetics (bioavailability >70% in animals, with plasma levels exceeding thein vitro EC50 for anti-HIV activity for at least 6 hr after oral dosing) (2). Recent reports have shown ritonavir to produce encouraging clinical decreases in plasma viral RNA and increases in CD4 cells in HIV-positive patients (3,4,5). These positive results have led to the recent approval of the drug by the FDA. This report describes the metabolism and disposition of [14C]ritonavir in rats and dogs after single oral, iv, or intraduodenal (id) dose administration and in humans after administration of a single oral dose.

Structure of ritonavir.
* indicates the position of the carbon-14 label.
Materials and Methods
Materials.
[14C]Ritonavir, uniformly 14C-labeled in the valine moiety of the molecule, was synthesized at Abbott Laboratories (Abbott Park, IL) via a five-step procedure beginning with [UL-14C]L-valine. The radiochemical purity of the [14C]ritonavir was ≥97% by HPLC and the specific activity was 64 μCi/mg. Unlabeled ritonavir, synthetic standards of metabolites M-1 (A-98498), M-2 (A-136478), andM-11 (A-155679) and the liquid oral solution used for human administration were all prepared at Abbott Laboratories. Drug was formulated for animal studies as the p-toluenesulfonic acid salt in 20% ethanol/30% propylene glycol/50% dextrose (5%) for iv dosing or in 10% ethanol/90% propylene glycol for oral or id dosing.
Human Study.
Five healthy male volunteers (weight 140–177 lb, age 20–33) received a single 600-mg liquid oral dose of [14C]ritonavir corresponding to 7.5 ml and 100 μCi per subject. Written informed consent and approval by the Peninsular Testing Corporation (Miami, FL) Institutional Review Board were obtained. Venous blood samples (10 ml) from each of the five subjects were collected into heparinized blood collection tubes just prior to dosing (0 hr) and at the following times after dosing: 0.25, 0.5, 1.0, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 48, 72, 96, 120, and 144 hr. The blood samples were centrifuged and the plasma was transferred into appropriately labeled tubes and frozen at −20°C. Urine was collected separately, as voided, from each of the five subjects and pooled according to the following time ranges: 0–12, 12–24, 24–48, 48–72, 72–96, 96–120, and 120–144 hr after dosing. Feces excreted by each subject were collected as eliminated for 6 days after dose administration and were pooled by day for each subject. All urine and fecal samples were stored at −20°C.
Rat Studies.
Four male and four female Sprague-Dawley rats, weighing 220–270 g and about 6–10 weeks old, were obtained from Sasco Animal Laboratories (Oregon, WI). Four male and four female 6- to 9-wk-old Sprague-Dawley rats, weighing 210–250 g and each bearing a chronically implanted jugular vein cannula, were obtained from Hilltop Lab Animals, Inc. (Scottdale, PA). The rats were fasted overnight before use and for 8 hr after dosing. Water was allowed ad libitum. The rats were housed individually in stainless steel metabolism cages. [14C]Ritonavir was administered orally by gavage to four male and four female rats, where two rats of each sex had jugular vein cannulas, at a dose of 20 mg/kg, corresponding to 1 ml/kg and 10–13 μCi per rat. Similarly, four male and female rats were given a 5 mg/kg iv dose of [14C]ritonavir, corresponding to 1 ml/kg and 8.1–10 μCi per rat. For iv injections the rats were anesthetized with carbon dioxide and diethyl ether, a small incision was made on the medial surface of the hind leg, and the dose solution was injected into the femoral vein. After the incision had been closed with wound clips, the rats were allowed to recover from the anesthetic (5–10 min). Urine and feces excreted by the two noncannulated males and the two noncannulated females from each of the dosing groups were collected daily for 3 days. During the first 24 hr, the urine was collected over dry ice. In addition, the cages were washed daily with a small amount of 70% ethanol which was collected and saved for radioassay. All excreta samples were stored at −20°C. Heparinized blood samples (0.4–0.5 ml) were withdrawn from the jugular vein cannula of each cannulated rat at 0.1 (iv dose only), 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12, 24, 48, and 72 hr after dosing. The blood samples were centrifuged and refrigerated. Cannulas were flushed with heparin lock flush (Abbott Laboratories) immediately after each sampling to maintain patency.
Four male and four female 6- to 10-wk-old Sprague-Dawley rats, weighing 230–250 g and each bearing a chronically implanted bile duct cannula, were obtained from Hilltop Lab Animals, Inc. Radiolabeled ritonavir was administered intravenously to two rats of each sex at a dose of 5 mg/kg, corresponding to 1 ml/kg and 5.5–6.0 μCi/rat. The remaining two rats of each sex received [14C]ritonavir intraduodenally via the distal bile duct cannula at a dose of 20 mg/kg, corresponding to 1 ml/kg and 8.2–10.0 μCi/rat. Immediately after drug administration, a solution of taurocholic acid in saline (0.0278 g/ml) was continuously infused via the distal bile duct cannula into each rat at a rate of 1 ml/hr for 24 hr. All four rats were placed in acrylic restrainers with access to food and water for the duration of the study. Bile was collected from 0–2, 2–4, 4–6, and 6–24 hr after drug administration, while urine was collected from 0–24 hr. All samples were stored at −20°C until analysis.
Dog Studies.
Four male and two female beagle dogs, weighing 6–12 kg and between 9 months and 1.5 yr old, were obtained from Marshall Research Animals (North Rose, NY). The dogs were fasted overnight before use but were given free access to water. For oral and iv drug administration, food was given 12 hr after dosing and daily thereafter. During the study the animals were housed individually in stainless steel metabolism cages. [14C]Ritonavir was administered orally by gavage to two male and two female dogs at a dose of 20 mg/kg, corresponding to 1.0 ml/kg and 22.9–24.5 μCi/dog. Two weeks later, radiolabeled ritonavir was administered intravenously to the same dogs as a slow bolus over 1–2 min at a dose of 5.0 mg/kg, corresponding to 1.0 ml/kg and 18.6–19.7 μCi/dog. Blood samples (4–5 ml) were withdrawn from the jugular vein of the dogs using heparinized Vacutainers (Becton-Dickinson) at the following times after drug administration: 0, 0.1 (iv dose only), 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, and 72 hr. Duplicate aliquots of whole blood samples taken at 0.25, 1, and 3 hr were saved for radioassay. All blood samples were then centrifuged at about 4°C and the plasma was transferred to clean tubes and frozen at −20°C. Urine and feces were collected from the dogs daily for 5 days. For the first 24 hr, the urine was collected in containers surrounded by dry ice. In addition, the cages were washed daily with a small amount of 70% ethanol which was collected and saved for radioassay. All excreta samples were stored at −20°C.
Two male dogs were anesthetized with an initial sodium pentobarbital iv dose and maintained under anesthesia with a slow iv drip infusion of sodium pentobarbital delivered via the cephalic vein using a butterfly infusion set. A midline incision was made in the abdomen, the gall bladder was clamped off, and the bile duct was cannulated with polyethylene tubing. [14C]Ritonavir was given intravenously, via the cephalic vein, to one dog at a dose of 5 mg/kg, corresponding to 1 ml/kg and 13.9 μCi. A second dog received a 20 mg/kg id dose of [14C]ritonavir, corresponding to 0.5 ml/kg and 17.8 μCi. Bile was collected from 0–2, 2–4, and 4–6 hr after dosing. After euthanasia with T-61 Euthanasia Solution (Hoechst Roussel, Somerville, NJ), urine was collected from the bladder. Individual volumes were recorded for the bile and urine samples, which were then stored at −20°C until analysis.
Rat Tissue Distribution.
Fifteen male and 21 female Sprague-Dawley rats (Sasco), weighing 180–260 g and 6–11 wk old were fasted overnight before use and for 4 hr after dosing. Water was allowed ad libitum. During the study the rats were housed individually in stainless steel metabolism cages. Radiolabeled ritonavir was administered orally by gavage to all rats at a dose of 50.0 mg/kg, corresponding to 2.0 ml/kg and 19–21 μCi/rat (males) or 10–13 μCi/rat (females). Groups of three rats of each sex were sacrificed at 1, 3, 6, 9 (females only), 12 (females only), 24, and 48 hr. The animals were anesthetized with carbon dioxide and exsanguinated by cardiac puncture. Aliquots of whole blood were saved for radioassay, then the plasma was separated from the cells by centrifugation and stored at −20°C. Tissues were excised, blotted dry, and weighed prior to radioassay.
Sample Radioanalysis.
Prior to radioassay, fecal samples and tissue samples were homogenized in 70% ethanol in water using a mechanical high frequency homogenizer. Duplicate aliquots of the plasma, bile, and urine samples were radioassayed directly in Insta-Gel (Packard Instruments, Meriden, CT) scintillation cocktail. Duplicate aliquots of the blood samples, tissue homogenates, and fecal homogenates were placed in Combusto-Cones (Packard) containing cellulose powder (Whatman) and burned in a Tri-Carb® (Packard) Model 306 or 307 Sample Oxidizer. Each small organ that was not homogenized was placed in a Combusto-Cone containing cellulose powder and burned intact as a single sample. The resultant 14CO2 was trapped in Carbo-Sorb® (Packard) and radioassayed in Permafluor® (Packard) scintillation fluid. All samples were counted in a Tri-Carb (Packard) Model 1500TR, 1900TR, or 2500TR Liquid Scintillation Spectrometer and corrected for quenching with automatic external standardization. The results were based on standards that were prepared in duplicate from the dose solution and radioassayed in a manner similar to the samples.
Sample Preparation.
Frozen plasma samples were allowed to thaw at ambient temperature. Three volumes of acetonitrile were added to plasma and the mixture was vortexed and centrifuged. The resulting supernatant was concentrated to dryness under a nitrogen stream, the residue reconstituted in HPLC mobile phase, and the mixture filtered by centrifugation (Millipore 0.45 μm filters) prior to HPLC analysis. Plasma samples from all four orally-dosed rats were combined by time point to afford single injection samples containing enough radioactivity for radioflow detection. Recovery of rat and dog plasma radioactivity was >89% by this precipitation procedure. Human plasma samples were treated similarly except that the protein pellet was rinsed with acetonitrile, followed by methanol and, after concentration, the residue was reconstituted in 200 μl of 15% acetonitrile/15% methanol/70% 25 mM ammonium acetate (adjusted to pH 4.8 with trifluoroacetic acid). Recovery of human plasma radioactivity was 97% by this procedure. Composite fecal homogenates for each human subject (24–96 hr for Subjects 501, 506, and 514; 24–120 hr for Subject 508; and 48–120 hr for Subject 504) or animal were prepared by combining proportionate aliquots of the individual fecal homogenates containing the highest percentages of radioactivity based on total volume. Fecal homogenates were centrifuged, the pellets rinsed twice with ethanol, and the supernatants filtered through Acrodisc (Gelman) 0.45 μm filters prior to analysis. This procedure afforded ≥90% recovery of fecal radioactivity. Bile samples were simply filtered through Acrodisc filters prior to HPLC assay. Urine samples were concentrated under a nitrogen stream or via lyophilization, reconstituted in HPLC mobile phase, and centrifugally filtered (Millipore) for HPLC analysis.
Enzymatic hydrolyses of potential glucuronide conjugates in bile were conducted by combining 40–50 μl of rat or dog 0–2 hr bile, or individual purified bile metabolites with either 260 μl of 0.1 M phosphate buffer, pH 6.8, or 200 μl of 1 M sodium acetate buffer, pH 5.0. β-Glucuronidase (Type IX-A, Sigma) was added to the phosphate buffer solution to give a final concentration of 5 mg enzyme/ml and 60 μl of a β-glucuronidase/sulfatase mixture (Type H-2, Sigma) was added to the acetate buffer solution. The mixtures were incubated at 37°C in a shaking water bath for 17 hr. Control samples consisting of bile in pH 6.8 phosphate buffer or pH 5.0 acetate buffer were also incubated without enzyme. After incubation, samples were centrifugally filtered (Millipore, 0.45 μm), the filtrates concentrated to dryness under a nitrogen stream and the residues reconstituted in 200 μl of HPLC mobile phase for analysis.
HPLC.
All analyses were performed using a Perkin-Elmer Series 410 liquid chromatography pump, an Hitachi Model AS-2000 autosampler, and an Applied Biosystems Model 1000S photodiode array detector operated at 220 nm. Separations were achieved at ambient temperature with a Beckman Ultrasphere 5 μm 4.6 × 250 mm C18 column connected to a Nucleosil 5 μm C18 cartridge guard column (Alltech Associates, Deerfield, IL). A linear gradient of 25–60% acetonitrile in 0.1% trifluoroacetic acid (adjusted to pH 4.8 with ammonium acetate) over 50 min was used as column eluent at a flow rate of 1 ml/min. Radioactivity in the column effluent was monitored with a Flo-One/Beta Model A-500 radioactivity flow detector (Packard) equipped with a 0.5 ml flow cell. A ratio of column effluent to liquid scintillator (Flo-Scint III, Packard) of 1:3 was standard. Preparative HPLC isolation of metabolites from rat and dog bile was performed on a Nucleosil 5 μm 10 × 190 mm C18 column (Alltech) connected to a Nucleosil 5 μm C18 cartridge guard column (Alltech). A linear gradient of 25–60% acetonitrile in 0.1% trifluoroacetic acid (adjusted to pH 4.8 with ammonium acetate) over 50 min was used as column eluent at a flow rate of 3 ml/min. HPLC column effluent was collected in 1-min fractions and mobile phase was removed from appropriate fractions by vacuum evaporation (Labconco RapidVap). The resulting residues were taken up in acetonitrile and stored at −20°C until analysis.
Mass Spectrometry.
Low resolution fast atom bombardment mass spectrometry (FABMS) mass spectra were acquired with a Finnigan (Finnegan MAT, San Jose, CA) MAT95 mass spectrometer using a primary ion beam of 8 kV xenon atoms, a mass resolution of 1700 and glycerol as the liquid matrix. High resolution FABMS mass spectra were acquired with a Kratos MS-50 mass spectrometer (Kratos Analytical, Manchester, UK) as described above except that a mass resolution of 10,000 was used. MS/MS spectra were acquired by electrospray ionization (ESI) using a Finnigan MAT TSQ700 mass spectrometer. Samples were infused with 2-methoxyethanol. A nitrogen sheath gas and −6 eV argon collision gas at 3.2 mTorr were used.
Protein Binding.
Heparinized blood was obtained from male and female rats, beagle dogs, and cynomolgus monkeys (at least two of each sex), that had been fasted overnight. Blood was also obtained from adult human volunteers (two of each sex), who had fasted at least 8 hr, had taken no medication except aspirin during the preceding 7 days, and had taken no salicylates during the preceding 48 hr. Binding was determined in a Spectrum Equilibrium Dialysis System (Spectrum Medical Industries, Los Angeles, CA) using 1-ml cells and a Spectra/Por 2 membrane with a molecular weight cut-off of 12000–14000. Before use, the membranes were soaked in distilled water for at least 15 min and then in 30% aqueous ethanol for 20 min. After rinsing to remove the residual ethanol, the membranes were placed in dialyzing buffer (0.02 M phosphate buffer, pH 7.4, containing 0.6% NaCl) for at least 15 min. Dialysis cells were rotated at about 20 rpm for 3 hr in a water bath maintained at approximately 37°C. Duplicate aliquots of all plasma and buffer samples were radioassayed. The protein binding was calculated according to the following formula:
Pharmacokinetic Parameters.
The peak plasma total radioactivity concentration (Cmax) and the time to reach the peak plasma concentration (Tmax) were taken directly from the plasma total radioactivity concentration-time data. The area under the plasma radioactivityvs. time curve (AUC) for total radioactivity and metabolites was calculated by the linear trapezoidal rule. Cell to plasma (C/P) ratios were calculated from radioactivity concentrations in blood (Cb) and plasma (Cp) as follows:
Results
Excretion.
Male and female rats and dogs given a 5 mg/kg iv dose and a 20 mg/kg oral dose of [14C]ritonavir displayed high fecal excretion (>92%) of the radiolabeled dose within 3–5 days, indicating that the drug was cleared predominantly by hepatobiliary processes in both species (table 1). Urinary excretion accounted for ≤ 4% of the dose in rats and dogs. In male humans, 11.3% of a 600-mg oral dose was excreted in urine and 86.3% was recovered in feces within 6 days after dosing. Most (7.7%) of the dose recovered in human urine was excreted within 24 hr after dosing.
Mean excretion of radioactivity after oral or iv administration of [14C]ritonavir to rats, dogs, and humans
The importance of biliary elimination to the clearance of ritonavir was verified in bile-exteriorized male rats and dogs where 85.5% (rat) and 39.8% (dog) of a 5 mg/kg iv dose was excreted in the bile within 6 hr (dog) and 24 hr (rat) after dosing, accompanied by minimal dose recovery in urine (<3%) (table 2). Intraduodenal administration of a 20 mg/kg dose resulted in the recovery of 10% of the dose in the 0–6 hr bile of dog and 25.9% in male rat 0–24 hr bile. Iv administration of a 5 mg/kg dose of [14C]ritonavir to chronically bile-exteriorized female rats resulted in excretion of 55.6% of the labeled dose in the bile within 24 hr. Not only was the percentage of total dose excreted significantly less than that of male rats, but the percentage of total dose excreted in the first 2 hr after dosing was significantly lower in female rats. After id dosing, female rats again excreted a significantly lower percentage of the total dose in the 0–2 hr bile compared to males, although the total dose excreted over 24 hr was similar.
Excretion of radioactivity in bile and urine of rats and dogs after a 5 mg/kg iv or a 20 mg/kg intraduodenal (id) dose of [14C]ritonavir
Tissue Distribution.
[14C]Ritonavir was widely distributed in the tissues of rats after oral administration of a 50 mg/kg dose. Concentrations of radioactivity were highest in most tissues and plasma at 3 hr after dosing (table 3). Highest radioactivity concentrations were found in the liver, with 133 μg eq/g present at 3 hr, which was 12.4 times greater than the corresponding plasma concentration of 11.2 μg eq/g. The next highest drug concentrations at 3 hr were found in the adrenals (72.6 μg eq/g), pancreas (41.1 μg eq/g), kidneys (37.0 μg eq/g) and thyroid (27.4 μg eq/g) where respective tissue to plasma (T/P) ratios of 6.55, 3.81, 3.34 and 2.30 were calculated (table4). Maximum radioactivity concentrations in all other tissues except eyes (T/P = 0.18) and brain (T/P = 0.08) were roughly similar to the 3-hr plasma-14C concentration, affording T/P ratios of 0.64–1.90. The negligible14C-concentrations found in the brain suggest that ritonavir does not appreciably penetrate the blood-brain barrier in rats.
Mean concentrations of radioactivity in tissues of rats after oral administration of a 50 mg/kg dose of [14C]ritonavir
Mean tissue to plasma ratios in rats after oral administration of a 50 mg/kg dose of [14C]ritonavir
After 3 hr, the levels of radioactivity in most tissues and plasma of rats generally declined until 12 hr after dosing. The plasma levels declined slowly from a peak concentration of 11.2 μg eq/g at 3 hr to 5.84 μg eq/g at 9 hr. At 12 hr plasma radioactivity concentrations declined to 0.07 μg eq/g, accompanied by similarly large decreases in tissue radioactivity levels. However, at 24 hr slightly higher concentrations of radioactivity were found in plasma (0.17 μg eq/g) and several tissues. The majority of the 48-hr tissue radioactivity was contained in the liver where a concentration of 2.70 μg eq/g corresponded to 0.24% of the 14C-dose. At 1 hr after dosing, 52.1% of the dose was found in the stomach and contents and 32.1% was found in the small intestine and contents. Liver, muscle, skin, and fat contained 5.86, 4.95, 1.55 and 1.14% of the dose, respectively, while the remaining tissues each contained ≤0.59% of the dose at 1 hr. The observation of 5.42–8.01 μg eq/g of dose radioactivity in the brachial, lumbar, mediastinal, and submaxillary lymph nodes at 1 hr indicates that ritonavir readily distributes into lymphatic tissue, which is believed to be the primary site of HIV replication in vivo (6). After 3 hr, tissue to plasma ratios generally increased or remained roughly constant over time for all tissues, indicating slower clearance of drug from tissues than from plasma. Liver, pancreas, kidneys and lungs had the highest T/P ratios at 24 hr after dosing and all tissues had T/P ratios >1.0 at 24 hr. The residual carcass contained 6.94% of the dose at 48 hr.
Metabolite Identification.
HPLC radiochromatograms of 0–2 hr rat and dog bile collected after iv dosing of [14C]ritonavir are shown in fig.2. Major radioactive peaks were isolated from rat and dog bile by preparative HPLC and characterized by mass spectral analysis. Positive ion ESI/MS/MS and positive ion FABMS were used to analyze each bile metabolite sample. High resolution mass spectra were obtained for several metabolites by positive ion FABMS using a peak matching technique with a resolution (10% valley) of 10,000. All measured metabolite masses were within ± 1.0 millimass unit of calculated masses for the proposed metabolite structures. The identities of major human metabolites of [14C]ritonavir were established by coinjection of metabolites with synthetic standards and with characterized metabolites obtained from human liver microsomal incubations (7). Additionally, HPLC coinjection was used to establish probable structural identity between individual purified rat bile metabolites, dog bile metabolites and human excreta metabolites. The proposed structures of the major metabolites of ritonavir are shown in fig. 3 and MS fragmentation data for ritonavir and select metabolites are shown in fig.4. Ritonavir eluted at 42–43.5 minutes and displayed a protonated molecular ion at m/z 721.

HPLC radiochromatograms of rat and dog 0–2 hr bile collected after a 5 mg/kg iv dose of [14C]ritonavir.
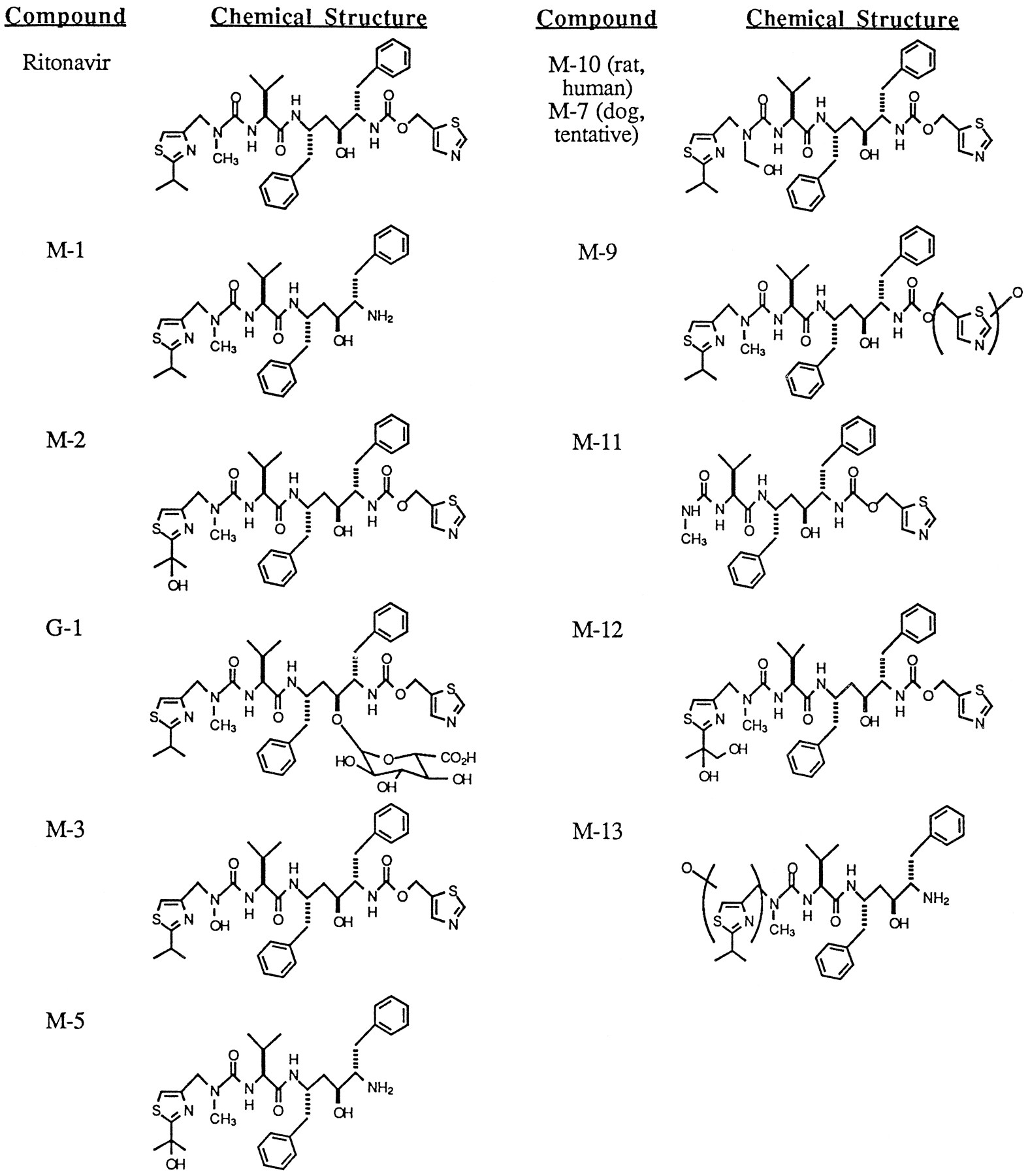
Proposed structures of the major metabolites of ritonavir.

Characteristic MS fragmentations of ritonavir and select metabolites.
M-1.
The peak eluting at 34.7 min yielded a protonated molecular ion at m/z 580. High resolution MS and MS/MS daughter ions at m/z 140, 171, 197, 268, and 296 pointed to loss of the thiazolyl carbamate moiety, C5H3NO2S. The structure ofM-1 was verified by HPLC coelution with the synthetic standard A-98498.
M-2.
A protonated molecular ion at m/z 737 was also obtained by FABMS and ESIMS for the peak eluting at 28.8 min. High resolution MS indicated addition of one oxygen atom to parent drug. ESI/MS/MS of this ion resulted in daughter ions at m/z 312, 426, and 551, indicating that the oxygen atom resided in theN-methyl-isopropylthiazolylmethyl terminal end of the molecule. Consideration of likely sites of oxidation within this region pointed to hydroxylation of the isopropyl group of the isopropylthiazolyl moiety. MS/MS analysis of the sodium adduct ofM-2 at m/z 759 afforded a major daughter ion at m/z 701. This loss of 58 amu was quite facile, suggesting that the isopropyl moiety of M-2 is hydroxylated at the central methine carbon, not on the pro-S or pro-R methyl carbons of the isopropyl moiety. This structure was verified by HPLC coelution ofM-2 with the synthetic standard A-136478.
M-3.
A protonated molecular ion was observed at m/z 723 for the peak eluting at 25.9 min. High resolution MS indicated a methyl group loss (−14 amu) and oxidation (+16 amu) to afford a net gain of 2 amu compared to parent drug. MS/MS daughter ions at m/z 199, 270, and 298 indicated that the functional group changes both occurred on theN-methyl urea isopropylthiazolyl terminus, while daughter ions at m/z 181, 252, and 280 indicated a facile loss of H2O from this region of the molecule. Additionally, a large daughter ion at m/z 705 was observed, again indicating facile loss of water from M-3. MS/MS spectra of other ritonavir metabolites containing a hydroxylated isopropyl group or oxygenated methyl isopropylthiazolyl moiety displayed no prominent [M+H-18]+ daughter ion, suggesting that the site of oxygen addition was on the urea nitrogen. Thus, M-3 was tentatively identified as the hydroxylamine resulting fromN-demethylation of the N-methyl urea nitrogen followed by oxidation of that same nitrogen.
M-5.
The metabolite eluting at 20.0 min displayed an [M+H]+ at m/z 596. This is 16 amu higher than the [M+H]+ observed for M-1 where the thiazolyl carbamate moiety has been lost from parent drug. MS/MS analysis of M-5 displayed daughter ions at m/z 410 and 285, indicating that oxidation occurred at a site within the isopropylthiazolyl moiety. Furthermore, observation of the loss of 58 amu from the protonated molecular ion (596 to 538) and from a fragment ion (212 to 154) suggests that the isopropyl group ofM-5 is hydroxylated at the methine carbon. Thus,M-5 appeared to be the product of methine-carbon hydroxylation of the isopropyl group of M-1 or of loss of the thiazolyl carbamate moiety of M-2.
M-9.
The peak eluting at 29.9 min yielded a protonated molecular ion at m/z 737, indicating the addition of one oxygen atom to parent drug. MS/MS daughter ions at m/z 197, 296, 442, and 567 pointed to the presence of this oxygen on the methylthiazolyl moiety of ritonavir although the position of oxygen addition (i.e. at the sulfur, nitrogen, aromatic carbons or methylene carbon) could not be discerned.
M-10/7.
The peak eluting at 27.7 min in the rat bile chromatogram and designated M-10 afforded a protonated molecular ion at m/z 737 and a sodium adduct at m/z 759. MS/MS analysis of the sodium adduct resulted in daughter ions at m/z 209, indicating that the additional oxygen atom is on the isopropylthiazolyl N-methyl urea moiety (m/z 171 + 16 + Na), at m/z 179, indicating a loss of 30 amu from the m/z 209 ion, and at m/z 729, indicating a loss of 30 amu from the [M+Na]+ adduct. The loss of 30 amu suggests loss of CH2O, which could occur either from a hydroxylated methyl group on the isopropyl moiety or from a hydroxylatedN-methyl group in the urea moiety. The intensity of the m/z 729 daughter ion suggested that this −30 amu fragmentation is quite facile so M-10 was tentatively identified as theN-hydroxymethyl urea metabolite of parent drug. The metabolite designated M-7 in dog bile was produced by hydrolysis of the glucuronide metabolite G-2 with β-glucuronidase. Previous MS analysis of G-2 had shown the compound to have a molecular weight 192 amu higher than that of Abbott-84538, consistent with the presence of glucuronic acid (176 amu) and one additional oxygen (16 amu). The M-7 thus produced had the same HPLC retention time as that of rat M-10, suggesting that the two metabolites were structurally identical. However, failure to obtain good ESI/MS/MS data for M-7prohibited confirmation of identity.
M-11.
ESIMS analysis of the metabolite eluting at 23.3 min displayed one major ion at m/z 582. MS/MS of the m/z 582 ion produced daughter ions at m/z 426 and 526, suggesting loss of the isopropylthiazolylmethyl moiety to afford the terminal N-methyl urea metabolite. The structure of M-11 was verified by HPLC coelution with the synthetic standard A-155679.
M-12.
The HPLC peak eluting at 23.3 min in rat and dog bile also displayed a very minor [M+H]+ at m/z 753 by FABMS and ESIMS, suggesting addition of two oxygen atoms to parent drug. MS/MS of the m/z 775 sodium adduct displayed a prominent daughter ion at m/z 701, indicating a loss of 74 amu, which is consistent with hydroxylation of both the methine and one of the two methyl groups of the isopropyl moiety. No prominent daughter ions corresponding to loss of 30 amu (as for M-10) or 58 amu (as for M-2) were produced, respectively indicating that the isopropyl group of M-12 was not monohydroxylated and the methyl group of the N-methyl urea moiety was not oxidized. Thus, M-12 was tentatively identified as the product of oxidation of the isopropyl group to a 1,2-diol.
M-13.
The HPLC peak eluting at 23.3 min in dog bile also displayed a minor [M+H]+ at m/z 596 by FABMS and ESIMS. High resolution MS of this ion indicated loss of C5H3NOS (-125 amu) from parent drug. MS/MS of the m/z 596 ion, designatedM-13, resulted in daughter ions at m/z 285 and m/z 410, indicating loss of the methylthiazolyl carbamate moiety, and at m/z 155, 187, 213 and 312, indicating oxidation on the isopropylthiazolylmethyl moiety. No evidence for loss of 30 amu or 58 amu was seen, indicating that the isopropyl group was unchanged and that the oxygen atom is probably on the methylthiazolyl group, perhaps in the form of an N- or S-oxide.
G-1.
The peak eluting at 20.9–22.7 min in dog bile displayed a protonated molecular ion at m/z 897, indicating the addition of 176 amu to parent drug. High resolution MS indicated addition of C6H8O6 to the molecular formula for ritonavir, strongly pointing to a glucuronic acid conjugate of parent drug. This was verified by hydrolysis of purified G-1 with β-glucuronidase enzyme, which afforded complete conversion of the metabolite to ritonavir. The hydoxyl group in the interior of the ritonavir structural backbone would seem to be the most logical site of glucuronic acid conjugation and, indeed, purified G-1 was stable to mild acid and base hydrolysis conditions (1 M acid or base, 37°C, 1 hr), consistent with an ether-type glucuronide.
G-2 (structure not shown in fig. 3).
A protonated molecular ion was observed at m/z 913 for the peak eluting at 14.7 min in dog bile. The apparent addition of 192 amu to parent drug suggests both a glucuronide moiety (+176) and an additional oxygen (+16). This was verified by hydrolysis of purified G-2 with β-glucuronidase, resulting in the complete disappearance of theG-2 peak at 14.7 min and the appearance of a peak at 28.1 min, designated M-7. Although the amount of M-7isolated via G-2 hydrolysis prohibited MS characterization, M-7 did coelute by HPLC with ratM-10. Thus, the structure of G-2 is consistent with the glucuronide conjugate of the N-hydroxymethyl urea metabolite of parent drug. However, it is unknown whether glucuronidation occurs at the hydroxy group within the interior to the molecule or at the N-hydroxymethyl group.
Metabolite Profiles in Bile.
The 0- to 6-hr bile metabolite profiles in rat and dog after iv and id dosing are characterized by extensive metabolism of ritonavir to a large number of metabolites, with no individual metabolite formed preferentially in either species (table 5). After iv dosing in male rats, M-2 (12.6% of 0–6 h biliary-14C), M-9 (10.7%), and M-11(15.2%) contributed the largest individual percentages of biliary radioactivity, while the secondary metabolite M-5 became more prominent (14.4%) after id administration. The small amounts ofM-12 and M-13 detected by MS in theM-11 metabolite peak were not resolvable fromM-11 by the HPLC method used in the rat and dog metabolism studies so they were quantitated as a single radio-HPLC peak. NoG-1 or G-2 were observed in rat bile after dosing by either route. Bile from female rats contained significantly more unchanged parent drug, M-2 and M-10 than seen in male rat bile. In dog, M-2, M-5, M-6,M-11, G-1 and G-2 each made up 7.9–10.1% of the biliary-14C after iv dosing.M-6 was a relatively polar metabolite eluting at 12 min that resisted several attempts at structural characterization viaESI/MS. Necessarily, no tentative structure can currently be proposed for this metabolite seen only in dog bile. However, M-6 did not appear to be a glucuronic acid conjugate, remaining stable to β-glucuronidase hydrolysis and chemical hydrolysis (1M NaOH or 1 M HCl, 60°C, 17 hr). After id dosing, M-6 (15.2%) andG-1 (18.1%) contributed larger percentages of the biliary radioactivity in dog than after iv dosing. Unchanged parent drug (≤1.9% of biliary-14C), M-6 and characterized metabolites together accounted for 61–70% of the total dose radioactivity recovered in male rat and dog bile after iv and id dosing, with the remaining radioactivity consisting mainly of a large number of minor, poorly-resolved, polar radio-HPLC peaks.
Distribution of parent drug and metabolites in 0- to 6-hr bile of rats and dogs following a 5 mg/kg iv or a 20 mg/kg intraduodenal (id) dose of [14C]ritonavir
Metabolite Profiles in Feces.
Radioactivity in rat and dog feces consisted predominantly of unchanged parent drug (45.9–61.9% of total dose radioactivity) after oral administration, accompanied by lesser amounts of M-1,M-2, M-3, M-5, M-10, andM-11 (table 6). An unknown metabolite,M-8, was a major fecal component present in dog, contributing 29.9% of the total dose radioactivity after iv administration and 8.3% after oral dosing. M-8 was not observed to any significant extent in dog bile, suggesting that this compound may be produced from bile metabolites by GI microbes. In rat,M-2, M-11, and M-1 were the major fecal metabolites observed after iv dosing. Despite its nearly complete absence in rat and dog bile, ritonavir contributed 12.9–21.2% of the total dose radioactivity in feces collected from both species after iv dosing. Unlike bile, no significant differences in fecal metabolite profiles were observed between male and female rats.
Distribution of parent drug and metabolites in rat, dog, and human feces after iv and oral administration of [14C]ritonavir
In humans, unchanged ritonavir was the major radioactive fecal component after oral dosing, making up 33.8% of the mean total dose radioactivity. However, the extent of ritonavir excretion in feces varied noticeably between subjects, with extremes of 17.4% and 47.0% of the dose excreted as ritonavir in Subjects 3 and 2, respectively.M-2 was the most abundant radioactive metabolite in feces, contributing 24.0% of the total dose radioactivity. This was accompanied by lesser amounts of M-11 (9.4% of total dose radioactivity), M-5 (3.9%), M-1 (3.5%), andM-3 (1.7%). No M-9, G-1, orG-2 were observed in human feces.
Metabolite Profiles in Urine.
The small amounts of radioactivity recovered in the urine of both animal species consisted mainly of M-1 and M-2, accompanied by a lesser amount of G-1 in dog andM-9 in rat (table 7). No ritonavir was detected in urine for either rat or dog.
Distribution of parent drug and metabolites in rat, dog, and human urine after iv and oral administration of [14C]ritonavir
After oral dosing in humans, M-2 was the major radioactive component of urine, contributing 57.5% of the mean urinary radioactivity and 6.4% of the mean total dose radioactivity. Ritonavir accounted for 30.5% of the urinary radioactivity and 3.5% of the total dose radioactivity. Significantly lesser amounts ofM-11, M-1, M-10, and M-5were also observed, each making up <1% of the total dose radioactivity. HPLC coinjection of dog G-1 with human urine showed no glucuronide to be present.
Total Radioactivity in Plasma.
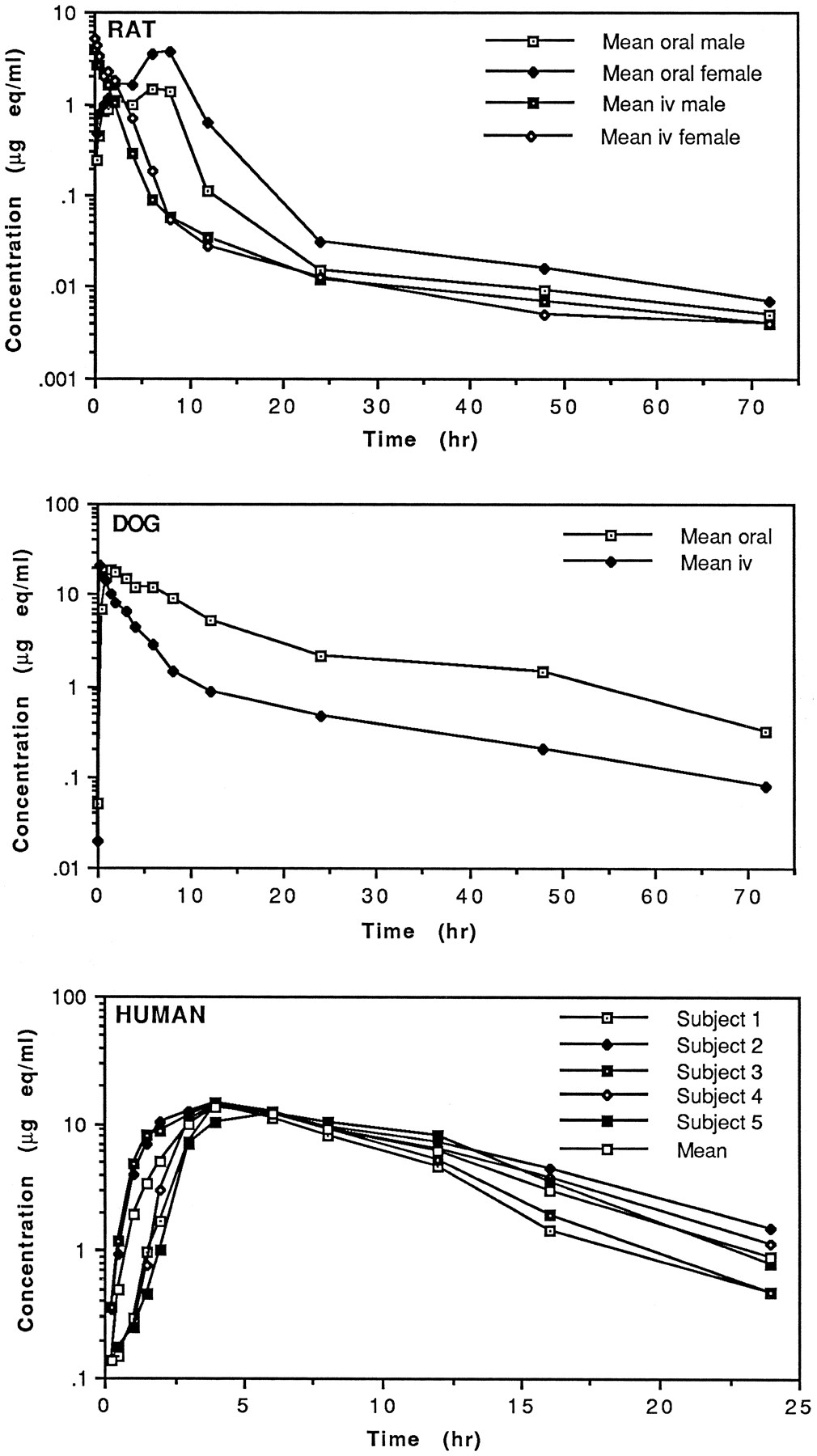
After oral administration of a 20 mg/kg dose of [14C]ritonavir to rats, total plasma radioactivity Cmax values of 1.433 μg eq/ml in males and 3.70 μg eq/ml in females were not attained until 8 hr after dosing (fig.5, table 8). In male rats this peak level was preceded by a smaller concentration maximum of 1.43 μg eq/ml at 2 hr. Although plasma concentration-time profiles were qualitatively similar between sexes, the mean total radioactivity AUC value for female rats was 2- to 3-fold higher than that of male rats. After a 5 mg/kg iv dose, female rats displayed a mean total radioactivity AUC value about 1.5-fold higher than male rats. In dogs, a plasma total radioactivity Cmax of 18.82 μg eq/ml was observed at 1 hr after a 20 mg/kg oral dose. Unlike rat, no sex difference was observed in total radioactivity AUC values after oral or iv dosing in dog.

Plasma concentration-time profiles of total radioactivity after administration of a 5 mg/kg iv or 20 mg/kg oral dose of [14C] ritonavir to rats and dogs or a 600 mg oral dose of [14C] ritonavir to humans.
Pharmacokinetic parameters of total radioactivity, ritonavir and metabolites in rat, dog, and human plasma after iv and oral administration of [14C]ritonavir
In male human subjects, a mean peak plasma radioactivity level of 14.0 μg eq/ml was observed at 4.4 hr after oral administration. In four of the five subjects, Cmax and Tmax values varied little, ranging from 14.3–14.6 μg eq/ml at 4 hr. Subject 5 displayed a lower Cmax of 12.2 μg eq/ml at 6 hr after dosing. A mean AUC0–24 of 135 μg eq·h/ml (range 106–161 μg eq·h/ml) was determined for the five subjects. Radioactivity levels fell below the limits of detection (0.15 μg eq/ml) in all plasma samples collected after 24 hr.
Metabolite Profiles in Plasma.
In rat, only unchanged parent drug was detected in pooled plasma samples collected between 0.25 and 8 hr after oral administration. Out to 2 hr after iv administration, M-1 was the only metabolite detected in plasma, with levels increasing over time to 0.037–0.08 μg eq/ml at 1.5–2 hr. Comparison of the AUC0–2 values for ritonavir and M-1 indicated a 48-fold and 135-fold excess of parent drug to M-1 in male and female rat plasma, respectively, in the first 2 hr after iv dosing. Plasma samples collected later than 2 hr after iv dosing did not contain sufficient radioactivity for profiling. M-1 is essentially inactive against HIV in an in vitro MT4 cell assay (8).
In dog, unchanged parent drug was the major circulating radioactive component during the first 6 hr after oral dose administration. This was accompanied by 3 minor metabolites, M-1, M-2, and G-1. Maximum levels (17.91 μg eq/ml) of ritonavir were reached at 1 hr, then levels gradually decreased over the next 5 hr. Mean plasma levels of M-1, the most abundant plasma metabolite, were roughly constant (0.53–0.70 μg eq/ml) from 1–6 hr after oral dosing. The mean AUC0–6 values calculated for ritonavir and the 3 plasma metabolites show the AUC of parent drug to be about 21-fold larger than the AUC of M-1 and about 15-fold larger than the combined AUCs of all three metabolites (4.64 μg eq·hr/ml). During the first 3 hr after iv dosing, unchanged parent drug was the major circulating radioactive component, again accompanied primarily by M-1, along with lesser amounts ofM-2 and G-1. Plasma levels of M-1generally increased after iv dosing, with 1.17 μg eq/ml ofM-1 present at 3 hr. The mean AUC0–3 values calculated for ritonavir and the plasma metabolites show the AUC of parent drug to be about 9-fold larger than the AUC of M-1and about 7-fold larger than the combined AUCs of all metabolites (3.34 μg ·hr/ml).
Only plasma samples collected between 2 and 12 hr contained enough radioactivity for radio-HPLC analysis for all five human subjects. After oral dose administration, M-2 was essentially the only metabolite seen in human plasma, achieving a mean Cmax of 0.67 ± 0.46 μg eq/ml at a mean Tmax of 8.8 ± 3.0 hr. Ritonavir reached a mean Cmax of 13.70 ± 0.92 μg eq/ml at a mean Tmax of 4.4 ± 0.9 hr. Comparison of the mean AUC2–12 values calculated for ritonavir andM-2 indicated a mean 30-fold (range = 12–164-fold) excess of parent drug over metabolite in systemic circulation between 2 and 12 hr after oral dosing. M-2 is equipotent to ritonavir against HIV in an MT4 cell assay (8).
Protein Binding.
The in vitro plasma protein binding of [14C]ritonavir was very high, especially for dog and human. At ritonavir concentrations of 0.01, 0.1, 1.0, 10, and 30 μg/ml, mean plasma protein binding percentages ranged from 97.2–99.2% for rat, 98.9–99.4% for dog, 96.2–99.2% for monkey, and 99.3–99.5% for human. Decreases in the extent of protein binding with increasing drug concentration were observed only for rat and monkey, where binding decreased from ∼99% at 0.1–1.0 μg/ml to 96–97% at 30 μg/mL. The extent of binding in human and dog plasma did not decrease significantly with increasing drug concentration, even at 30 μg/ml. No significant sex differences in the degree of protein binding were observed for any of the four species at any concentration.
Partitioning in Blood.
Radioactivity levels in blood and plasma samples collected from rats and dogs afforded cell to plasma (C/P) ratios of 0.11–0.29 in female rat, 0.48–0.51 in male rat, and 0.03–0.30 in dog. Negative C/P values were calculated from blood and plasma radioactivity concentrations measured in the human study. In addition, an in vitropartitioning study with human whole blood from male (N= 18) and female (N = 3) volunteers resulted in C/P values of 0–0.28, with no sex difference in C/P values seen (data not shown). Thus, the distribution of ritonavir and metabolites into the formed elements of blood was minimal in both animals and humans.
Discussion
The large percentages of single iv and oral dose radioactivity recovered in the feces of rats, dogs, and humans indicates that ritonavir is cleared primarily via hepatobiliary elimination in these species. This conclusion was verified in bile-exteriorized rats and dogs. Ritonavir is metabolized by several oxidative pathways in rat, dog, and human, with some of these pathways being species-specific, and by conjugation with glucuronic acid in dog only (fig. 6). Parent drug is converted in all three species to at least four primary oxidative metabolites; the des-thiazolyl carbamate product, M-1, the isopropylthiazolyl oxidation product, M-2, the des-isopropylthiazolyl product,M-11, and the N-hydroxymethyl urea metabolite, designated M-10 (rat and human) or M-7 (dog, assignment tentative). The thiazolyl heteroatom oxidation product,M-9, is produced only in rat, while the unknown metaboliteM-6 and the glucuronide of parent drug, G-1, appear only in dog. Several secondary metabolites are then produced by further biotransformation of some of these primary metabolites. Isopropyl thiazolyl oxidation or thiazolyl carbamate oxidative cleavage of M-1 or M-2, respectively, producesM-5 in all three species. A second oxidation of the isopropyl moiety of M-2 generates the glycol M-12primarily in rat, while oxidation of the methylthiazolyl moiety ofM-1 affords M-13 in dog. Loss of formaldehyde from M-7/10 would presumably afford theN-desmethyl urea product, which does not appear to be a major rat or dog bile metabolite and was not observed in human excreta. Instead, this N-desmethyl compound must undergo oxidation to yield the N-hydroxyurea metabolite, M-3, in all three species. M-7 also undergoes glucuronidation in dog to afford G-2. Except for M-1 (inactive),M-2 (active), and M-11 (active)(8), the in vitro antiviral activity of these metabolites remains undetermined. Ritonavir and M-2 accounted for 37.3% and 30.4%, respectively, of the total dose recovered in human feces and urine within 6 days after oral dosing. Unchanged parent drug,M-2, M-11 (9.7%), M-1 (4.1%),M-5 (4.0%), M-3 (1.7%), and M-7/10(0.1%) together accounted for about 90% of the total dose radioactivity recovered in human excreta after oral dosing. A previous study has shown that the formation of M-1 andM-11 by human liver microsomes is mediated by the CYP3A subfamily of enzymes, whereas both CYP3A and CYP2D6 contribute toM-2 formation (7).

Proposed pathway of ritonavir metabolism in rats, dogs, and humans.
A significant sex difference in plasma drug levels was apparent in rats, but not in dogs. In rats, plasma levels of radioactivity were generally higher in females than in males, presumably owing to gender specificity in the enzymes responsible for ritonavir metabolism (9,10). This gender specificity, commonly seen in rats but not in dogs or humans, was also apparent in rat bile, where bile from females contained higher percentages of ritonavir than did male bile. Plasma metabolite levels were exceedingly low in all three species, withM-1 being the major plasma metabolite seen in rat and dog and M-2 being the major human plasma metabolite. In male humans, 96.8% of the mean total radioactivity AUC2–12consisted of ritonavir.
In rat, M-9 was a major radioactive component of bile collected after iv dosing and only trace amounts of unchanged parent drug were detected in bile. However, fecal metabolite profiles obtained after iv dosing showed that 21.2% of the fecal radioactivity consisted of parent drug, while no M-9 was detected. This would seem to indicate that M-9 undergoes conversion in the rat gastrointestinal tract, either owing to chemical factors or to microbial metabolism, to regenerate ritonavir. Since in vivomicrobial reduction of N-oxides and S-oxides back to parent drug has been reported (11, 12, 13, 14) it is reasonable thatM-9, which has been shown by ESI/MS/MS to be oxidized on the methylthiazolyl moiety, bears an oxygen atom on the nitrogen or the sulfur atom of the thiazole ring. Additionally, the high levels of ritonavir in rat feces relative to bile may also be partially a result of transintestinal transport (exsorption) of circulating ritonavir, a phenomenon associated with a number of other compounds (15). In dog, the presence of ritonavir in feces collected after intravenous dosing, despite the absence of ritonavir in dog bile, suggests that hydrolysis of the G-1 glucuronide and/or reduction of various oxidized bile metabolites is occurring in the GI tract.
The proposed pathway of ritonavir metabolism in rats, dogs, and humans contains several interesting structural features. Regarding formation of the N-methylurea-functionalized metaboliteM-3, oxidative N-demethylation of secondary and tertiary amine or amide substrates is generally believed to proceedvia formation of an N-hydroxymethyl intermediate, which goes on to lose formaldehyde to afford the amine or amide product. In most cases, the existence of the N-hydroxymethyl intermediate cannot be demonstrated owing to its poor stability. However, there are a few literature examples (16) of stable, isolableN-hydroxymethyl intermediates in which the nitrogen involved is adjacent to a carbonyl group, as is the situation with the urea moiety of M-3. Also relatively uncommon in the literature is the hydroxylation of an amide nitrogen atom, as proposed for the metabolite M-10. However, a structurally analogous HIV protease inhibitor BMS-187071 has been reported (17) to yield anN-hydroxy amide metabolite, providing some precedence for the N-hydroxyurea structure proposed for M-10. The decarbamoylation of ritonavir to form the des-thiazolyl carbamate product, M-1, is also a rather unique biotransformation. This conversion would presumably involve oxidation of the carbon α to the ether oxygen, followed by intramolecular rearrangement to liberateM-1, carbon dioxide, and thiazole-2-aldehyde (7). Formation of the glucuronide of ritonavir (G-1) in dog was also rather unexpected considering the steric hindrance near the hydroxy group caused by the two benzyl groups within the interior “core” of the molecule. However, the HIV protease inhibitor L-689,502, which has an interior “core” structure very similar to ritonavir, has also been reported to form an ether glucuronide at the hydroxy group of the central hydroxyethyl isostere during incubation with rat liver slices (18). It should be noted that although G-1 could sucessfully be produced in vitro during ritonavir incubations with dog liver microsomes suitably fortified with UDPGA and detergent, no evidence of ritonavir conversion to G-1 was observed during extensive in vitro experiments with rat and human liver microsomes, hepatocytes, or slices (data not shown). This result is consistent with the observed absence of G-1 in rat and humanin vivo metabolism profiles.
Acknowledgments
The authors would like to acknowledge the following contributors to this work: Dr. Dale Sharp and Ms. LeAnn Eastman of Corning Hazleton (rat tissue distribution study, human study), Ms. Janet Lamm of Abbott Laboratories (human study), and Ms. Ruth Bruce of Peninsular Testing Corporation (human study). The authors also wish to thank Dr. Barbara Bopp of TAP Pharmaceuticals for helpful discussions throughout the course of this work and Dr. Gondi Kumar of Abbott Laboratories for a critical review of this manuscript.
Footnotes
-
Send reprint requests to: Dr. Jon F. Denissen, Biotransformation Department, D46V, Bldg AP9, Abbott Laboratories, 100 Abbott Park Road, Abbott Park, IL 60064.
- Abbreviations used are::
- ritonavir
- 10-Hydroxy-2-methyl-5-(1-methylethyl)-1-[2-(1-methylethyl)-4-thiazolyl]-3,6-dioxo-8,11-bis(phenylmethyl)-2,4,7,12-tetraazatridecan-13-oic-acid, 5-thiazolylmethyl ester, [5S -(5R*,8R*,10R*11R*)]
- HIV
- human immunodeficiency virus type-1
- AIDS
- acquired immune deficiency syndrome
- Received October 3, 1996.
- Accepted December 18, 1996.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}