


Abstract
There is a need for methodology to predict clinically significant drug-drug interactions so that clinical studies can be directed toward interactions which are likely to be clinically relevant. To this end, we evaluated selective assays for the seven drug-metabolizing cytochrome P450 (P450) isozymes 1A2 (caffeineN3-demethylation), 2A6 (coumarin 7-hydroxylation), 2C9 (tolbutamide hydroxylation), 2C19 (S-mephenytoin 4-hydroxylation), 2D6 (dextromethorphan O-demethylation), 2E1 (chlorzoxazone 6-hydroxylation), and 3A4/5 (dextromethorphanN-demethylation). Using initial rate conditions, we determined the Km andVmax values of each reaction in human liver microsomes from three individuals. Because organic solvents (usually methanol) are frequently used as solubilization aids for drugs/inhibitors, we also screened several solvents for inhibitory activity. Methanol was the least inhibitory toward P450s 2A6, 2D6, and 3A4, dimethylformamide was the least inhibitory toward P450s 1A2 and 2C9, and acetonitrile was the least inhibitory toward P450s 2C19 and 2E1. Using substrate concentrations close to the determinedKm and an appropriate solvent (where necessary), we used the selective inhibitors furafylline (1A2), 8-methoxypsoralen (2A6), sulfaphenazole (2C9),S-mephenytoin (2C19), quinidine (2D6), diethyldithiocarbamate (2E1), and troleandomycin (3A4) to assess the limitations of each probe assay as an indicator of the P450 isoform in question. Our results were consistent with these inhibitors and probes, being selective tools for studying P450 drug metabolism.
Toxicity or therapeutic failure has long been recognized as a product of drug-drug interactions when polytherapy is instituted. With increasing use of polytherapy, as in the treatment of acquired immunodeficiency syndrome and its associated opportunistic infections, there is a need for rapid, simple, and cost-effective in vitro methods to predict clinically significant drug-drug interactions. Once validated, such methods can be used to identify and prioritize the further study of drug-drug interactions that are most likely to be significant in the clinic.
As part of an ongoing series of studies on drug-drug interactions associated with anti-acquired immunodeficiency syndrome and anti-opportunistic infection (OI) drugs (Palamada et al., 1995), we sought to determine the capacity of these drugs to inhibit the microsomal cytochrome P450 monooxygenase (P4503) enzymes most commonly involved in xenobiotic metabolism. To do so, we first evaluated the selectivity of various in vitro probes and the suitability of various organic solvents for the measurement of human cytochrome P450 activities in human liver microsomes. We chose to focus our studies on the P450 enzymes because these enzymes are involved in the clearance of the vast majority of the anti-OI drugs. In humans, these enzymes are present at the highest concentration in the liver but are also present in other tissues such as the intestinal mucosa. They exist as a superfamily of isozymes, of which there are more than 37 individual members in humans (Nelson et al., 1996). However, relatively few isozymes are responsible for most metabolic biotransformations involving the clearance of drugs from the body. These are P450s 1A2, 2A6, 2C8/9, 2C19, 2D6, 2E1, and 3A4/5, which collectively account for more than 70% of all P450 isozymes present in the human liver (Shimada et al., 1994).
We selected our assays based on previous accounts of the selectivity of each drug probe for each specific P450 isozyme of interest. We also considered the ease with which each assay could be implemented on a simple HPLC system with either UV or fluorescence detection. As a result, we chose caffeine N3-demethylation as a probe for CYP1A2 (Butler et al., 1989; Fuhr et al., 1992; Guet al., 1992; Tassaneeyakul et al., 1992), coumarin 7-hydroxylation for CYP2A6 (Maurice et al., 1991;Miles et al., 1990; Yamano et al., 1990), tolbutamide 4-methyl-hydroxylation for CYP2C8/9 (Miners et al., 1988; Brian et al., 1989; Relling et al., 1990; Doecke et al., 1991; Srivastava et al., 1991), S-mephenytoin 4-hydroxylation for CYP2C19 (Srivastava et al., 1991; Goldstein et al., 1991), dextromethorphan O-demethylation for CYP2D6 (Kupferet al., 1986; Dayer et al., 1991), chlorzoxazone 6-hydroxylation for CYP2E1 (Peter et al., 1990), and dextromethorphan N-demethylation for CYP3A4/5 (Jacqz-Aigrainet al., 1993; Gorski et al., 1994). The concentration of each probe drug used in each assay was chosen with the aim that the assay would be isozyme specific while providing enough sensitivity in the assay to allow facile detection of the isozyme-specific metabolite. Because it was our aim to have a battery of isozyme-specific assays and isozyme-selective inhibitors that were internally consistent, we tested each assay with one or more selective inhibitors. In addition, as many drugs are poorly soluble in water or buffer, solvents are frequently used to aid drug solubility in in vitro drug metabolism experiments. With the exception of the cytochrome P450 2E1 (Peter et al., 1990), there is little data regarding the consequences of the use of organic solvents on the cytochromes P450 even though concentrations of 1% solvent have been reported in previous inhibitor selectivity studies (Newton et al., 1995). Therefore, we also assessed the effect of the organic solvents (1% v/v) methanol, dimethylformamide (DMF), isopropanol, DMSO, acetone, and acetonitrile in these microsomal P450 assays.
Materials and Methods
Materials.
Caffeine, chlorzoxazone, coumarin, dextromethorphan, diethyldithiocarbamate (DDC), 8-methoxypsoralen, NADPH, 4-nitrophenol, quinidine, tolbutamide, troleandomycin (TAO), and umbelliferone (7-hydroxycoumarin) were purchased from Sigma. Furafylline, 4-hydroxy-S-mephenytoin, sulfaphenazole, and 4-methylhydroxytolbutamide were purchased from RBI (Natick, MA). Dextrophan tartrate, 3-hydroxymorphinan, and 3-methoxymorphinan were provided by Hoffmann-La Roche (Nutley, NJ). S-Mephenytoin was provided by Dr. Rene H. Levy, and 6-hydroxychlorzoxazone was provided by Dr. R. M. Peter. Other reagents were purchased from Sigma or JT Baker (Phillipsburg, NJ), and HPLC grade solvents were purchased from Baxter Healthcare (Muskegon, MI). Solvents for the inhibition experiments were purchased from Sigma (isopropanol, molecular biology grade 99+%, dimethyl sulfoxide, ACS reagent andN, N-dimethylformamide, HPLC grade 99.9+%), Baxter (methanol, B & J Brand), JT Baker (acetone, 99.4+%), and Fisher Scientific (acetonitrile, HPLC grade 99.9+%).
Isolation of Human Liver Microsomes.
Human liver tissue was procured, prepared, and stored as described previously (Rettie et al., 1989). Human liver microsomes were prepared by differential centrifugation as described previously (Rettie et al., 1989) with the following modifications. Microsomes were prepared from 10–15 g of thawed tissue, which was homogenized in 5 volumes of 250 mM sucrose, 100 mM potassium phosphate, pH 7.4, 1mM EDTA, using a Virtis Vertishear probe homogenizer (The Virtis Company, Inc., Gardiner, NY) at 50% speed for 2 × 30 sec. The final microsome pellet was resuspended in homogenization buffer (without glycerol), and protein concentrations were estimated using the Bradford assay with bovine serum albumin standard as commercially available from Bio-Rad. The microsomes from three human livers, designated HL139 (15-year-old female with history of phenytoin therapy), HL140 (63-year-old male with a history of alcohol use), and HL141 (59-year-old male with a history of alcohol use who quit 14 years prior to tissue collection) were used in this study.
Microsomal Incubations for the Determination of Cytochrome P450 Monooxygenase Activities.
Microsomal incubations were carried out in a total volume of 100 μl and in the presence of 1mM NADPH (freshly prepared). Microsomes (25 μl in 250 mM sucrose, 100 mM potassium phosphate and 1mM EDTA, pH7.4) were preincubated for 5 min at 37° with 100 mM potassium phosphate, 1mM EDTA buffer, pH 7.4 (55 μl) and substrate (10 μl) prepared in water with minimal use of methanol or DMF [caffeine (water), coumarin (0.01% methanol), tolbutamide (0.2% DMF), S-mephenytoin (0.1–0.375% methanol), dextromethorphan (2D6, 0.0025% methanol; 3A4/5, 0.25% methanol), and chlorzoxazone (final equivalent of aqueous 0.06 mM KOH)]. The reactions were started by the addition of 10 μl of freshly prepared 10 mM NADPH. Reactions were terminated by the addition of 10 μl of 2 M HCl and cooling on ice (5–60 min). Samples were then centrifuged (20,000g, 5 min, 4°), and the supernatant was injected directly onto the HPLC. The time of incubation and concentration of microsomal protein were chosen for each substrate such that rates of metabolite formation were demonstrated to be linear (table 1). Unless otherwise stated, the concentration of the substrate, the incubation time, and the protein concentration used in each assay were as specified in table 1. All microsomal incubations were conducted in triplicate. Controls included incubations without microsomes, without NADPH, or without substrate.
A summary of the reaction conditions and Michaelis-Menten parameters of drug biotransformation reactions used as selective probes for human cytochrome P450 in vitro
Determination of Michaelis-Menten Parameters for Cytochrome P450 Isozyme-Specific Activities.
Enzyme velocity experiments were carried out as described above over a range of concentrations of the P450 substrates caffeine (50–30,000 μM), coumarin (0.25–10 μM), tolbutamide (12.5–800 μM),S-mephenytoin (10–150 μM), dextromethorphan-2D6 (1–50 μM), chlorzoxazone (5–500 μM), and dextromethorphan-3A4/5 (50–1200 μM). The parameters Km andVmax, with standard errors, were estimated by fitting the Michaelis-Menten equation to the data using nonlinear regression analysis (PCnonlin). Initial estimates for nonlinear regression were chosen based on substrate concentration (S) verses reaction velocity (V) plots and Eadie-Hofstee plots (V/S verses V).
Inhibition of Specific P450 Isozymes by Selected Solvents.
To investigate the inhibitory capacity of the solvents acetone, DMF, DMSO, isopropanol, methanol, and acetonitrile, microsomal incubations were conducted as above, except 10 μl of 10% solvent (v/v) in water (or water as control) was added to each preincubation (1% v/v final concentration in assay). The volume of 100 mM phosphate buffer with 1 mM EDTA was adjusted to 45 μl to maintain the final total volume of 100 μl.
Inhibition of Specific P450 Isozymes by Selected Compounds.
The concentration of each inhibitor was selected such that the inhibition of each target P450 activity was estimated to be greater than or equal to 90% based on previous studies (Broly et al., 1989; Chang et al., 1994; Chiba et al., 1993; Kunze and Trager, 1993; Maenpaa et al., 1993; Minerset al., 1988; Peter et al., 1990; Tassaneeyakulet al., 1993). To investigate the inhibitory capacity of 8-methoxypsoralen (5 μM), sulfaphenazole (2.16 μM),S-mephenytoin (360 μM), quinidine (0.45 μM), 4-nitrophenol (540 μM), and chlorzoxazone (900 μM), microsomal incubations were conducted as above, except 10 μl of each inhibitor (at 10 times the final concentration) was added to each preincubation. The volume of 100 mM phosphate buffer with 1 mM EDTA was adjusted to 45 μl to maintain the final total volume of 100 μl. Furafylline (10 μl of 300 μM in 5% solvent4) was preincubated with microsomes (25 μl) and phosphate buffer (45 μl) for 3 min as described above except without substrate. NADPH (10 μl of 10 mM in water) was then added and incubated for a further 3 min prior to the addition of enzyme substrate (10 μl) to start the reaction. TAO (25 μl of 200 μM in 5% solvent4) was incubated with microsomes (125 μl), phosphate buffer (75 μl), and NADPH (25 μl of 10 mM in water) for a 30-min inactivation period at 37°. A 50 μl aliquot of this TAO/microsome incubation was added to a mixture of substrate (10 μl), NADPH (10 μl of 10 mM in water), and phosphate buffer (30 μl) to start the reaction. DDC was used in the same way as TAO, except DDC was soluble in water (25 mM), the concentration in the inactivation incubation was 25 μM, and the inactivation took place over 15 min. The concentration of solvent4 (v/v) in the final incubation was as follows: furafylline (0.5%), 8-methoxypsoralen (0.02%), coumarin (0.05%), sulfaphenazole (0.2%),S-mephenytoin (0.5%), quinidine (<0.01%), DDC (0%), chlorzoxazone (0%), p-nitrophenol (0.5%), and TAO (0.25%). Control incubations were conducted with the appropriate concentration of solvent.4
Analysis of Microsomal Incubates by HPLC.
Isocratic HPLC was sufficient for all the separations described using a Waters 501 or 510 HPLC pump with either a Waters 712 WISP autosampler or Gilson model 323 bio sample injector. Reversed phase HPLC columns (Microsorb MV, Rainin Instrument Company, CA) were used with a 0.2 μm precolumn filter (Upchurch Scientific, Oak Harbor, WA) for all the assays described below. The mobile phase conditions used to separate each substrate from the metabolite of interest without interference resulting from the other components or metabolites in the assay were as follows. The product of N3-demethylation of caffeine, 1,7-dimethylxanthine (17X), was separated from caffeine, 1,7-dimethyluric acid, 1,3-dimethylxanthine, and 3,7-dimethylxanthine using a C18 column (4.6 × 250 mm, 5-μm particle size) and methanol:water:acetic acid:triethylamine 12:88:1:0.02 (v/v) as mobile phase at 1 ml·min-1. A linear gradient to methanol:water 50:50 (v/v) over 1 min held for 1 min with a linear gradient back to mobile phase over 1 min was used to hasten the elution of the substrate, caffeine, after the elution of 17X. The product of 7-hydroxylation of coumarin was separated from coumarin using a C18 column (4.6 × 100 mm, 3-μm particle size) and methanol:20mM sodium phosphate buffer (pH 4.4) 27:73 (v/v) as mobile phase at 0.8 ml·min-1. The product of the hydroxylation of tolbutamide was separated from tolbutamide using a C8 column (4.6 X 250 mm, 5-μm particle size) and acetonitrile:water:acetic acid:triethylamine 35:65:1:0.02 (v/v) as mobile phase at 1ml·min-1. The tolbutamide HPLC assay conditions were also used for the separation of 4-hydroxy-S-mephenytoin from S-mephenytoin and the separation of the dextromethorphan metabolites, 3-hydroxymorphinan, dextrophan, and 3-methoxymorphinan from dextromethorphan. Chlorzoxazone was separated from 6-hydroxychlorzoxazone using a C8 column (4.6 × 250 mm, 5-μm particle size) and acetonitrile:water:acetic acid:triethylamine 30:70:1:0.02 (v/v) at 1ml·min-1. Analytes were detected using variable wavelength absorbance (Waters 481 or 484 detector) for caffeine (270 nm), coumarin (324 nm), tolbutamide (240 nm),S-mephenytoin (240 nm), and chlorzoxazone (297nm). Dextromethorphan and its metabolites were detected by fluorescence (Hewlett Packard HP1046A programmable fluorescence detector) using excitation and emission wavelengths of 235 nm and 310 nm, respectively, with a 280 nm filter. In each case, data were recorded using Waters Maxima HPLC analysis software. Quantitation of metabolites in unknown samples was possible by comparison of peak area with calibration lines generated by injections of authentic standards.
Results
We present data suggesting that a system of seven selective P450 assays and several selective inhibitors are suitable for use inin vitro metabolism studies directed at the prediction of metabolic drug-drug interactions involving P450 isozymes. We have determined suitable incubation conditions for the following enzyme activities: caffeine N3-demethylation (1A2), coumarin 7-hydroxylation (2A6), tolbutamide 4-methyl-hydroxylation (2C8/9),S-mephenytoin 4-hydroxylation (2C19), dextromethorphanO-demethylation (2D6), chlorzoxazone 6-hydroxylation (2E1), and dextromethorphan N-demethylation (3A4/5). Each enzyme activity was characterized in terms of linearity with respect to time and with respect to protein concentration. The reaction times, protein concentration used, and the Michaelis-Menten parameters determined for each isozyme activity in the human liver microsomes are summarized in table 1. The effects of six organic solvents at a final concentration of 1% (v/v) on each enzyme activity compared with activity in the absence of the additional solvent were then determined, and these results are summarized in fig. 1. Using this information, we then investigated the selectivity of the inhibitors, furafylline (1A2), 8-methoxypsoralen (2A6), coumarin (2A6), sulfaphenazole (2C9), S-mephenytoin (2C19), quinidine (2D6), DDC (2E1), p-nitrophenol (2E1), chlorzoxazone (2E1), and TAO (3A4) on each P450 activity. These results are summarized in fig.2.

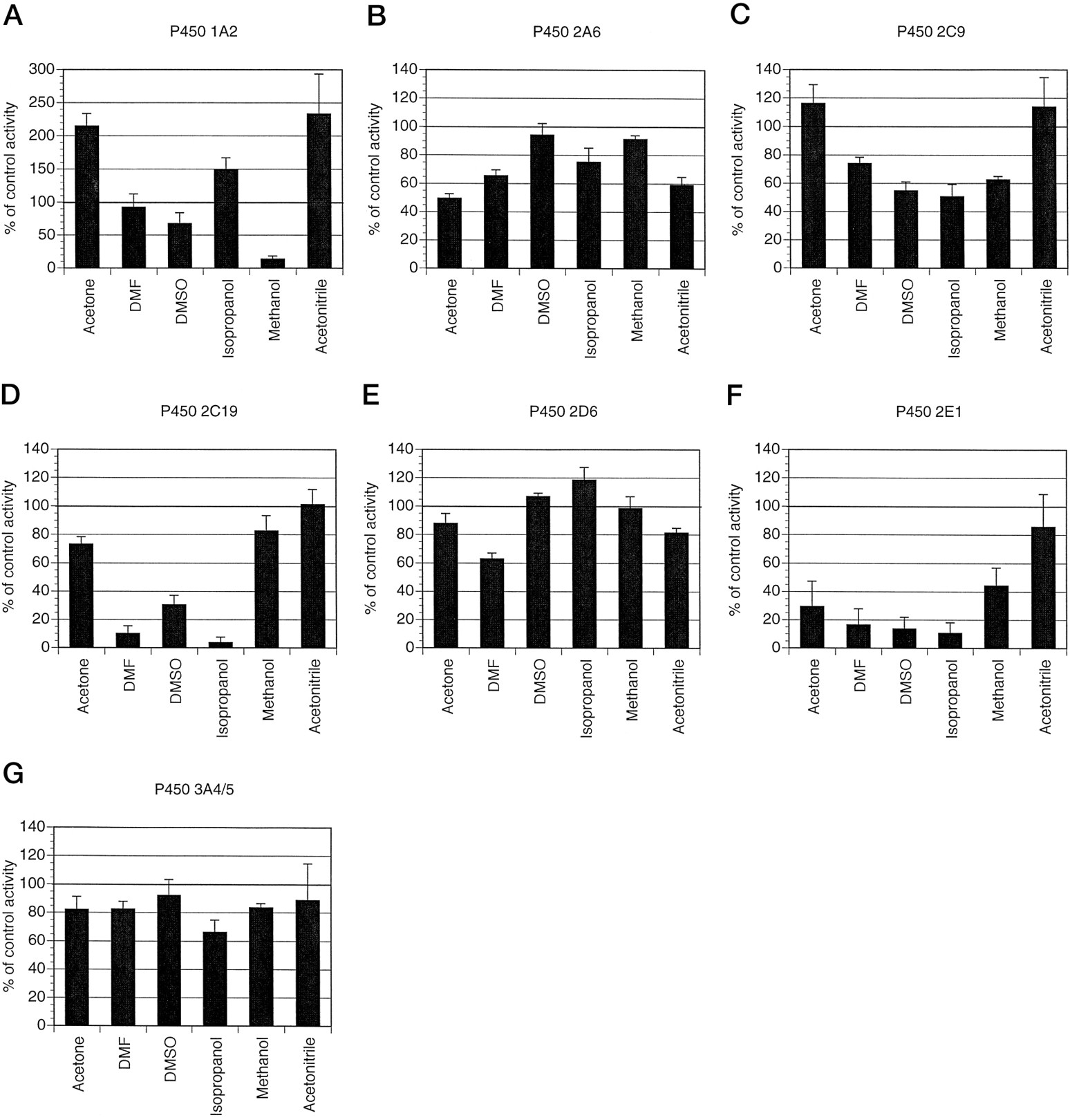
The effect of several organic solvents on the activity of P450-mediated biotransformations. The effect of acetone, DMF, DMSO, isopropanol, methanol, and acetonitrile on caffeine N3-demethylation (A), coumarin 7-hydroxylation (B), tolbutamide 4-methyl-hydroxylation (C), S-mephenytoin 4-hydroxylation (D), dextromethorphan O-demethylation (E), chlorzoxazone 6-hydroxylation (F), and dextromethorphan N-demethylation (G) was determined by coincubation of each solvent (1% v/v) with human liver microsomes, 1 mM NADPH, and caffeine (1000 μM), coumarin (5 μM), tolbutamide (100 μM), S-mephenytoin (75 μM), dextromethorphan (5 μM), chlorzoxazone (50 μM), or dextromethorphan (500 μM), respectively. All activities were measured under initial rate conditions and compared with incubations in the absence of additional solvent. Eachbar represents the mean of experiments conducted on microsomes prepared from three different livers that have been expressed individually as a percent of control activity. Theerror bars indicate standard deviations. Due to substantial activation of caffeine N3-demethylation, data for this activity are presented on an expanded y axis compared with the other solvent inhibition experiments.
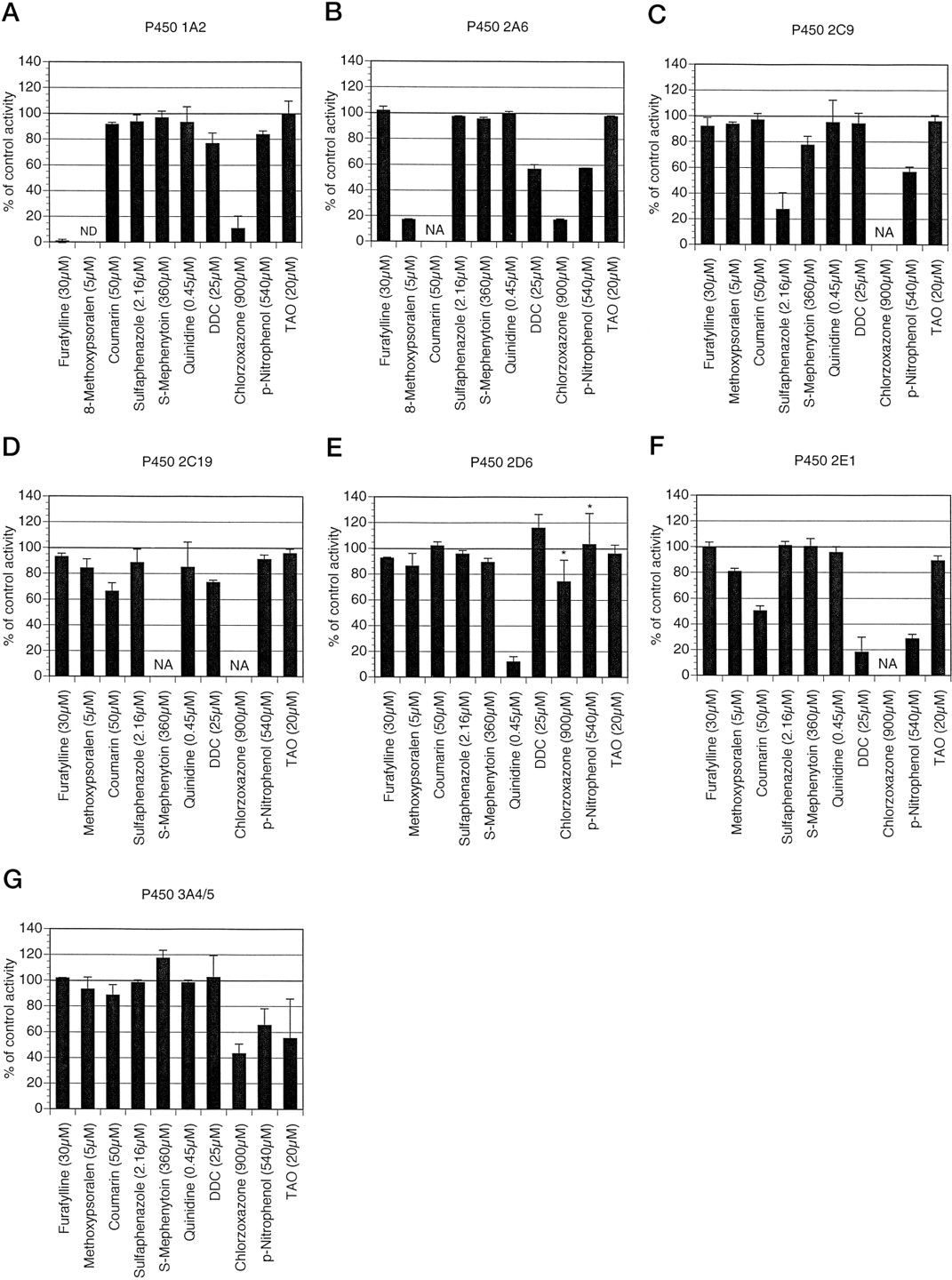
The effect of several selective P450 inhibitors on the activity of P450-mediated drug biotransformations. The effect of inhibitors furafylline (30 μM preincubation for 3 min), 8-methoxypsoralen (5 μM), sulfaphenazole (2.16 μM),S-mephenytoin (360 μM), quinidine (0.45 μM), DDC (25 μM preincubation for 15 min), p-nitrophenol (540 μM), and TAO (20 μM preincubation for 30 min) on caffeine N3-demethylation (A), coumarin 7-hydroxylation (B), tolbutamide 4-methyl-hydroxylation (C), S-mephenytoin 4-hydroxylation (D), dextromethorphan O-demethylation (E), chlorzoxazone 6-hydroxylation (F), and dextromethorphan N-demethylation (G) was determined by coincubation of each inhibitor (as described inMaterials and Methods) with human liver microsomes, 1 mM NADPH, and caffeine (1000 μM), coumarin (5 μM), tolbutamide (100 μM), S-mephenytoin (75 μM), dextromethorphan (5 μM), chlorzoxazone (50 μM), or dextromethorphan (500 μM), respectively. All activities were measured under initial rate conditions and compared with control incubations in the absence of inhibitor. Each bar represents the mean of experiments conducted on microsomes prepared from three different livers that have been expressed individually as a percent of control activity. Theerror bars indicate standard deviations. NA, not applicable or not determined due to interference of the assay by inhibitor or metabolites of the inhibitor; ND, not detectable. * denotes a large standard deviation due to an unrepresentative response of dextromethorphan O-demethylation activity to both chlorzoxazone and p-nitrophenol in HL140 or inter-liver variability in the inhibition of dextromethorphanN-demethylation by troleandomycin.
Discussion
In vitro metabolic probes for each of the principle drug-metabolizing P450 microsomal oxidases have been chosen so that at a suitable concentration its conversion to the metabolite of interest is mediated by a single form, or in some cases two closely related forms, of these microsomal enzymes. Where possible, we have chosen substrate concentrations close to that of theKm for the P450 isozyme of interest for subsequent inhibition studies. This is in contrast to the evaluation byNewton and colleagues (1995) where the concentration of the P450 substrates were 5–10 times the Km as in, for example, phenacetin (100 μM), tolbutamide (500 μM), chlorzoxazone (500 μM), bufuralol (100 μM), and testosterone (250 μM). Although it can be argued that concentrations approachingVmax (as used by Newton) offer certain advantages, such as increased sensitivity of assay and reduced effect of Km on apparent velocity,Vmax concentrations are less sensitive to inhibition by both specific and inadvertant inhibitors (mechanism-based inhibitors excepted). In this study, a concentration approximately equal to the Km was chosen as a practical compromise between sensitivity of the assay for measurement of the metabolite (higher concentrations allow for more reliable detection of metabolite) and both selectivity of the P450 assay for a single isozyme and sensitivity of the enzyme velocity toward inhibition by selected compounds (concentrations below Km preferred). Each specific P450 activity (and its selective inhibitor) is discussed in turn.
Caffeine N3-Demethylation (1A2).
Caffeine N3-demethylation is a relevant in vitro measure of cytochrome P450 1A2 activity (Butler et al., 1989; Guet al., 1992; Tassaneeyakul et al., 1992). We observed that caffeine N3-demethylation (50–30,000 μM caffeine) followed biphasic kinetics in human liver microsomes, which is in agreement with several previous studies (Campbell et al., 1987; Fuhr et al., 1992; Gu et al., 1992;Tassaneeyakul et al., 1992). TheKm estimates for the low affinity component were 20.5 mM and 22.3 mM for microsomes prepared from HL139 and HL141, respectively. Furthermore, this low affinity component was effectively abolished by preincubation of the human liver microsomes with TAO (as described in Materials and Methods), which is in agreement with a previous study indicating that P450 3A4 is responsible for this low affinity activity (Tassaneeyakul et al., 1992). Using a two-enzyme Michaelis-Menten model, we calculated that this low affinity enzyme (3A4) would be responsible for less than 5% of caffeine N3-demethylation at a caffeine concentration of 1000 μM, which is the estimated Km of the high affinity enzyme (table 1). The preincubation of the human liver microsomes with furafylline, a specific mechanism-based inhibitor of 1A2 (Kunze and Trager, 1993; Newton et al., 1995;Sesardic et al., 1990; Tassaneeyakul et al., 1994), confirmed this prediction (fig. 2A). In addition, this result indicated a minimal role for the related isozyme 1A1 in caffeine N3-demethylation in the three livers tested, as 1A1 is less sensitive to inhibition by furafylline (Tassaneeyakul et al., 1994). Furthermore, furafylline preincubation did not inhibit any of the other six P450 activities investigated here by more than 10% compared with controls (fig. 2, B–G) and thus proved to be a specific inhibitor for 1A2 under the conditions used. This is in contrast to a previous study where incubations with higher, and presumably less selective, concentrations of furafylline (200 μM) indicated that furafylline was not a specific inhibitor of 1A2 (Onoet al., 1996).
Our finding that caffeine N3-demethylation at 1000 μM caffeine is modulated specifically by 1A2 is somewhat at odds with previous findings (Tassaneeyakul et al., 1992, 1994). They estimate a substantially lower Km (200–240 μM) and a lower Vmax (0.018 nmol/min/mg protein) for the high affinity caffeine N3-demethylation, resulting in their recommendation that a lower concentration of caffeine (<200 μM) be used to retain 1A2-specific caffeine N3-demethylation. These differences might be explained in part by the interindividual variability in Km estimates for caffeine N3-demethylation; here, 600-1600 μM caffeine was estimated for the three livers, whereas 220-1200 μM was estimated by others (Bloomeret al., 1995) for six livers. In addition, our estimates ofVmax for the high affinity enzyme (1A2) ranged from 0.110 to 0.295 nmol/min/mg protein, which compared well with other researchers (Bloomer et al., 1995; Campbellet al., 1987) whose estimates range from 0.140 to 0.570 and 0.115 to 0.450 nmol/min/mg protein, respectively. TheVmax estimates obtained by both Gu and colleagues (1992) and Tassaneeyakul and colleagues (1992, 1994) are 0.023 and 0.018 nmol/min/mg protein, which is an order of magnitude lower contributing to the greater relative importance of the lower affinity enzyme to caffeine N3-demethylation, at low concentrations of caffeine, in their experiments. The reasons for these differences between studies are unknown, but differences in buffer constituents and/or solvent usage remain distinct possibilities given the sensitivity of P450 enzymes to specific experimental conditions.
The results of our solvent inhibition study with caffeine N3-demethylation (fig. 1A) also indicated that methanol was an unsuitable solvent in any study in which P450 1A2 is implicated. For instance, the use of furafylline to investigate the importance of 1A2 in the metabolism of an investigational compound would be confounded by the use of methanol even if relevant controls containing methanol were included. Methanol at 1% (v/v) would decrease the relative importance of 1A2 by 90% (fig. 1A), and a hypothetical biotransformation in which 1A2 contributed 50% under normal conditions (i.e. no solvent) would only contribute 5% in the presence of methanol at 1% v/v and thus 1A2 would be erroneously ruled out as significant in metabolism. Therefore, on the basis of our results, we chose a minimal concentration of DMF as a solvent where necessary. It is also interesting to note that acetone and acetonitrile at 1% (v/v) activated caffeine N3-demethylation by more than 2-fold, on average, in our three liver microsome preparations (fig. 1A). In addition, isopropanol at 1% (v/v) activated caffeine N3-demethylation by 1.5-fold, on average. If confirmed for other 1A2 probe activities, and therefore presumably due to a general effect of these solvents on 1A2 activity, acetone, acetonitrile, and isopropanol activation might be diagnostic of processes involving P450 1A2 as α-naphthoflavone is for P450 3A4 (Shimada et al., 1994; Shou et al., 1994).
Coumarin 7-Hydroxylation (2A6).
Coumarin 7-hydroxylation is catalyzed by CYP2A6 (Maurice et al., 1991; Miles et al., 1990; Yamano et al., 1990). This reaction followed monophasic kinetics consistent with the involvement of a single enzyme (0.25–10 μM coumarin). The mean estimates of Km andVmax for coumarin 7-hydroxylation (table 1) were similar to the 2.3 μM Km and the 0.03–1.35 nmol/min/mg protein Vmaxestimated in human liver microsomes by Yamano and colleagues (1990). Due to limitations in assay sensitivity, we used 5 μM coumarin for examining the effect of solvents and inhibitors on coumarin 7-hydroxylation activity so that we could reliably detect at least 10% of the control activity.
The results of our solvent inhibition study indicated that CYP2A6 was relatively resilient to solvent inhibition (fig. 1B). Methanol and DMSO at 1% (v/v) did not inhibit 2A6 activity and therefore proved the most suitable of those tested. Given that methanol was most often a preferred solvent for use with other P450 enzymes, further experiments were conducted using a minimal amount of methanol where solvents were necessary. 8-Methoxypsoralen at 5 μM inhibited coumarin 7-hydroxylation greater than 80%. This compound has previously been reported to have a Ki of 1.5 μM for human CYP2A6 (Maenpaa et al., 1993). With the exception of CYP1A2, 8-methoxypsoralen was relatively specific for CYP2A6, inhibiting no other activity more than 20%, indicating that 8-methoxypsoralen is a more selective inhibitor of P450 2A6 than has been indicated previously (Ono et al., 1996). However, this inhibitor abolished any detectable caffeine N3-demethylation activity consistent with previous reports of CYP1A2 inhibition bothin vivo (Mays et al., 1987) and in vitro (Fuhr et al., 1992; Ono et al., 1996). Because coumarin is a substrate for CYP2A6, we also investigated the utility of coumarin itself as a potential selective inhibitor of 2A6 by looking at its profile of inhibition of the other P450 enzymes. Although 18 times the Km (orKi ) concentration would be estimated to inhibit CYP2A6 activity 90% when a substrate is present atKm concentration, we used 50 μM coumarin (50 times the Km ) in an attempt to avoid inhibitor depletion by metabolism of coumarin by CYP2A6. Mostly, coumarin (50 μM) was less selective for CYP2A6 than 8-methoxypsoralen. Coumarin (50 μM), however, was not inhibitory toward CYP1A2, 2C9, and 2D6 (fig. 2, A, C, andE). Therefore, inhibition of any biotransformation mediated by an unknown P450 by 8-methoxypsoralen (5 μM) alone does not implicate CYP2A6. We postulate, however, that inhibition by coumarin (50 μM) in a separate experiment would strongly implicate the 2A6 isozyme.
Tolbutamide 4-Methyl-Hydroxylation (2C8/9).
The hydroxylation of tolbutamide at the 4-methyl position is mediated by cytochrome P450s of the 2C subfamily (Miners et al., 1998). Specifically, tolbutamide has been shown to be a substrate for the expressed human P450s 2C8, 2C9, and 2C10 (Brian et al., 1989; Relling et al., 1990; Srivastava et al., 1991; Veronese et al., 1993). Tolbutamide hydroxylation in human liver microsomes prepared from HL139–141 (12.5–800 μM tolbutamide) was consistent with the involvement of a single enzyme or more than one enzyme with a similar Km as previously indicated by Miners and colleagues (1988). Our estimates ofKm (133–348 μM) andVmax (0.387–0.434 nmol/min/mg protein) are also in agreement with this previous study.
Tolbutamide hydroxylation was somewhat sensitive to inhibition by the solvents (1% v/v) isopropanol, DMSO, and methanol (fig.1C). Conversely, acetone and acetonitrile at 1% (v/v) resulted in a moderate increase in tolbutamide hydroxylation activity to 118 and 114% of the control activity, respectively. Therefore, DMF, which inhibited this activity less than 27%, was chosen as the preferred solvent where solvents were necessary for further experiments.
Sulfaphenazole has been previously shown to be a potent inhibitor of 2C enzymes (Doecke et al., 1991; Miners et al., 1988; Rettie et al., 1992; Veronese et al., 1990). In our studies, sulfaphenazole was used at a concentration of 2.16 μM, which is 18-fold higher than theKi reported previously (Miners et al., 1988). Sulfaphenazole (2.16 μM) inhibited tolbutamide hydroxylation (100 μM tolbutamide) 59 to 85% in our human liver microsomes (HL140>141>139). It is interesting to note that the microsomes with the highest Km (HL139) were also most resistant to sulfaphenazole inhibition. This is likely due to a significant contribution of 2C8 in this liver, as this isozyme has both a 5-fold higher Km for tolbutamide hydroxylation than 2C9 and is also more resistant to sulfaphenazole inhibition (Veronese et al., 1993).
Sulfaphenazole inhibition was also very selective toward tolbutamide hydroxylation, as the P450-specific assays for 1A2, 2A6, 2D6, 2E1, and 3A4 were not inhibited (fig. 2C). S-Mephenytoin 4-hydroxylation (75 μM S-mephenytoin) was only marginally inhibited (on average 12%; range 3–23%). A recent study demonstrates that sulfaphenazole concentrations up to 100 μM do not inhibit 1A2, 2D6, 2E1, or 3A4 activities (Newton et al., 1995), suggesting a higher concentration of sulfaphenazole could be used without loss of selectivity toward tolbutamide hydroxylation, although this would have to be tested for coumarin 7-hydroxylation andS-mephenytoin 4-hydroxylation.
S-Mephenytoin 4-Hydroxylation (2C19).
The 4-hydroxylation of S-mephenytoin is mediated by cytochrome P450 2C19 (Goldstein et al., 1994). Substrate concentration velocity experiments indicated thatS-mephenytoin 4-hydroxylation (10–150 μMS-mephenytoin) was mediated by a single enzyme in human liver microsomes prepared from HL139 and HL141. Our estimates ofKm were 29.5 and 75.8 μM, and our estimates of Vmax were 0.066 and 0.055 nmol/min/mg protein, respectively, for the two livers tested. We did not determine these values for HL140 due to low turnover ofS-mephenytoin in this liver microsome preparation.S-Mephenytoin 4-hydroxylation was inhibited by the solvents (1% v/v) isopropanol>DMF>DMSO>acetone> methanol>acetonitrile (fig. 1D). Methanol, which inhibited this activity less than 20% compared with the control (75 μM S-mephenytoin), was chosen where solvents were necessary for further experiments. In the absence of any putative specific inhibitors of P450 2C19 other thanS-mephenytoin itself and because a direct measurement of inhibition of S-mephenytoin 4-hydroxylation by itself is not possible, we assumed that Km was equal toKi for this enzyme and chose 360 μMS-mephenytoin as our 2C19 inhibitor. This concentration was predicted, using the Michaelis-Menten equation in the presence of a competitive inhibitor, to inhibit 2C19 activity by 85 and 65% in HL139 and HL141, respectively, if the substrate was present at itsKm concentration. S-Mephenytoin (360 μM) inhibited P450 2C9 by 22% compared with control but inhibited the other P450-specific assays (1A2, 2A6, 2D6, 2E1, and 3A4/5) only marginally (less than 10% inhibition) or not at all.
Dextromethorphan O-Demethylation (2D6).
Dextromethorphan O-demethylation activity correlates well with debrisoquin 4-hydroxylase (Kupfer et al., 1986; Schmidet al., 1985) and is a good in vitro probe activity for P450 2D6 (Gorski et al., 1994; Jacqz-Aigrainet al., 1993). Substrate concentration velocity experiments indicated that dextromethorphan O-demethylation activity (1–50 μM) was mediated by a single enzyme in human liver microsomes prepared from HL139 through 141. Our estimates ofKm ranged from 2.2 to 8.5 μM andVmax from 0.097 to 0.566 nmol/min/mg protein, which were consistent with several previous studies [Km , 7.5 μM;Vmax, 0.07 nmol/min/mg protein (Jacqz-Aigrain et al., 1993);Km , 3.4 μM;Vmax, 0.17 nmol/min/mg protein (Dayeret al., 1989); Km , 4.6 μM;Vmax, 0.07 nmol/min/mg protein (Brolyet al., 1989)]. Dextromethorphan O-demethylation activity was inhibited by the solvents (1% v/v) DMF>acetonitrile>acetone (fig. 1E) and moderately increased by the solvents (1% v/v) isopropanol >DMSO. Methanol at 1% (v/v) had, on average, no effect on dextromethorphanO-demethylation (5 μM substrate) and was used in subsequent experiments. Quinidine is a potent and selective inhibitor of P450 2D6 (Broly et al., 1989). Based on a previously published Ki of 0.025 μM (Broly et al., 1989), we used 0.45 μM (18 timesKi ) for our inhibition experiments. As predicted for a 2D6 activity with substrate concentration at theKm , we observed 90% inhibition of dextromethorphan O-demethylation (fig. 2E). Quinidine (0.45 μM) was also a selective inhibitor of 2D6 inhibiting the P450s 1A2, 2A6, 2C9, 2C19, 2E1, and 3A4/5 marginally (less than 20%) or not at all in our three liver microsome preparations.
Chlorzoxazone 6-Hydroxylation (2E1).
Chlorzoxazone 6-hydroxylation activity is a relevant measure of P450 2E1 activity (Peter et al., 1990). Substrate concentration velocity experiments indicated that this activity (5–500 μM chlorzoxazone) was mediated by a single enzyme in human liver microsomes prepared from HL139 through 141. Our estimates ofKm ranged from 51 to 64 μM andVmax from 2.75 to 5.59 nmol/min/mg protein, which are consistent with previous studies [Km , 22–49 μM;Vmax, 1.1–5.9 nmol/min/mg protein (Peteret al., 1990)]. Chlorzoxazone 6-hydroxylation activity was inhibited by the solvents (1% v/v) isopropanol>DMSO>DMF>acetone> methanol>acetonitrile (fig.1F). Methanol (1% v/v), which inhibited this activity approximately 55% compared with the control (50 μM substrate), was used in subsequent experiments as methanol (0.1% v/v) only marginally inhibited chlorzoxazone 6-hydroxylation (10%) compared with the control (data not shown). However, given the apparent tolerance of chlorzoxazone 6-hydroxylation activity to acetonitrile, this solvent may prove a useful alternative to methanol in future studies involving P450 2E1.
DDC is a mechanism-based inhibitor of P450 2E1 (Guengerich et al., 1991). However, there have been reports that DDC is both a selective inhibitor (Newton et al., 1995) and a poorly selective inhibitor (Chang et al., 1994; Ono et al., 1996) for this isozyme. Therefore, in addition to DDC, we also used p-nitrophenol as a potential selective inhibitor for P450 2E1. We chose a concentration of 25 μM DDC and a preincubation time of 15 min based on a previous concentration-dependent study (Newton et al., 1995), which indicated that this treatment would inhibit P450 2E1 greater than 60% while inhibiting P450s 1A2, 2C9, 2D6, and 3A4 less than 10%. Based on a previously published Km forp-nitrophenol of 30 μM (Tassaneeyakul et al., 1993), we used 540 μM (18 times Km ) for our p-nitrophenol inhibition experiments.
DDC proved to be a potent inhibitor of P450 2E1 inhibiting chlorzoxazone 6-hydroxylation by greater than 80% (fig.2F). DDC (125–200 μM) has previously been shown to be a poorly selective inhibitor of P450 2E1 (Chang et al., 1994;Ono et al., 1996). However, our data indicate that DDC used at a concentration of 25 μM is a useful and selective inhibitor of 2E1 inhibiting 1A2 and 2C19 less than 25%, on average, and not inhibiting P450s 2C9 and 3A4/5 in our three liver microsome preparations. P450 2D6 activity was apparently increased by DDC (118% of control). p-Nitrophenol (540 μM) inhibited P450 2E1 approximately 70%, which is less than the 90% inhibition predicted for an inhibitor at 18 times Ki .p-Nitrophenol was less selective than DDC, inhibiting the P450 2C19, 2D6, and 1A2 activities less than 20% but inhibiting the 2C9 and 3A4/5 activities approximately 40–50%. Both DDC andp-nitrophenol inhibited P450 2A6 activity around 50%. We did not pursue the P450 2E1 substrate chlorzoxazone (900 μM) as a potential inhibitor as it inhibited the P450 1A2, 2A6, and 3A4/5 activities greater than 50%, 2D6 activity 25%, and this inhibitor interfered with the 2C9 and 2C19 HPLC assays. This finding was in agreement with in vivo and in vitro interaction studies with caffeine and chlorzoxazone (Berthou et al., 1995), which demonstrated a marked inhibition of caffeineN-demethylation activities by chlorzoxazone. However, the inhibition of P450 1A2 activity by chlorzoxazone does not by itself implicate chlorzoxazone as a metabolically significant substrate for this isozyme. Indeed, given that furafylline did not inhibit chlorzoxazone 6-hydroxylation activity in human liver microsomes (fig.2F), it is unlikely that P450 1A2 is involved in this activity at therapeutic concentrations of chlorzoxazone (30–60 μM), which is suggested by the expressed enzyme studies of Ono and colleagues (1995).
Dextromethorphan N-Demethylation (3A4/5).
Dextromethorphan N-demethylation activity is a selectivein vitro probe activity for P450 3A4/5 (Gorski et al., 1994; Jacqz-Aigrain et al., 1993). Substrate concentration velocity experiments indicated that this activity (50–1200 μM dextromethorphan) was mediated by a single enzyme in human liver microsomes prepared from HL139 through HL141. Our estimates of Km ranged from 133 to 369 μM andVmax from 0.33 to 2.89 nmol/min/mg protein, which compared with previous studies [Km , 520–710 μM; Vmax, 0.375–0.812 nmol/min/mg protein (Gorski et al., 1994); 450–830 μM, 0.078–147 nmol/min/mg protein (Jacqz-Aigrain et al., 1993)]. Dextromethorphan N-demethylation activity was inhibited by the solvents (1% v/v) isopropanol>acetone= DMF = methanol>acetonitrile>DMSO (fig. 1G). Methanol (1% v/v), which inhibited this activity less than 20% compared with the control (500 μM substrate), was used in subsequent experiments.
TAO is a mechanism-based inhibitor of P450 3A4 (Murray, 1987). We used a concentration of 20 μM TAO and a preincubation time of 30 min based on a previous concentration-dependent study (Newton et al., 1995), which indicated that this treatment would inhibit P450 3A4/5 greater than 60% while inhibiting P450s 1A2, 2C9, 2D6, and 2E1 less than 20%.
TAO proved to be a selective inhibitor of P450 3A4/5, inhibiting dextromethorphan N-demethylation by 20–80% (fig.2F) but inhibiting the other P450 enzymes studied less than 10%, on average, in our three liver microsome preparations. We believe that the large variation in percent inhibition of dextromethorphanN-demethylation activity by the TAO pretreatment is due to a variable contribution of P450 3A5, which is possibly more resistant to TAO inhibition when compared with P450 3A4 as indicated by Ono and colleagues (1996).
We validated a comprehensive battery of selective P450 assays and inhibitors for use in in vitro microsomal drug metabolism experiments. We discovered that certain solvents, even when present at modest concentrations of 1%, can substantially inhibit P450 activity. For concentrations of solvent that are below a final concentration of 0.1%, the choice of solvent may not be critical. Where concentrations of solvent must approach or exceed 1% in the incubation, the choice of solvent may be more critical. In these cases, a solvent may be selected on a basis of minimal inhibition, with methanol used when determining the activity of CYPs 2A6, 2D6, and 3A4/5, DMF with the CYPs 1A2 and 2C8/9, and acetonitrile with the CYPs 2C19 and 2E1. If none of these solvents are suitable and a solvent not examined here is to be used, we recommend that the inhibitory profile toward the various P450s be ascertained prior to use. Not doing so could lead to masking the contribution of certain P450s toward the metabolism of the substrate of interest. In addition, our studies have shown that we can confidently use the P450 assays described above for screening drugs for potential inhibitory metabolic interactions in human liver microsomes and in correlational studies designed to assess P450 isozyme involvement in the biotransformation of a new drug. Furthermore, the inhibitors can be utilized as described above for identifying specific isozymes responsible for xenobiotic biotransformations that have not as yet been characterized with respect to P450 isozyme involvement. The demonstrated lack of cross-inhibition for each inhibitor will allow confident assignment of specific P450 isozymes to studied biotransformations. Correlational analysis and specific isozyme inhibition after determination of the Michaelis-Menten parameters for a biotransformation will provide a solid foundation that can be supported by other evidence such as demonstration of expressed enzyme activity and immunoinhibition experiments.
Footnotes
-
Send reprint requests to: Jashvant D. Unadkat, Department of Pharmaceutics, University of Washington, H272 Health Sciences, Box 357610, Seattle, WA 98195.
-
↵1 Present address: D-46V Biotransformations, AP-9, Abbott Laboratories, Abbott Park, IL 60064-3500.
-
↵2 Deceased.
-
This work was supported by National Institutes of Health grants AI27664 and DK41978.
-
↵4 When necessary, methanol was used to aid the solubility of the inhibitors used in the coumarin,S-mephenytoin, dextromethorphan, and chlorzoxazone assays. DMF was used with the caffeine and tolbutamide assays.
- Abbreviations used are::
- DMT
- dextromethorphan
- DDC
- diethyldithiocarbamate
- DMF
- dimethylformamide
- TAO
- troleandomycin
- DMSO
- dimethyl sulfoxide
- HPLC
- high performance liquid chromatography
- P450
- cytochrome P450
- Received May 20, 1997.
- Accepted November 18, 1997.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}