


Abstract
The pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide has been evaluated in 12 patients with metastatic breast cancer undergoing high-dose chemotherapy followed by bone marrow transplantation. Each patient received an initial dose of 4 g/m2 of cyclophosphamide over 90 min to prime peripheral blood progenitor cells (the first course), and 3 weeks later, 6 g/m2of cyclophosphamide with 800 mg/m2 of thiotepa by 96-hr infusion before marrow stem cell infusion (the second course). Whole blood cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide concentrations were measured by a GC-EIMS method using deuterium labeled compounds as internal standards. In addition, plasma and urine cyclophosphamide concentrations were determined by a GC assay. Whole blood concentrations of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide vs. time data and urinary excretion of cyclophosphamide data from the first course were co-modeled using a one-compartment model with Michaelis-Menten saturable elimination in parallel with first-order renal elimination (N = 7) or first-order metabolic and renal elimination (N = 5) for cyclophosphamide and one-compartment model with first-order elimination for 4-hydroxycyclophosphamide/aldophosphamide. The parallelism between cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide disposition curves implies that the pharmacokinetics of 4-hydroxycyclophosphamide/aldophosphamide is formation limited; only the fractional 4-hydroxycyclophosphamide/aldophosphamide clearance rate (Clmet/Fmet) can be estimated. The mean Vmax andKm for cyclophosphamide were 0.78 μM/min and 247 μM, respectively. The mean nonrenal clearance (Clnr) of cyclophosphamide for five patients with apparent first-order elimination of cyclophosphamide was 67 ml/min. The mean Clmet/Fmet of 4-hydroxycyclophosphamide/aldophosphamide was 2982 ml/min. The mean renal clearance (Clr) of cyclophosphamide was 29 ml/min and 24 ml/min for the first course and the second course, respectively. The correlations between cyclophosphamide AUCs and 4-hydroxycyclophosphamide/aldophosphamide AUCs were sought for both drug courses. Blood and plasma cyclophosphamide concentrations were remarkably similar, indicating that cyclophosphamide partitions equally in the red cell and plasma volume. Computer simulation of the effect of potential alterations in Michaelis-Menten saturable elimination and renal clearance on 4-hydroxycyclophosphamide/aldophosphamide has been used to illustrate the complex relationship between the exposure to parent compound and active metabolite.
Cyclophosphamide is one of the most frequently used antitumor agents in patients receiving high-dose chemotherapy prior to bone marrow transplantation. It is a prodrug that undergoes complex metabolic activation and detoxification. The current understanding of cyclophosphamide activation is presented in fig.1. Cyclophosphamide is enzymatically metabolized to 4-hydroxycyclophosphamide/aldophosphamide, which exists in equilibrium with its open ring tautomer, aldophosphamide (1-3). 4-Hydroxycyclophosphamide/aldophosphamide serve as the transport forms for the toxic species of phosphoramide mustard and acrolein (1-3). They also undergo enzymatic oxidative detoxification to form inactive urinary excretion products, 4-ketocyclophosphamide and carboxyphosphamide (1, 3). The extent of metabolism by these pathways depends, in part, on the activity of aldehyde dehydrogenase in the tissues; this may account for the tissue selectivity as well as drug resistance (2-6). Since the active metabolites are formed from the 4-hydroxycyclophosphamide/aldophosphamide equilibrium mixture, measurement of these intermediates is crucial to determine the relationship between the drug exposure and the therapeutic outcome in patients.
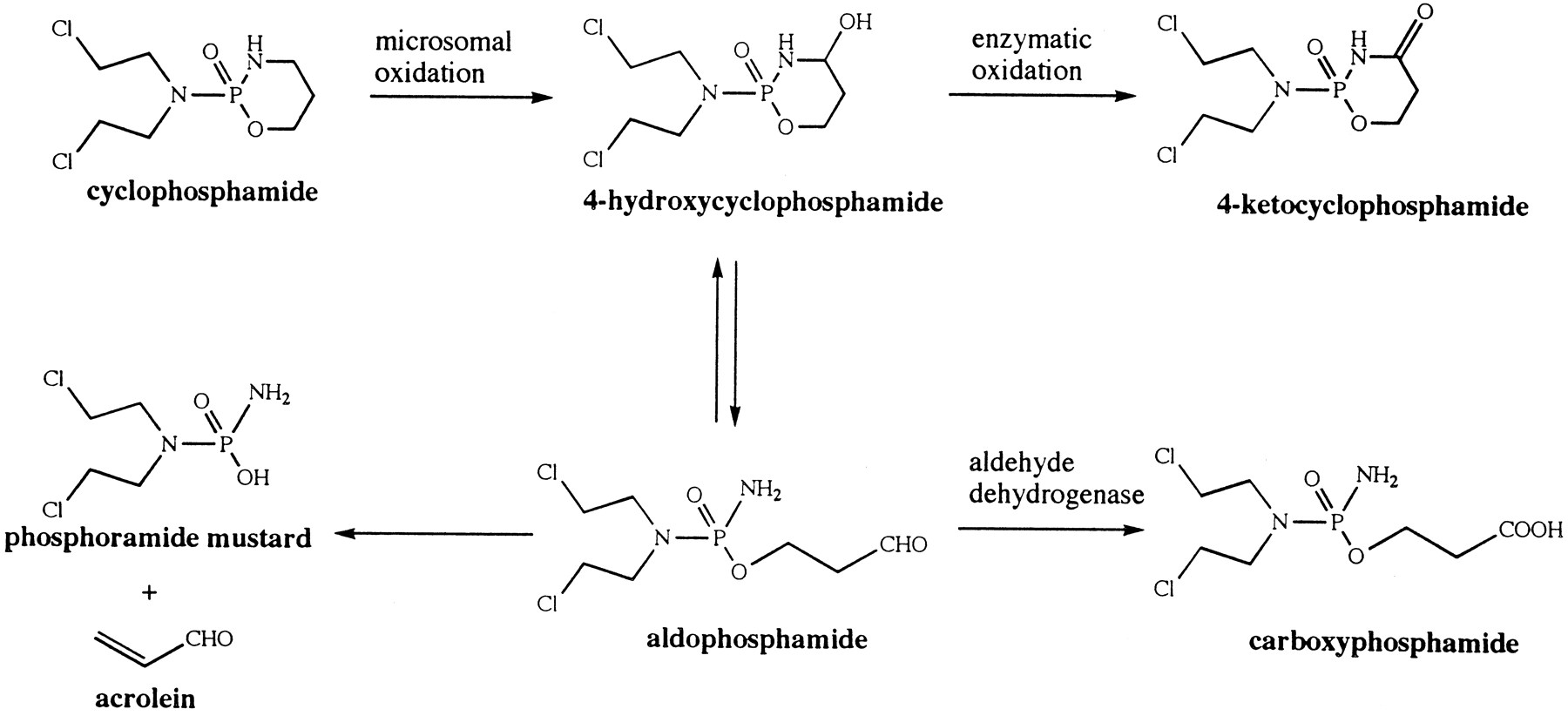
Metabolic pathways of cyclophosphamide.
We have published nonlinear pharmacokinetics of high-dose cyclophosphamide in patients (7), but co-modeling with its circulating metabolite 4-hydroxycyclophosphamide/aldophosphamide has not been reported. The primary limitation of the assessment of 4-hydroxycyclophosphamide/aldophosphamide exposure has been the evanescent nature of this chemical, which has a t1/2 of 6 min in human blood at 37°C (8). Thus, although several investigators reported the disposition of cyclophosphamide and its metabolites in patients receiving moderate doses of cyclophosphamide, the relative degradation prior to assay was not quantified (3, 9, 10). Slattery et al. (11) recently described a new LC-MS method, which included a solid phase extraction procedure without using an internal standard, for quantitation of plasma 4-hydroxycyclophosphamide/aldophosphamide only. The development of a GC-EIMS method using deuterium labeled compounds as internal standards with a pre-assay stabilizing procedure to simultaneously quantitate cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide (12, 13) has facilitated a formal description of the metabolic process and disposition of cyclophosphamide and its circulating metabolite, 4-hydroxycyclophosphamide/aldophosphamide in patients.
We have carried out a comprehensive pharmacokinetic analysis of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide in 12 patients with metastatic breast cancer undergoing high-dose chemotherapy with alkylating agents followed by autologous bone marrow transplantation. Eleven of 12 patients’ cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide AUCs have been reported previously (13), but no pharmacokinetic modeling was presented. The purpose of this study was (a) to co-model whole blood cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide concentrationvs. time data after a 90-min infusion of cyclophosphamide alone using suitable kinetic models; (b) to analyze the correlation between cyclophosphamide AUC and 4-hydroxycyclophosphamide/aldophosphamide AUC in whole blood after a 90-min infusion of cyclophosphamide alone and after a 96-hr infusion of cyclophosphamide concurrently with thiotepa; and (c) to compare cyclophosphamide concentrations in whole blood with that in plasma; and (d) to discuss the variability of disposition of cyclophosphamide and its circulating metabolite, 4-hydroxycyclophosphamide/aldophosphamide, in patients and illustrate this variability through computer simulation. The clinical responses and toxicities of this treatment will be the subject of a separate report.
Patients and Methods
Patient Population and Study Design.
Women with stage IIIB or IV breast cancer undergoing autologous bone marrow transplantation were eligible for this study. Patients were required to have a histologically documented breast cancer responsive to conventional therapy, an age between 18 and 60 years old, an Eastern Cooperative Oncology Group (ECOG) performance status of less than 2, normal hematopoietic function, and adequate cardiac (LVEF>45%), pulmonary (FVC and FEV1>65% of predicted for patient’s height and weight), renal (serum creatinine concentration <2.0 mg/dl) and hepatic (serum AST concentration <60 IU/ml and serum bilirubin concentration <1.5 mg/dl) functions. Creatinine clearance (Cr Cl) was calculated for each patient using the method of Cockcroft and Gault (14). The study was approved by the Joint Committee for Clinical Investigation of the Johns Hopkins Hospital and written informed consent was obtained from each patient.
After the initial marrow harvest, patients received 4 g/m2of cyclophosphamide administered iv over 90 min, for mobilization of peripheral blood progenitor cells (the first course). Three weeks later, the patients received a combination of cyclophosphamide (6 g/m2) and thiotepa (800 mg/m2) administered simultaneously as a 96-hr continuous iv infusion (the second course). Novobiocin (2 g every 12 hr orally for 14 doses starting 36 hr prior to the chemotherapy) was added to inhibit the development of alkylating agent resistance (15). Ondansetron (0.15 mg/kg loading dose followed by 1 mg/min continuous iv infusion) and lorazepam (1 mg every 4 hr iv) were administered from the time chemotherapy infusion started until 24 hr after treatment finished. Prochlorperazine (5 mg every 3 hr iv) was given as needed.
Blood and urine specimens were collected for the determination of whole blood cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide concentrations, and plasma and urine cyclophosphamide concentrations. For the first course, blood samples were obtained at 0, 45, and 80 min, and 2, 3, 4, 5, 8, 10, 16, 24, 27 hr from the beginning of the infusion. For the second course, blood samples were obtained at 0, 3, 6, 12, 18, 24, 30, 42, 54, 66, 78, 90, 96 hr during the infusion, and 1, 3, 5, 8, and 24 hr after the end of the infusion. Two aliquots of blood samples were drawn: a 1 ml of blood drawn in a 1 ml TB syringe was immediately (<1 min) placed in a pre-weighted tube containing the trapping agent for stabilizing 4-hydroxycyclophosphamide/aldophosphamide (12), and each tube was re-weighted to obtain the exact amount of whole blood added; a 5 ml of blood was drawn into a heparinized Vacutainer for plasma collection. Urine was collected up to 32 hr and 120 hr after the infusion began for the first course and second course, respectively. Plasma and urine aliquots were stored at −20°C until analysis.
Analytical Methods.
Whole blood cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide concentrations were simultaneously quantitated by a GC-EIMS assay (12, 13). In this method, the trapping agent o-(2, 3, 4, 5, 6-pentafluorobenzyl)hydroxylamine reacted with aldophosphamide and, indirectly, with its tautomer, 4-hydroxycyclophosphamide, to provide a stable oxime. This oxime represented a combined concentration of aldophosphamide and any species (most notably, 4-hydroxycyclophosphamide) with which it spontaneously interconverts. The oxime and unchanged parent drug, cyclophosphamide, were extracted from the biological fluid and assayed by a GC-EIMS method. Deuterium labeled oxime and cyclophosphamide were used as internal standards (12, 13). Plasma (first course only) and urine cyclophosphamide levels were measured by a gas chromatography assay described previously (7).
Pharmacokinetic Analysis.
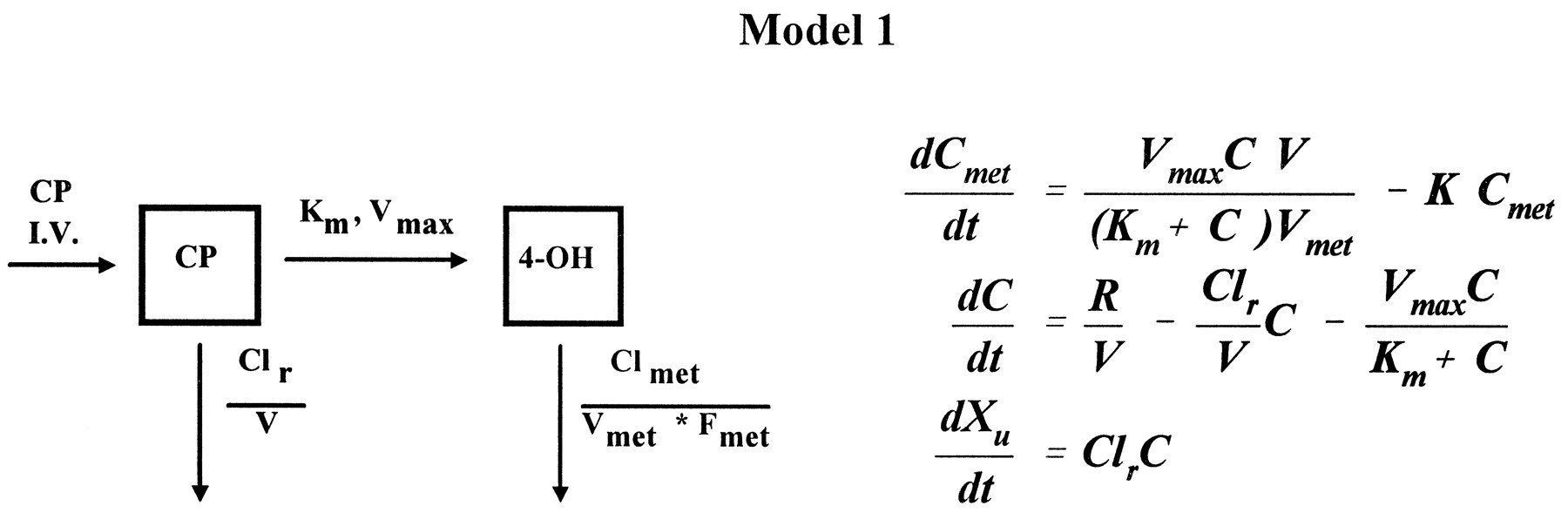
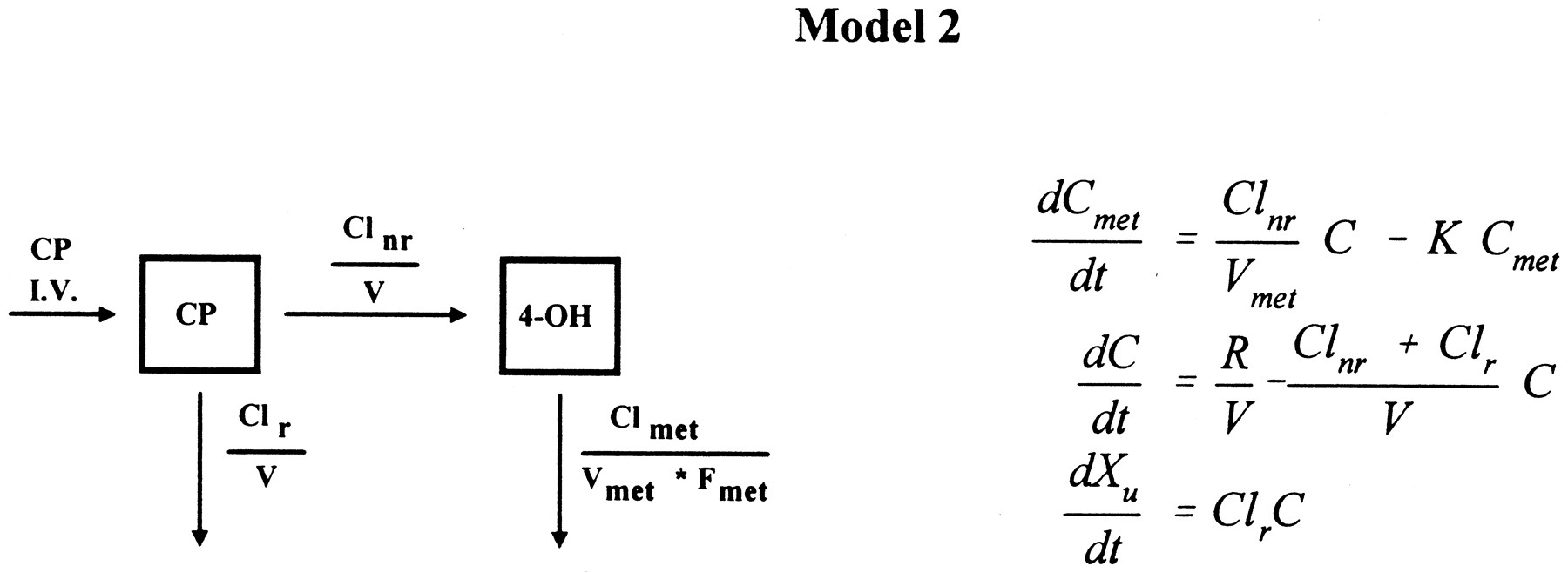
Blood cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide disposition curves from the first course were first examined visually to assess suitable models. If both cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide elimination curves exhibited convex-downward curves on a semilog scale, a Michaelis-Menten saturable elimination process was anticipated (16). We evaluated the fit of a one-compartment model with Michaelis-Menten metabolic elimination coexisting with first-order renal elimination for cyclophosphamide (CP) and a one-compartment model with first-order elimination for 4-hydroxycyclophosphamide/aldophosphamide (4-OH) (16):
where dCmet/dt and dC/dt are the rate of change of drug concentration at time t for 4-hydroxycyclophosphamide/aldophosphamide and cyclophosphamide, respectively, R is the rate of infusion (t>infusion time,R = 0), Cmet and C are whole blood concentrations for 4-hydroxycyclophosphamide/aldophosphamide and cyclophosphamide, respectively, Vmet and V are the apparent volume of distribution for 4-hydroxycyclophosphamide/aldophosphamide and cyclophosphamide, respectively, K is the elimination rate constant of 4-hydroxycyclophosphamide/aldophosphamide, Clr is the calculated renal clearance rate of cyclophosphamide, Vmax andKm are the theoretical maximum rate of the elimination process and Michaelis-Menten constant for cyclophosphamide, respectively, and Xu is the amount of urinary excretion of cyclophosphamide. A weight of 1/C was used in the iterative fitting process.
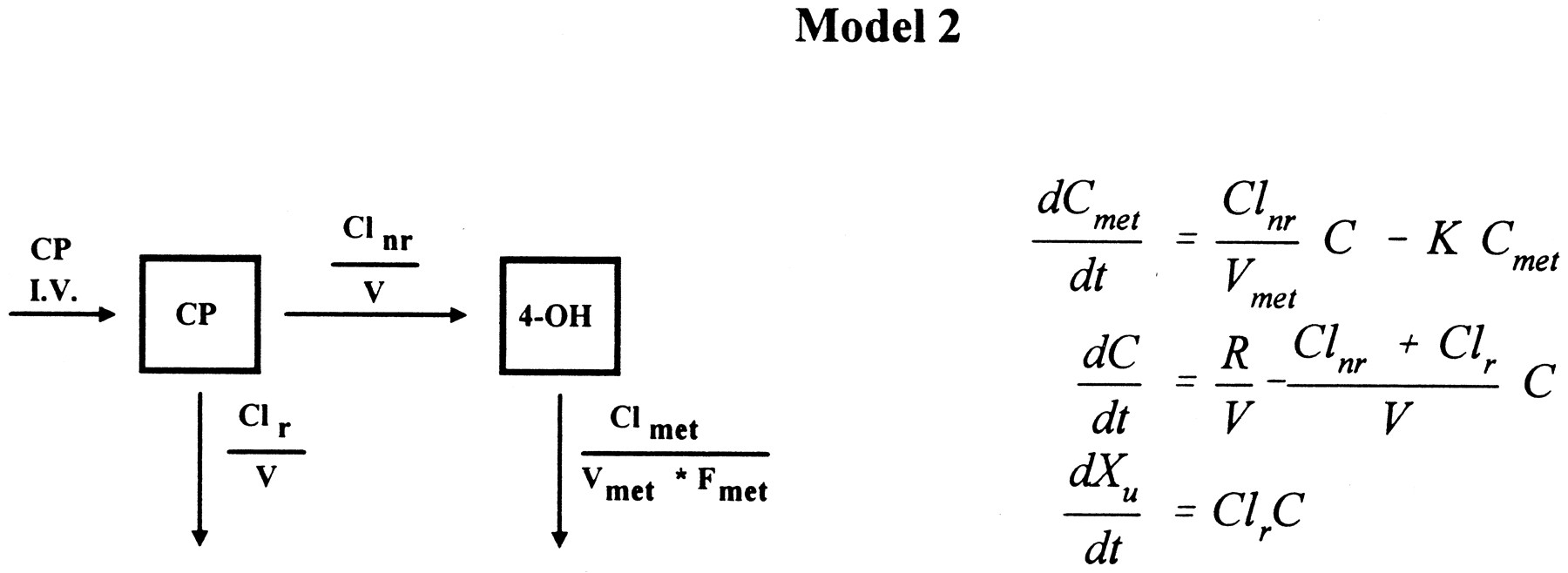
For data sets that were not well fit by the above models,i.e. when visual inspection of these disposition curves of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide were straight lines on a semilog scale, and when the asymptotic standard error for Km was >50% associated with estimates of Km>2*Cmax, a one-compartment model with constant input and first-order elimination for both cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide was used to fit the data (16):
where Clnr is the nonrenal clearance rate of cyclophosphamide.
AUC (AUCmet), the extrapolated area under the blood cyclophosphamide (4-hydroxycyclophosphamide/aldophosphamide) disposition curve was calculated using a combined linear and logarithmic trapezoidal rule (17) for data obtained from both courses. The percentage of urinary excretion of cyclophosphamide (Xu) and Clr for the second course were calculated from Xu/dose and Xu/AUC, respectively (16). Correlation between Cr Cl and cyclophosphamide Clr was sought for data obtained from the first course.
All pharmacokinetic modelings were performed by nonlinear regression analysis using PCNONLIN (Statistical Consultants, Apex, NC). The codes for Model 1 and Model 2 can be obtained from the authors of this report. Computer simulation was performed by varying one of these parameter estimates at a time, CLr,Vmax (CLnr), andKm, while holding others constant using mean values from this report. Areas of under simulated time-concentration curves of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide were calculated as described above and relationships between these two were sought. The mean, median, and SD of pharmacokinetic parameters were calculated using Quattro Pro for Windows (Borland International, Inc., Scotts Valley, CA). The statistical analyses were completed by using InStat (GraphPad Software, Inc., San Diego, CA).
Results
Twelve of 14 women with stage IIIB (2 patients) or IV (10 patients) breast cancer undergoing autologous bone marrow transplantation between March 1994 and July 1995 had pharmacokinetic sampling performed and are included in this report. The median age of the patients was 46.5 years (range 30–61 years). Two patients had liver metastases but normal liver enzymes. In the first course, all 12 patients had complete blood cyclophosphamide and 4-hydroxycyclo
phosphamide/aldophosphamide disposition curves and urinary excretion data, and 11 patients had complete plasma cyclophosphamide concentrations. Before the second course, two patients’ disease progressed; therefore, only 10 patients had complete cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide disposition curves and urinary excretion data in the second course.
After a 90-min infusion, blood cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide disposition curves were parallel. This phenomenon implies that the pharmacokinetics of 4-hydroxycyclophosphamide/aldophosphamide was formation limited (16). Since the bioavailability of 4-hydroxycyclophosphamide/aldophosphamide (Fmet) was unknown, only the fractional 4-hydroxycyclophosphamide/aldophosphamide clearance rate (Clmet/Fmet) could be estimated while an equal value for V and Vmet was assumed.
Blood cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide disposition curves from 7 of 12 patients were well fit by Model 1, a one-compartment model with Michaelis-Menten saturable elimination and first-order renal elimination in parallel for cyclophosphamide and first-order elimination for 4-hydroxycyclophosphamide/aldophosphamide. In this case, the input rate of 4-hydroxycyclophosphamide/aldophosphamide varied during the entire time course, i.e. when cyclophosphamide concentration was in the vicinity of Km, the input rate appeared zero-order, a constant rate regardless of the concentration of cyclophosphamide, and when the cyclophosphamide concentration was much lower than Km, the input rate was first-order, a rate proportional to the concentration of cyclophosphamide. This was represented by a change in the relationship of the differences between concentrations of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide (fig. 2).

Time course of blood cyclophosphamide (▪) and 4-hydroxycyclophosphamide/aldophosphamide (▴) concentrations during and after a 90-min infusion at dose of 4 g/m2for patient 6 with nonlinear fitting,Vmax = 0.63 μM/min,Km = 167 μM, Clr = 22 ml/min, CLmet/Fmet = 1987 ml/min. Lines represent the model fit.
The remaining five patients’ disposition curves did not appear convex, and were well fit by Model 2, a one-compartment model with first-order elimination for both cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide. In this case, the apparent input rate of 4-hydroxycyclophosphamide/aldophosphamide was first order during the entire time course. Therefore, the difference between cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide concentrations was constant (fig. 3).

Time course of blood cyclophosphamide (▪) and 4-hydroxycyclophosphamide/aldophosphamide (▴) concentrations during and after a 90-min infusion at dose of 4 g/m2for patient 4 with apparent linear fitting, Clnr = 76 ml/min, Clr = 57 ml/min, CLmet/Fmet = 4115 ml/min. Lines represent the model fit.
The volume of distribution (V = Vmet) and elimination parameter estimates from the model fitting are shown on table1. The mean Clr from the model fitting (29 ml/min) was very similar to that by direct calculation from Xu/AUC (28 ml/min), suggesting that the urine collection was nearly complete. The mean total body clearance of cyclophosphamide in patients with apparent linear fitting (N = 5) was 99 ml/min (t1/2 = 294 min), in which one-third was accounted for by the renal clearance (32 ml/min). The mean percentage of the total dose of cyclophosphamide excreted unchanged in the urine was 29% (16–43%). Eleven of 12 patients had calculated Cr Cl between 75 and 133 ml/min, and one patient had 209 ml/min. Within this limited range, there was no evident correlation between patients’ Cr Cl and cyclophosphamide Clr (r2 = 0.06608, p = 0.4199).
Volume of distribution and elimination parameter estimates for cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide in patients receiving cyclophosphamide at 4 g/m2/90 min
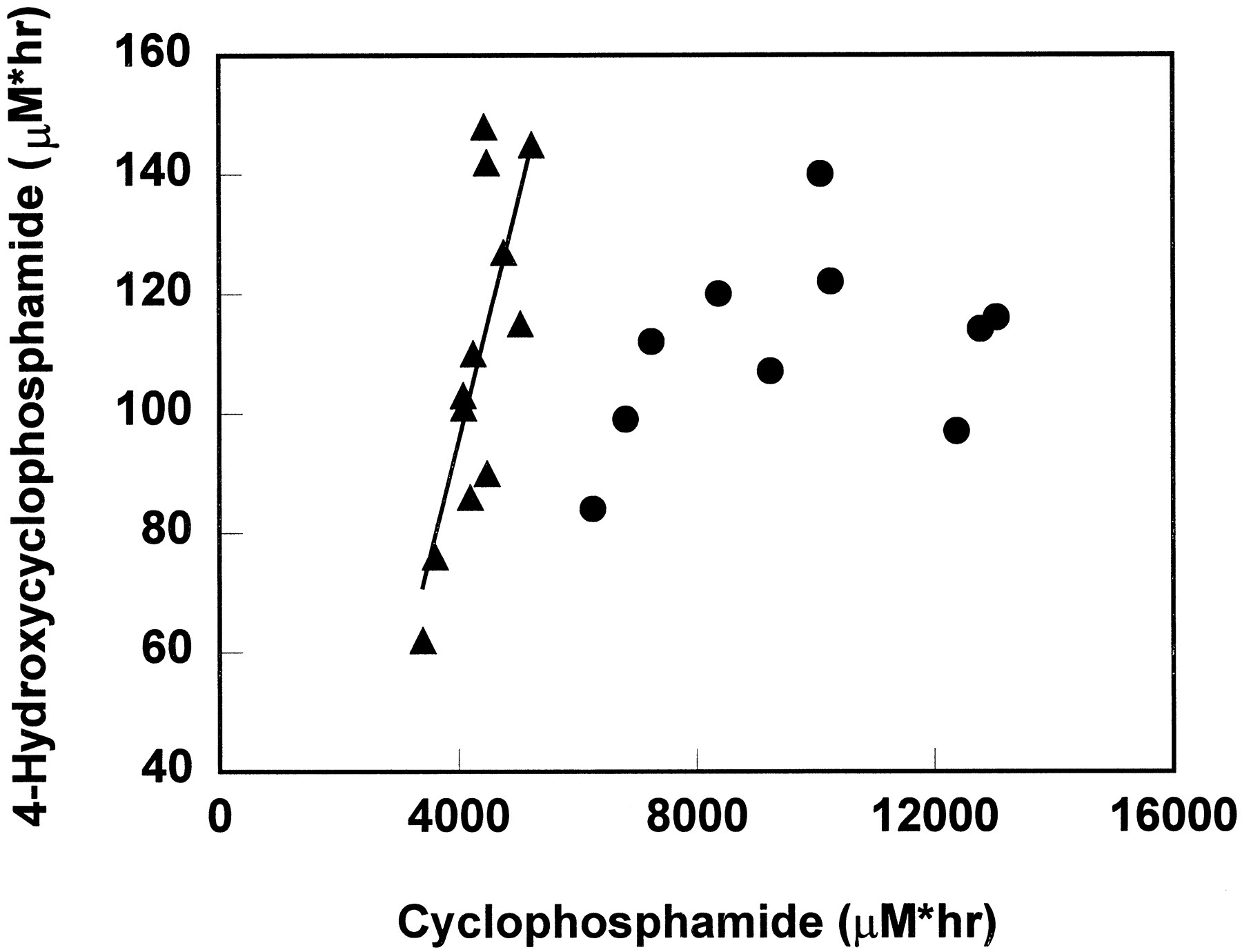
A positive correlation was found between AUC of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide in the first drug course (r2 = 0.588, p = 0.0036) (fig.4). This is consistent with the observed weak inverse correlation between AUC of 4-hydroxycyclophosphamide/aldophosphamide and percentage of total cyclophosphamide renal excretion (r2 = 0.1207, p = 0.2685). There was a 1.5-fold variability in cyclophosphamide AUC but a 2.4-fold range in 4-hydroxycyclophosphamide/aldophosphamide AUC.
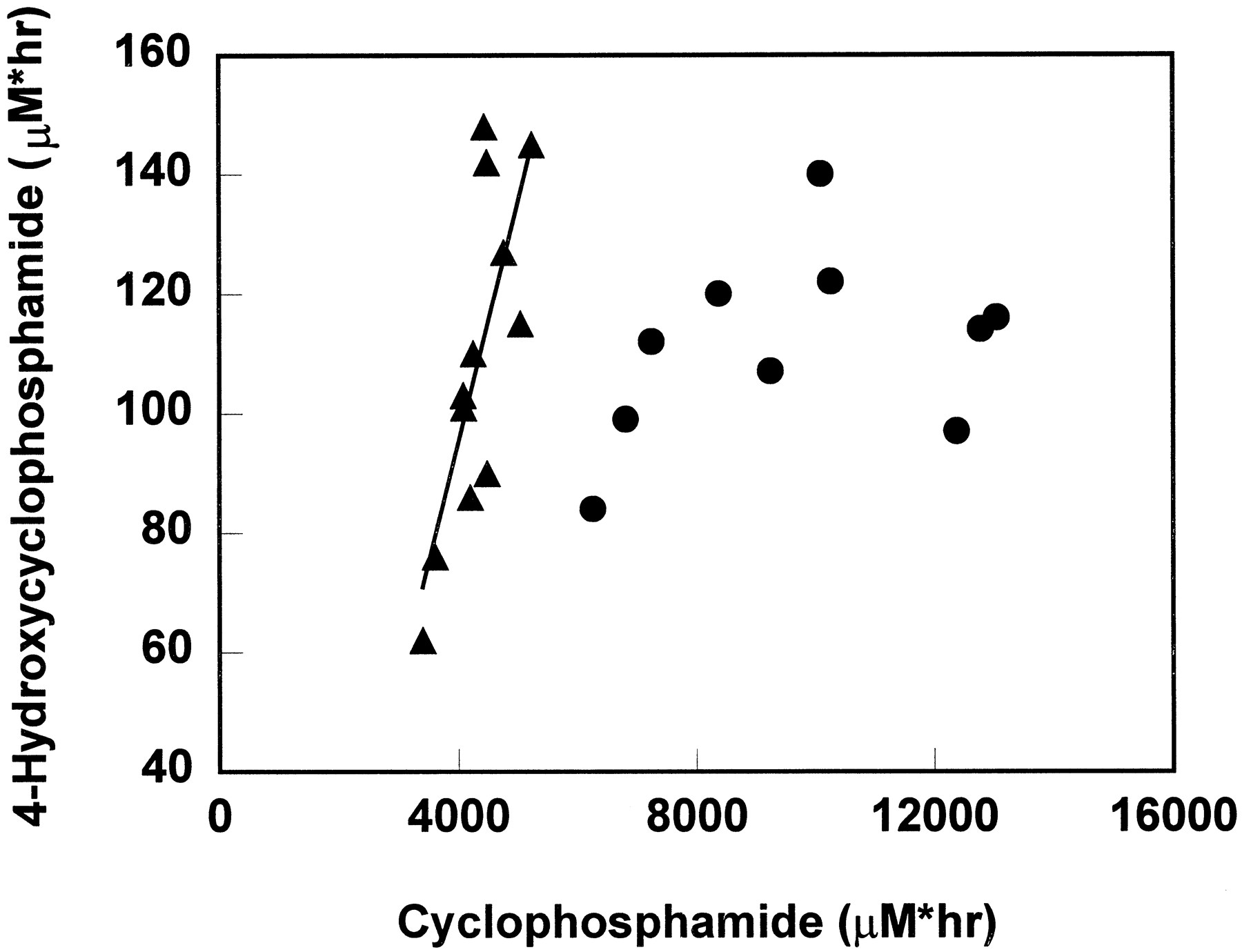
The correlation of cyclophosphamide AUC and 4-hydroxycyclophosphamide/aldophosphamide AUC when cyclophosphamide was given as a single agent (▴) and when cyclophosphamide was given concurrently with thiotepa (•).
The line represents the fit line for single injection data by least squares regression, y = 0.04x − 66.
In the second drug course, after a 96-hr infusion, whole blood cyclophosphamide concentrations during a 96-hr infusion did not reach steady state as previously reported, but showed evidence of induction of cyclophosphamide metabolism (7). However, 4-hydroxycyclophosphamide/aldophosphamide disposition curves did not demonstrate a consistent induction pattern, precluding co-modeling of the cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide data by using a time-variant model as described previously (7). The AUCs of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide data are presented in fig. 4. In contrast to the data obtained from the first course, there was no correlation between these two AUCs when cyclophosphamide was given concurrently with thiotepa (r2 = 0.1202, p = 0.3263). The mean urinary excretion and renal clearance of cyclophosphamide as a 96-hr infusion were 35% of the total dose administered and 24 ml/min, respectively.
The AUC of 4-hydroxycyclophosphamide/aldophosphamide in patients with nonlinear fitting was compared with that in patients with apparent linear fitting by a nonparametric method, Mann-Whitney test. There was no statistical difference between these two AUCs (two-tailed,p = 0.7551).
The cyclophosphamide concentrations in whole blood and in plasma (a total of 118 data points) were very similar with a regression line of y = 1.083 + 14.06, r2 = 0.9207,p<0.0001 (fig. 5).
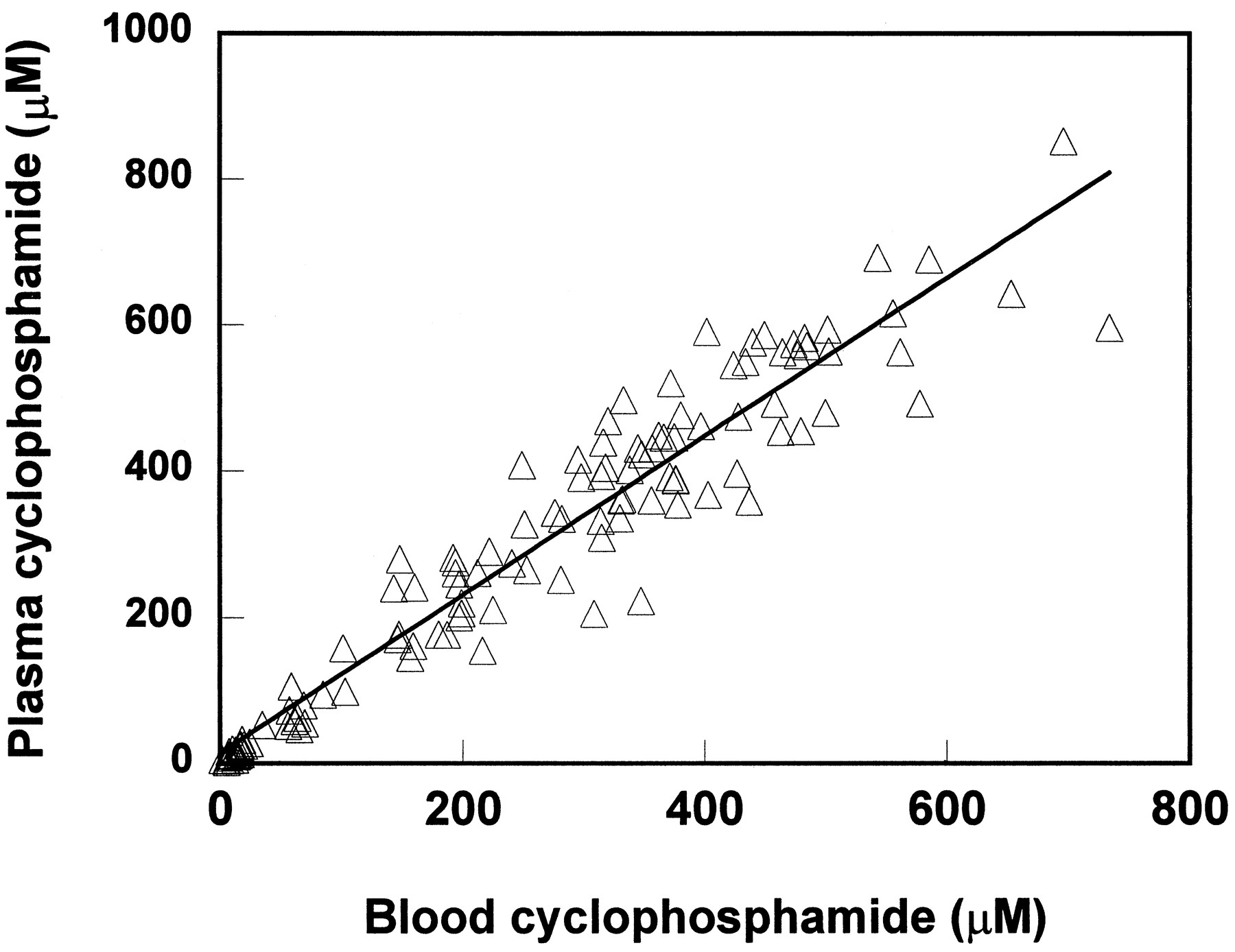
Cyclophosphamide concentrations in whole blood vs. in plasma.
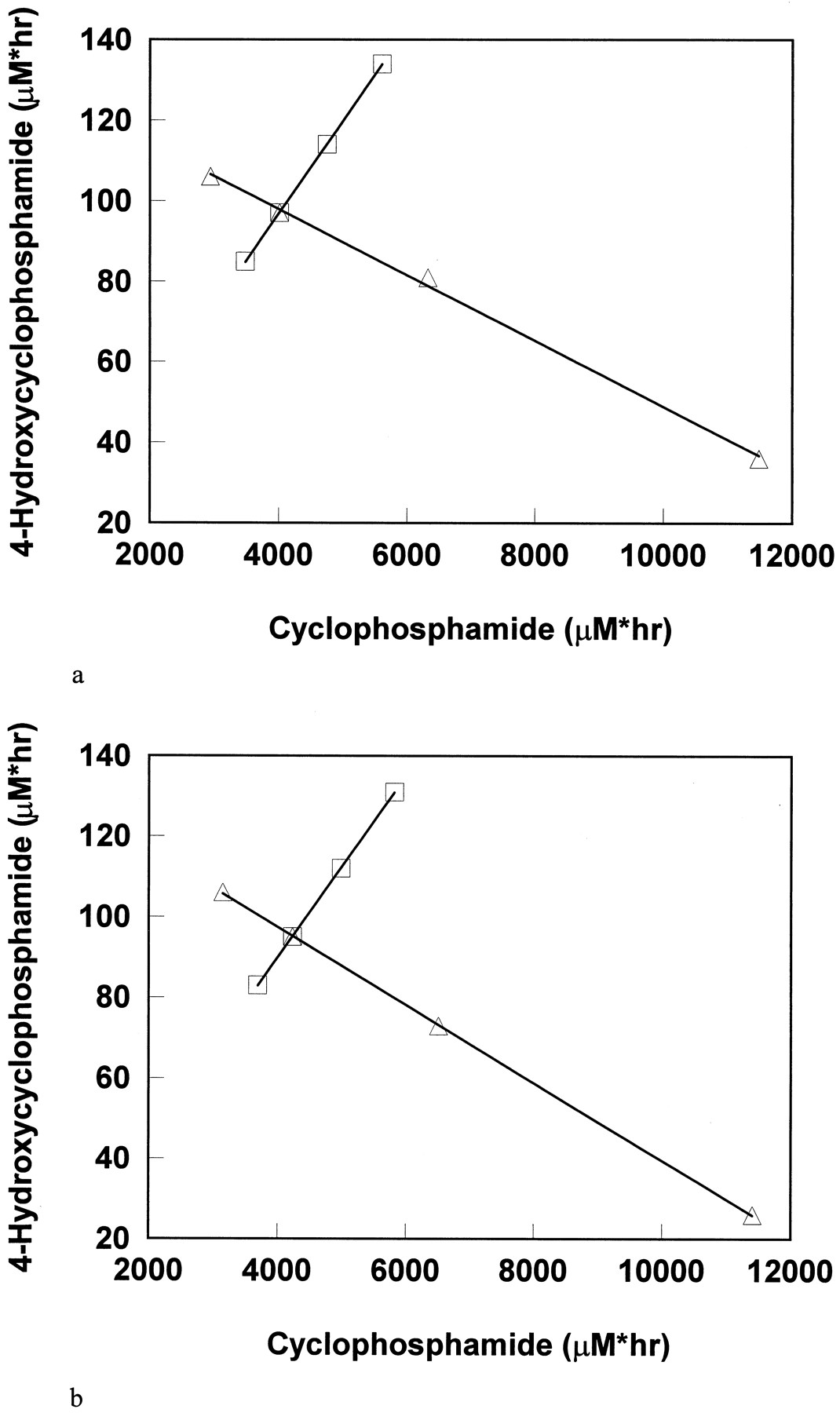
The relationship of simulated cyclophosphamide AUC and 4-hydroxycyclophosphamide/aldophosphamide AUC by varying CLr and Vmax (CLnr) were demonstrated in figs. 6a and b. AUC and Cmax values of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide simulated with variousKm are shown in table 2.
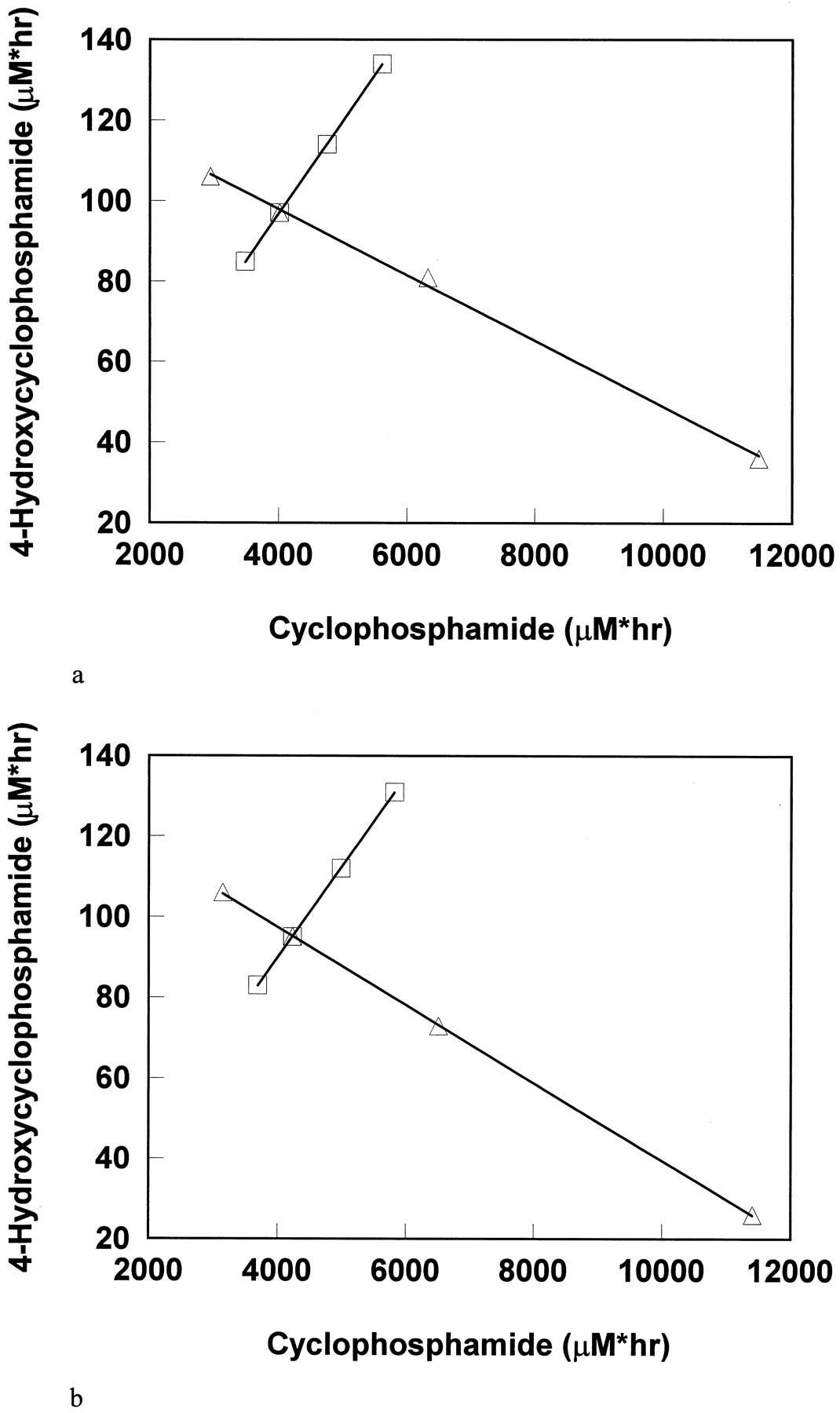
The correlation of cyclophosphamide AUCs and 4-hydroxycyclophosphamide/aldophosphamide AUCs by computer simulation.
Variables are CLr (□) and Vmaxor Clnr (▵). Points on the left side of the cross point represent 150% of the cross point variable value, and those on the right side represent 50% and 10% of the cross point variable value. (a) Simulation by using Model 1. Parameters used for the cross point: dose = 6387 mg, infusion time = 90 min, V = 45 liters, Vmax = 0.78 μM/min,Km = 247 μM, Clr = 29 ml/min, and CLmet/Fmet = 2982 ml/min. (b) Simulation by using Model 2. Parameters used for the cross point: dose = 6387 mg, infusion time = 90 min, V = L, Clnr = 67 ml/min, Clr = 29 ml/min, and CLmet/Fmet = 2982 ml/min.
AUC and Cmax data for cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide by computer simulation
Discussion
The presence of the nonlinear elimination of cyclophosphamide at high doses (concentration- and time-dependent kinetics) has been of growing interest in the past decade (18-25). The complexity and interpatient variability of the metabolic processing of cyclophosphamide in patients, like many drugs manifesting concentration-dependent elimination, may produce substantial variation in anti-tumor efficacy. A GC-EIMS assay for the simultaneous determination of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide (12, 13) has been a valuable tool for a complete description of cyclophosphamide and its circulating metabolite, 4-hydroxycyclophosphamide/aldophosphamide. Because of the instability of 4-hydroxycyclophosphamide/aldophosphamide in blood (8), a stabilizing procedure was undertaken that required placing the blood sample into a trapping agent at the bedside immediately after it was obtained. The oxime formed from 4-hydroxycyclophosphamide/aldophosphamide with the trapping agent was very stable (12), and no further sample processing was needed until GC-EIMS assay. This simple stabilizing procedure and the use of deuterium labeled compounds as internal standards are important to ensure the precise assessment of 4-hydroxycyclophosphamide/aldophosphamide concentrations in patients. Any conventional sample handling without the stabilizing step will result in highly variable underestimation of the 4-hydroxycyclophosphamide/aldophosphamide concentrations because these intermediates will be lost during both transporting and freezing-thawing processes prior to assay.
Several clinical and laboratory observations are worth noting. The capacity of hepatic biotransformation of cyclophosphamide varies enormously from patient to patient. This was evidenced by a wide range of Km values in patients receiving cyclophosphamide at 4 g/m2 in 90 min: saturable elimination occurred in one patient at blood cyclophosphamide concentration as low as 70 μM, while others showed an apparent linear elimination process at concentrations as high as 700 μM. Although patients with lowKm may not achieve high peak concentrations of 4-hydroxycyclophosphamide/aldophosphamide, the AUCs of 4-hydroxycyclophosphamide/aldophosphamide were similar whether the clearance pattern was obviously nonlinear or apparently linear.
In this cohort of patients, urinary excretion of cyclophosphamide was higher (29% for the first course and 35% for the second course) than in patients we reported previously (17% and 23%) (7). A review of the volume of total urine collected indicated no evidence of incomplete urine collection in the earlier study. Since the clinical management of these patients (i.e. iv hydration) was unchanged, the cause of this variability remains unclear.
In patients (N = 5) with apparent linear elimination, one-third of the total body clearance of cyclophosphamide was accounted for by the renal clearance. This figure is much higher than that reported previously (11%) when cyclophosphamide was given at low and moderate doses (3). For decades it has been widely accepted that impaired renal function would not alter the pharmacokinetic behavior of cyclophosphamide because the clearance of cyclophosphamide is primarilyvia nonrenal mechanisms. It is unclear whether this is valid when high doses of cyclophosphamide are used.
The parallel disposition curves of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide imply that the kinetics of 4-hydroxycyclophosphamide/aldophosphamide is formation limited. This is consistent with reports by Sladek et al. (8) and Honget al. (26). They found that the t1/2 of 4-hydroxycyclophosphamide/aldophosphamide in rats was much shorter than the apparent t1/2 of 4-hydroxycyclophosphamide/aldophosphamide when it was formed from cyclophosphamide, indicating a typical formation limited kinetics (16). Except for the Clmet/Fmet, it is not possible to calculate other pharmacokinetic parameter estimates for 4-hydroxycyclophosphamide/aldophosphamide because the bioavailability (Fmet) of 4-hydroxycyclophosphamide/aldophosphamide remains undefined.
Ayash et al. have previously reported an inverse correlation between cardiac toxicity and tumor response and cyclophosphamide AUC in patients receiving high-dose chemotherapy (cyclophosphamide 6 g/m2, thiotepa 500 mg/m2, and carboplatin 800 mg/m2 by 96-hr infusion) followed by autologous bone marrow transplantation (27). They found that patients who developed congestive heart failure and who also showed tumor response had a lower AUC of total cyclophosphamide. They have suggested that a lower cyclophosphamide AUC might be related to an increase in conversion of cyclophosphamide to its active alkylating metabolites and may enhance both end-organ and tumor cytotoxicity. Slattery et al. also reported an inverse relationship between cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide in patients receiving cyclophosphamide after busulfan or with total body irradiation (11). We compared cyclophosphamide AUC and 4-hydroxycyclophosphamide/aldophosphamide AUC in these 12 patients receiving cyclophosphamide as a single agent after a 90-min infusion and receiving cyclophosphamide concurrently with thiotepa after a 96-hr infusion. In contrast to the hypothesis by Ayash et al. and the laboratory finding by Slattery et al., we found that there was a positive correlation between cyclophosphamide AUC and 4-hydroxycyclophosphamide/aldophosphamide AUC in patients receiving cyclophosphamide as a single agent. However, in the clinical situation paralleling the treatment received by Ayash’s patients (96-hr infusion with thiotepa), no correlation was found between these two AUCs.
These clinical and laboratory findings have given rise to an important question: what is the relationship between total cyclophosphamide exposure and 4-hydroxycyclophosphamide/aldophosphamide exposure in patients with nonlinear elimination or apparent linear elimination. Based on Model 1 and Model 2 used in this report, we have performed computer simulation, which may provide possible explanations for the variability of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide disposition in patients with various physical (interpatient variability) and medical conditions (by disease or by drug interactions).
When the other kinetic parameter estimates are held constant (mean values from table 1) and Clr is varied, there is a positive correlation between cyclophosphamide AUCs and 4-hydroxycyclophosphamide/aldophosphamide AUCs, i.e. higher cyclophosphamide exposure is associated with higher 4-hydroxycyclophosphamide/aldophosphamide exposure (figs. 6aand b). Simulation revealed that a 90% reduction of the renal clearance of cyclophosphamide may be associated with 30% increase in 4-hydroxycyclophosphamide/aldophosphamide AUC. However, considering the much larger interpatient variability of AUC, our simulation suggests that no dose adjustment of cyclophosphamide will be necessary in patients with mild and moderate renal insufficiency. In patients with severe renal failure, more information is needed to define the necessity of dose adjustment.
Similarly, by varying Vmax (CLnr), there is an inverse relationship between cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide AUCs (figs. 6a and b).Vmax (Clnr) values of cyclophosphamide are directly related to the biovailability of 4-hydroxycyclophosphamide/aldophosphamide (Fmet),i.e. the higher Vmax(Clnr), the higher Fmet. In addition to the patients’ physical status and the liver function, multidrug administration may significantly influence theVmax (Clnr) or Fmet. Coadministration of drugs that induce specific cytochrome enzymes enhance the Vmax (Clnr) or Fmet, and coadministration of drugs that inhibit these enzymes would decrease the Vmax(Clnr) or Fmet. Therefore, a larger interpatient variability of Vmax(Clnr) or Fmet may be expected with coadministered drugs and with resultant increased interpatient variability in 4-hydroxycyclophosphamide/aldophosphamide AUC. Drugs involved in such interactions include thiotepa, phenobarbital, phenytoin, and cimetidine (3). The actual clearance of 4-hydroxycyclophosphamide/aldophosphamide is also affected by other factors including urinary excretion of 4-hydroxycyclophosphamide/aldophosphamide, the chemical reaction to form the active species, and the enzymatic reaction by aldehyde dehydrogenase to form other inactive products. Inhibition of aldehyde dehydrogenase has been associated with an increased plasma 4-hydroxycyclophosphamide/aldophosphamide AUC as well as untoward cyclophosphamide-induced toxicity (3). To date, whether the detoxification of 4-hydroxycyclophosphamide/aldophosphamide by aldehyde dehydrogenase follows genetic polymorphism as is observed in acetaldehyde oxidation in some ethnic groups has not been fully elucidated. If such polymorphism exists, the clearance of 4-hydroxycyclophosphamide/aldophosphamide may vary more among ethnic groups which represent varying degrees of metabolism.
Several drug interactions should be considered in this cohort of patients receiving bone marrow transplantation. There was no observed drug interaction between novobiocin and cyclophosphamide reported by Kennedy et al. (15), and no patient was on any anticonvulsant medication. Another possible drug interaction was the combination of antiemetic drugs with chemotherapy agents. It is a common practice that several antiemetic drugs (e.g.ondansetron, lorazepam, and prochlorperazine) are coadministered with the preparative chemotherapy regimen to patients before bone marrow transplantation. Agura et al. (28) conducted a pharmacokinetic study in patients receiving ondansetron and cyclophosphamide. They discovered that there was no significant mutual metabolic interaction between cyclophosphamide and ondansetronvia competition for the cytochrome P450 oxidase. As of this writing, no detailed pharmacokinetic data were found regarding the specific drug interaction between cyclophosphamide and other antiemetic drugs, lorazepam, and prochlorperazine.
Simulated AUC and Cmax values of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide by varying theKm value only are shown in table 2. This simulation suggests that Cmax is more affected by theKm value. When Kmdecreases 5-fold, Cmax of 4-hydroxycyclophosphamide increases 75% whereas AUC only increases 20%. TheVmax value but not the Kmvalue influences the total exposure of 4-hydroxycyclophosphamide/aldophosphamide. Although many investigators have alleged that the cytotoxic effects of cyclophosphamide are directly proportional to AUC values of 4-hydroxycyclophosphamide/aldophosphamide (3), the relationship of peak concentration of 4-hydroxycyclophosphamide/aldophosphamide and therapeutic efficacy or toxicity has not been well established. Earlier trials have shown that bolus injection of cyclophosphamide at 120 mg/kg caused fatal heart failure, but at a much slower infusion rate of the same dose, heart failure was rare.1 This observation suggested that there might be a direct correlation between peak concentration of active metabolite and cardiac toxicity.
In patients with normal hepatic and renal function, a positive relationship between cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide should be expected when cyclophosphamide is given as a single agent. This is because the kinetics of 4-hydroxycyclophosphamide are formation limited and plasma AUC values for 4-hydroxycyclophosphamide/aldophosphamide increase with an increase in the amount (exposure) of cyclophosphamide. Patients who are enrolled in the solid tumor bone marrow transplantation program are generally in fair physical condition. Therefore, it is not surprising that such a relationship was observed in this cohort of patients.
As the use of combination chemotherapy has become more frequent, drug interations have been a major concern in clinical management. We cannot elucidate the complex metabolic processing of cyclophosphamide in humans with the limited clinical data thus far available. Variations are expected and unpredictable. This may explain why different results have been reported by us and other investigators when cyclophosphamide was administered with various agents. More detailed pharmacokinetic-pharmacodynamic studies are warranted to define the relationship between cyclophosphamide and active metabolites in patients.
Acknowledgments
We are grateful to Drs. Dennis Noe, John Hilton, Susan Ludemen, Ellen Shulman-Roskes, and Norman Sladek for many helpful and scientific discussions, and Ms. Rosemary Clark for her excellent secretarial support.
Footnotes
-
Send reprint requests to: Dr. Tian-Ling Chen, The Johns Hopkins Oncology Center, Room 1–121, 600 North Wolfe Street, Baltimore, Maryland 21287.
-
↵1 O. M. Colvin, unpublished observation.
-
This study was supported in part by Grants CA15396 and CA63437 from the National Institutes of Health. Presented in part at the 87th Annual Meeting of American Association for Cancer Research, Washington, DC, April 20–24, 1996.
-
Dr. M. J. Kennedy is a recipient of an ACS Clinical Oncology Career Development Award.
- Received August 7, 1996.
- Accepted January 29, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}