


Abstract
Induction of cytochromes P450 (P450s) by drugs can lead to drug-drug interactions. Primary hepatocytes have been reported to retain inducible P450s. To optimize the use of primary hepatocytes for predicting induction of P450 (CYP 3A and 2B) expression in vivo, both culture conditions and expression of induction potentials were investigated. In rat hepatocytes, basal CYP 3A1/2 expression was better maintained in cells cultured on Matrigel compared with collagen when low concentrations of dexamethasone were used. However, CYP 3A1/2 induction was not affected by either matrix. In contrast, induction of CYP 2B1/2 by phenobarbital was markedly stronger in hepatocytes cultured on Matrigel. To further validate the in vitro model, Sprague-Dawley rats and isolated hepatocytes cultured on Matrigel were exposed to a series of compounds. In an attempt to minimize large variability between experiments, a novel approach for calculating induction potential was applied. In vitro results for CYP 3A1/2 and 2B1/2 induction correlated well with those observed in vivo. In contrast with rat hepatocytes, basal CYP 3A4 expression in human hepatocytes decreased rapidly in cells cultured on either Matrigel or collagen. However, CYP 3A4 inducibility was retained in cells cultured on either matrix. Interestingly, induction of CYP 3A4 in human hepatocytes by several model compounds did not correlate with the induction of CYP 3A1/2 in rat hepatocytes. This in vitro assay should facilitate the demand for a fast and reproducible method for addressing P450 induction by numerous compounds at the drug discovery stage.
Cytochromes P450 (P450s)2 form a gene superfamily that are involved in the metabolism of a variety of chemically diverse substances ranging from endogenous compounds to xenobiotics including drugs, carcinogens, and environmental pollutants. Although P450 regulation is still poorly understood, it is well known that some of the P450 genes are induced severalfold by specific drugs. This may cause variability in enzymatic activity with different groups of patients producing unexpected pharmacological activity of some drugs as a result of drug-drug interactions (Wadhwa et al., 1987). Knowledge of potential P450 inducibility by drug candidates, prior to drug development, would greatly enhance the ability to develop drugs that are free of P450-inducing properties. In the past, various animal species have been used as models to investigate P450 induction by drug candidates. The data obtained from such in vivo models has proven to be beneficial in assessing P450 induction. However, at the drug discovery stage this is difficult to carry out because of the large numbers of animals and the large amount of compound needed to conduct such experiments. In addition, there exist species differences in P450 induction (Kocarek et al., 1994b), making the extrapolation from animals to humans unreliable. Therefore, having a simple, robust, and reproducible in vitro model to study P450 induction would greatly facilitate the ability to develop drugs devoid of these possible negative traits. Such a model would offer the advantage of requiring less drug (<1 mg) and reducing the number of animals used. In addition, confounding factors such as bioavailability, blood, and liver levels of the drug would be avoided.
There are no known hepatoma cell lines able to express most of the major forms of adult P450. Induction of P450s have been documented to occur in primary cultured hepatocytes isolated from various species including man (Combalbert et al., 1989; Kocarek et al., 1994b; Schuetz et al., 1988). However, it is well known that primary cultured hepatocytes tend to lose expression of specific liver functions, including P450s, as they adapt to culture conditions over time (Schuetz et al., 1988). In an attempt to maintain the differentiated function of hepatocytes in culture, cell culturists have tried various approaches, including manipulation of culture medium with various hormonal supplementation (Decad et al., 1977; Jefferson et al., 1984) and cocultures of hepatocytes with other cells such as fibroblasts under the hypothesis that factors produced by nonparenchymal cells may be important in maintaining hepatocytes (Baffet et al., 1982;Guguen-Guillouzo et al., 1983; Morin and Normand, 1986). Others have focused on the substratum to which hepatocytes attach (Kleinman et al., 1985). In this latter case, instead of culturing hepatocytes on plastic dishes coated with a thin layer of collagen, more complex matrices have been investigated including fibronectin (Johansson and Hook, 1984), extracts from rat liver (Reidet al., 1980), and more recently Matrigel (Li et al., 1987; Schuetz et al., 1988). However, the best culture conditions for maintaining constitutive and inducible P450 expression in primary hepatocytes remains unclear.
The objective of this study was to refine culture conditions and validate the use of hepatocytes to study CYP 3A and 2B induction. Specifically, basal and inducible P450 expression were investigated in rat (CYP 3A1/2 and 2B1/2) and human (CYP 3A4) hepatocytes cultured on collagen or Matrigel. These two isozymes were targeted because in humans, CYP 3A4 is the most abundant form of liver P450 and is responsible for metabolism of >60% of all clinically used drugs (Cholerton et al., 1992; Molowa et al., 1986), and CYP 2B1 has been associated with rodent tumorogenesis (Lubetet al., 1989). Results obtained in rat hepatocytes were then contrasted with those from in vivo studies.
Materials and Methods
Materials.
Williams’ E media, dexamethasone, phenobarbital, phenytoin, and rifampicin were obtained from Sigma. Waymouth 752 media and penicillin G/streptomycin were from GIBCO (Grand Island, NY). Horseradish peroxidase-conjugated goat anti-rabbit antibody was purchased from Amersham. Anti-rat CYP 3A1 and CYP 2B1 antibodies were obtained from Human Biologics (Phoenix, AZ). Collagenase type I was purchased from Worthington (Freehold, NJ). Matrigel, dispase, and type I collagen-coated dishes were purchased from Collaborative (Bedford, MA). All other chemicals were of high purity grade and purchased from commercial sources.
Rat Hepatocyte Isolation and Culture.
Rat hepatocytes were isolated from male Sprague-Dawley rats (weighing 200–250 g) by a two-step collagenase perfusion (Moldeus et al., 1978). Only hepatocyte preparations attaining a cell viability of >85%, as assessed by trypan blue exclusion (0.2% trypan blue), were used. Hepatocytes were suspended in Williams’ E medium (1 × 106 cells/ml) containing 10-7 M dexamethasone and 10-6 M insulin and were seeded on 60-mm LUX culture dishes (3 × 106 cells/dish) previously coated with rat tail type I collagen or Matrigel. Culture dishes coated with Matrigel were prepared 1 hr prior to cell isolation by spreading 170 μl of freshly thawed Matrigel (1 mg/ml) in each plate (Schuetz et al., 1988). Culture dishes containing hepatocytes were then placed in a humidified incubator saturated with a gas mixture of 6% CO2 and 94% air at 37°C. Cells were allowed 2 hr to attach, at which time the medium was changed to remove unattached cells. Fresh media were replenished on a daily basis. The media used for rat hepatocyte cultures were chosen based on previous studies showing P450 induction by well characterized inducers (Schuetz et al., 1988).
Human Hepatocytes Isolation and Culture.
Fresh human liver tissue was obtained from consenting donors undergoing partial hepatectomies and from unused liver portions from patients undergoing liver transplants. Tissue samples were transported from the operating room in ice-cold University of Wisconsin solution. Hepatocyte isolation was conducted by a two-step collagenase perfusion of the liver sample as described by Li et al. (1992) with the following modifications: the final perfusion buffer contained 650 μg collagenase/ml, and the tissue (20–30 g) was perfused for approximately 25 min. Perfusion was via the single blood vessel, which gave the most blanching of the liver. Rapid perfusate outflow from large blood vessels on the cut surface of the tissue sample was minimized by inserting sterile 200-μl pipette tips. This allowed perfusate to rise up the tip, thereby increasing resistance but not completely blocking outflow. This modification was found to increase the yield of viable cells, especially from liver samples that were not fully encapsulated on all three sides. Cells were then sedimented at 50 g for 2 min and washed three times with ice-cold phosphate-buffered saline, pH 7.4. Typically, a viability >85% (as measured by trypan blue exclusion) was obtained. The cells were suspended (1 × 106 cells/ml) in Waymouth 752/L medium supplemented as previously described (Li et al., 1992). The cells were inoculated into 24-well plates (3 × 105 cells/well) coated with rat tail type I collagen or Matrigel (30 μl/well). Hepatocytes were then treated as described for rat hepatocytes. The media used for rat hepatocyte cultures were chosen based on previous studies showing P450 induction by well characterized inducers (Li et al., 1992).
Hepatocyte Treatment.
Isolated hepatocytes were cultured on collagen or Matrigel-coated dishes. To determine constitutive expression of CYPs 3A and 2B, cells were incubated in culture media and harvested every 24 hr for up to 96 hr, and P450-specific proteins were measured by Western blot immunochemical analysis. To determine the inducibility of P450s, hepatocytes were first allowed 48 hr to adapt to culture conditions followed by exposure to inducers for an additional 48 hr, at which time they were harvested. Inducers were dissolved in dimethyl sulfoxide and added to cultures at a final volume not exceeding 0.25%. The medium was changed 24 hr after treatment, and the cultures were retreated. At 96 hr, hepatocytes were harvested.
Harvesting of Hepatocytes.
Hepatocytes cultured on 60-mm dishes were overlaid with ice-cold phosphate-buffered saline containing 5 mM EDTA (4 ml/plate). Cells and gel were then removed with a cell scraper, transferred to 15-ml tubes, and placed on ice for approximately 45 min to solubilize the Matrigel. Cells were resuspended by repeated pipetting until cells separated from the substratum (∼10 times) and sedimented by centrifugation at 50g for 2 min. For hepatocytes cultured on 24-well plates, cells were harvested by incubating the cells with 25% dispase (400 μl/well) for approximately 1 hr at 37°C followed by addition of phosphate-buffered saline (1 ml) containing 5 mM EDTA. Cells were then transferred to Eppendorf tubes and sedimented by centrifugation at 50g for 2 min.
Preparation of Samples for Western Blot Analysis.
For rat hepatocytes cultured in 60-mm dishes, the cellular microsomal fraction was isolated. Briefly, cell pellets were suspended in 100 μl of ice-cold 0.1 M potassium phosphate, pH 7.4, and homogenized by sonication. The cell homogenate was centrifuged at 600g for 10 min, followed by ultracentrifugation of the supernatant for 30 min at 100,000g in a Beckman TL100 tabletop ultracentrifuge. The microsomal pellet was then resuspended in 0.1 M potassium phosphate, pH 7.4, and stored at −70°C until use.
For human hepatocytes cultured on 24-well plates, the cellular microsomal fraction was not routinely isolated. Instead, cells harvested from each well were resuspended in 100 μl of ice-cold 0.1 M potassium phosphate, pH 7.4, and homogenized by sonication. The cell homogenate was then used directly for Western blot analysis. Protein concentration was determined by the Bradford method using bovine serum albumin as the standard (Bradford, 1976).
Western Blot Analysis.
Microsomal protein fraction and cell homogenates (1–10 μg/well) were resolved on 10% polyacrylamide gels, as described by Laemmli (1970). After electrophoresis, proteins were transferred to nitrocellulose membranes using the method of Towbin et al. (1979). The blots were incubated with the following antibodies: polyclonal rabbit anti-rat CYP 3A1 or polyclonal rabbit anti-rat CYP 2B1. Specific analysis of CYP 3A1 and 2B1 was not possible because the antibody for CYP 3A1 cross-reacts with 3A2, and the antibody for CYP 2B1 cross-reacts with 2B2 (Parkinson and Gemzik, 1991). Hence, CYP 3A1 and 2B1 induction will be referred to as CYP 3A1/2 and CYP 2B1/2, respectively. For human hepatocytes, anti-rat-CYP 3A1 antibody was found to cross-react with CYP 3A4 to give one band. Proteins were visualized using ECL (Amersham). ECL detection reagents were used as per supplier instructions, and the signal generated was analyzed using a densitometer. P450 induction by drug candidates is expressed by formulating the densitometry readings as follows:
Northern Blot Analysis.
Northern blots hybridized with cDNA probes recognizing CYP 3A1 and CYP 2B1 were performed on rat hepatocytes cultured on Matrigel. The cells were incubated with inducers 48 hr after plating and harvested at 2, 4, 6, 8, 16, and 24 hr. Total RNA was extracted from cultured hepatocytes using TRIzol reagent (Life Technologies, Inc.). The RNA (20 μg) was size-fractionated by electrophoresis in 1% agarose formaldehyde gels and transferred to zetaprobe membranes (Bio-Rad). Hybridization was performed using oligonucleotides complementary to rat CYP 3A1 and 2B1, respectively. Oligonucleotides were synthesized on a model 377 Applied Biosystems DNA synthesizer as described by the manufacturer and had the following sequence: CYP 2B1, 5′-GGTTGGTAGCCGGTGTGA-3′ and CYP 3A1, 5′-CGGATAGGGCTGTATGAGATTC-3′. Prehybridization solution was 0.25 M sodium phosphate, pH 7.2, 7% sodium dodecyl sulfate, and hybridization was carried out in the same solution with added32P-kinased oligonucleotide probe (∼1 × 106 cpm/ml) at 48°C. Washes were done at 48°C for CYP 3A1 and at 42°C for CYP 2B1 in 20 M sodium phosphate, pH 7.2, 5% sodium dodecyl sulfate. Blots were then stripped and reprobed with a human G3PDH cDNA (Clontech).
In Vivo Treatments.
Sprague-Dawley male rats (four animals/treatment group) were treated po with 13 drug candidates from our current drug discovery program at 400 mg/(kg × day) for 4 days. The induced CYP 3A1 positive control group was treated with dexamethasone at 50 mg/(kg × day) for 3 days, whereas the induced CYP 2B1 positive control was treated with a mixture of phenobarbital/benzafibrate at 50 mg/(kg × day) for 4 days. A negative control group was treated with vehicle only, once a day for 4 days. At the end of the 4-day study, the animals were sacrificed and liver microsomes prepared as described by Lu and Levin (1972) with the following modifications. Livers were homogenized with 2 volumes of 0.1 M potassium phosphate, pH 7.4. An aliquot of the homogenate (100 μl) was diluted 10-fold for a total volume of 1 ml and centrifuged for 10 min at 9000g, and the supernatant layer then centrifuged for 30 min at 100,000g in a Beckman TL-100 ultracentrifuge. The resulting microsomal pellet was resuspended in 200 μl of 0.1 M potassium phosphate, pH 7.4, and stored at −70°C. P450 analysis was conducted as described above.
Results
Induction of CYP 3A1/2 and 2B1/2 in Rat Hepatocytes.
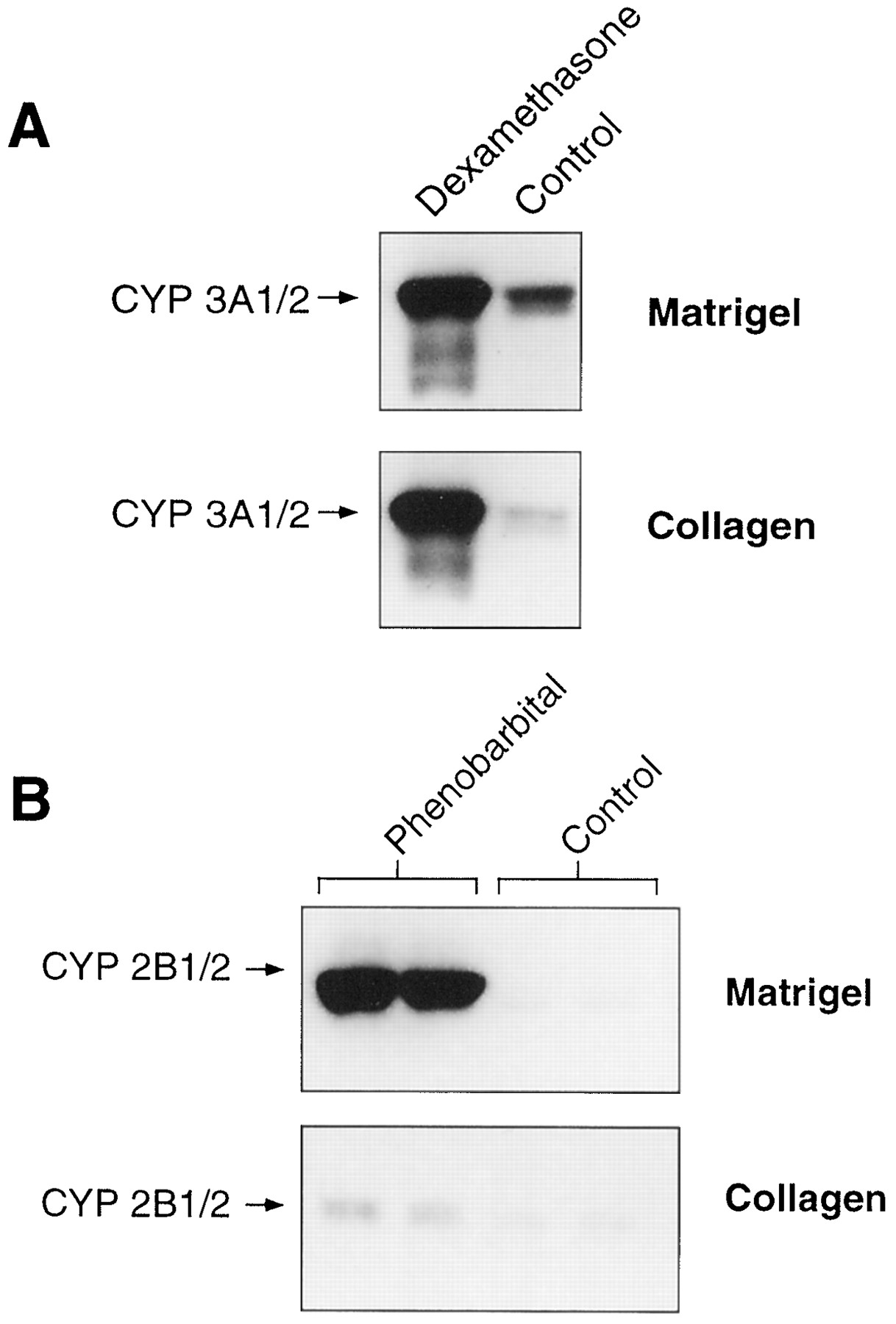
As shown in fig. 1A, dexamethasone (10 μM) caused a marked increase in CYP 3A1/2 protein in hepatocytes cultured on either matrix. An approximate 50-fold increase was observed in cells cultured on collagen compared with an approximate 10-fold increase in cells on Matrigel. Even though the -fold increase in CYP 3A protein was dependent on the cell matrix, the final protein levels after induction seemed similar. Higher doses of dexamethasone did not further increase CYP 3A1/2 protein accumulation (results not shown). However, as shown in fig.2, -fold induction of CYP 3A varied from one experiment to another. In these latter experiments, dexamethasone caused a 6.9- to 19.8-fold increase in CYP 3A protein with seven different hepatocyte preparations.

Immunoblot anlalysis of CYP 3A1/2 (A) and CYP 2B1/2 (B) protein in rat hepatocytes cultured on Matrigel and collagen. Cells cultured on Matrigel and on collagen were incubated in the absence or presence of 10 μM dexamethasone or 50 μM phenobarbital for 48 hr. P450s were analyzed as described in Materials and Methods.

Variability in CYP 3A1/2 induction in rat hepatocytes cultured on Matrigel. Cells from seven different preparations of hepatocytes were incubated in the absence or presence of 10 μM dexamethasone for 48 hr. Panel A represents -fold increase in CYP 3A protein from dexamethasone-treated cells over nontreated cells as calculated from the density readings of the Western blots shown in panel B. P450s were analyzed as described in Materials and Methods.
Fig. 1B represents blots of CYP 2B1/2 immunoreactive protein from hepatocytes treated with phenobarbital (50 μM). In cells maintained on Matrigel, CYP 2B1/2 was induced approximately 50-fold. Higher concentrations of phenobarbital did not further increase induction of CYP 2B1/2 expression (data not shown). In comparison, induction of CYP 2B1/2 by phenobarbital in hepatocytes cultured on collagen was markedly less.
We next examined expression of CYP 3A1 and 2B1 mRNAs in rat hepatocytes as a response to inducers. Fig. 3 shows an increase in CYP 3A1 and 2B1 mRNA within 2 hr of adding dexamethasone and phenobarbital, respectively. G3PDH was simultaneously probed to prove that equal amounts of mRNA were loaded in each lane.

Northern blot analysis of CYP 3A1 and 2B1 mRNAs in rat hepatocytes cultured on Matrigel. Hepatocytes maintained on Matrigel were incubated with 10 μM dexamethasone or 50 μM phenobarbital for up to 48 hr, and mRNA expression was analyzed. The blots were then reprobed with G3PDH cDNA to verify the loading of RNA.
Maintenance of Basal CYP 3A1/2 and 2B1/2 in Rat Hepatocytes.
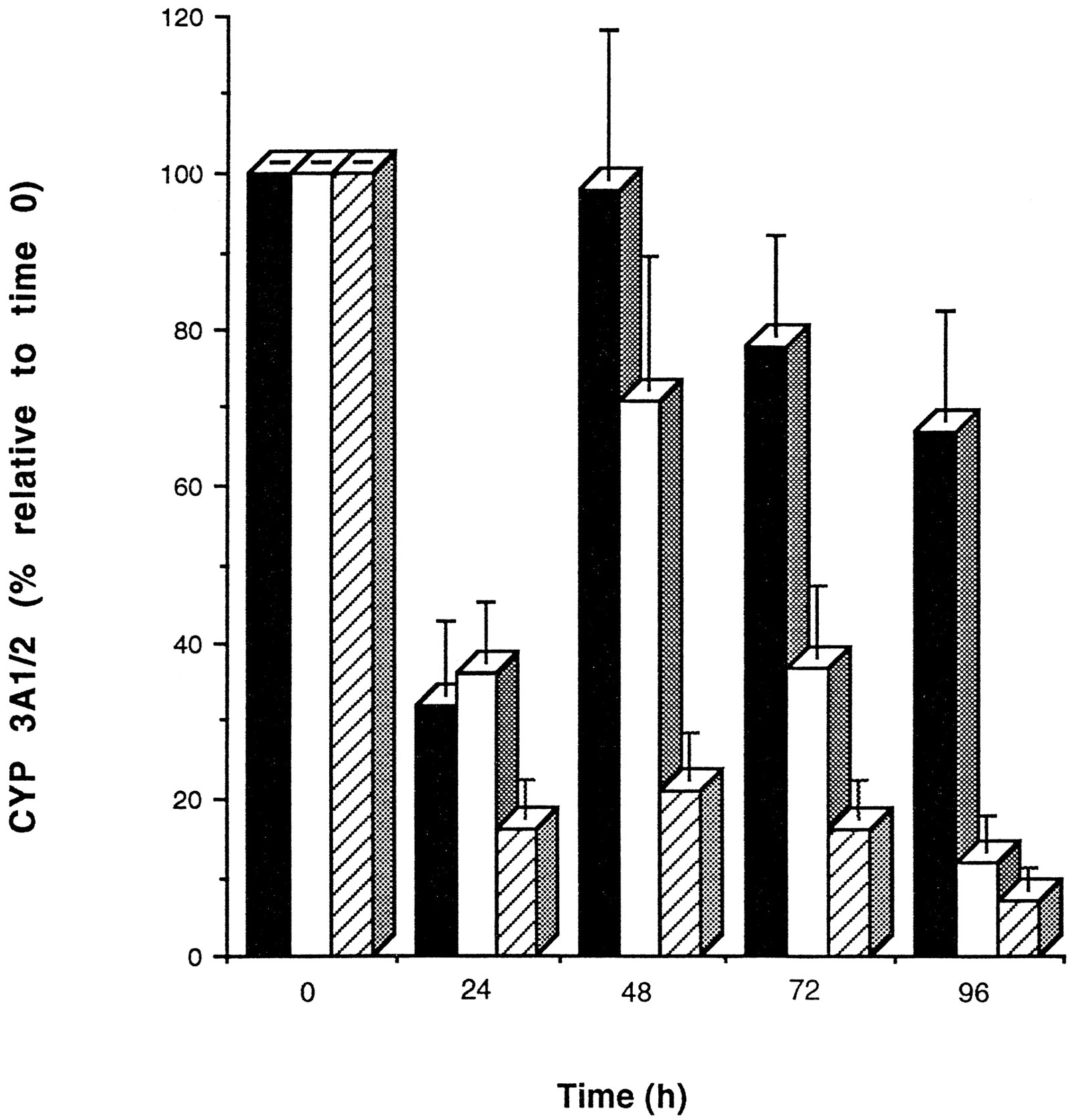
As shown in fig. 4, CYP 3A1/2 protein levels throughout the 4-day period differed between cells cultured on Matrigel from those cultured on collagen. A rapid decrease in CYP 3A1/2 protein was observed during the initial 24 hr in cells cultured on either matrix, followed by recovery to initial levels by 48 hr. Longer incubation periods caused a rapid decrease in CYP 3A1/2 protein levels in hepatocytes cultured on collagen (∼30% CYP 3A1/2 protein left after 96 hr compared with day 0). In contrast, CYP 3A1/2 protein levels were better maintained (∼80% CYP 3A1/2 left after 96 hr compared with day 0) in hepatocytes cultured on Matrigel. Interestingly, if dexamethasone (100 nM) was omitted from the media, the recovery and maintenance of CYP 3A1/2 protein previously observed did not occur (fig. 4).

Maintenance of CYP 3A1/2 protein in rat hepatocytes cultured on Matrigel and collagen. Cells were cultured for 96 hr on Matrigel (closed bar) or on collagen (open bar) dishes in media containing 100 nM dexamethasone and also on Matrigel in media devoid of dexamethasone (stippled bar). Immunochemical quantitation of CYP 3A1/2 protein was analyzed every 24 hr as described in Materials and Methods. Band densities were then expressed as percentages relative to a sample at time zero. Values shown are the means of three separate experiments (±SD).
Basal CYP 2B1/2 expression in rat hepatocytes cultured on collagen and Matrigel was also measured on each consecutive day. However, Western blot analysis demonstrated that CYP 2B1/2 protein was present at levels that were too low to be able to quantify (results not shown).
Induction of CYP 3A1/2 and 2B1/2 In Vitro vs. Induction In Vivo.
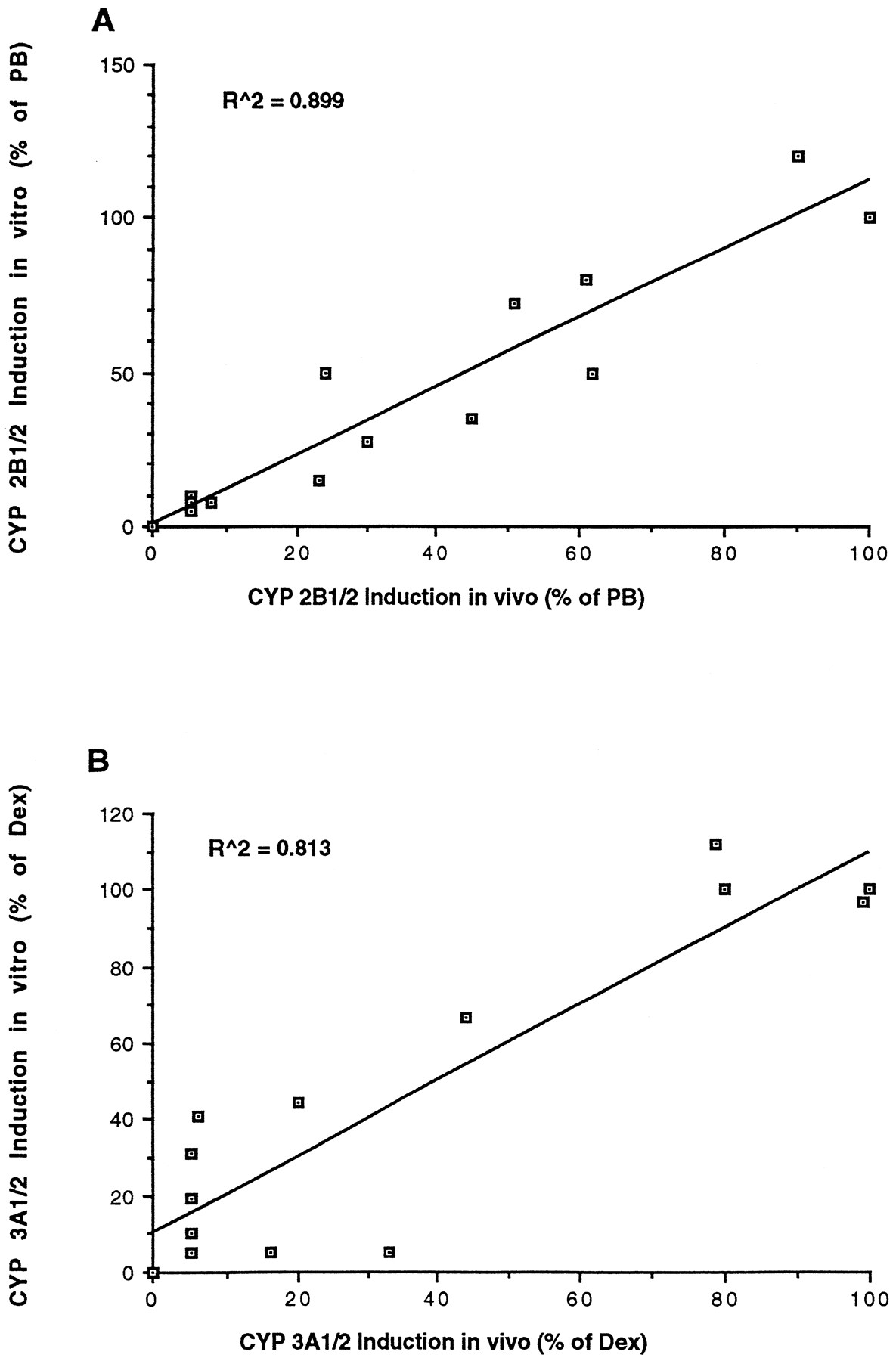
A series of 13 compounds from our current drug discovery program was used to further validate the in vitro model by directly comparing results obtained in vivo with those fromin vitro. Previous experiments in our laboratory with compounds from the same structure class suggested that a 50 μM concentration gave maximal effect (results not shown). Hence, hepatocytes were treated with 50 μM doses. Similarly, in vivo doses of 400 mg/kg were chosen to obtain sufficient drug blood levels to elicit maximum response. Induction of CYP 3A and 2B are defined as a percentage of a classic inducer present in each experiment, rather than -fold increase because of the variability observed between different preparations of hepatocytes. Fig.5 compares the results obtained fromin vivo with those from in vitro experiments. The correlation coefficients (R2) were 0.81 and 0.90 for CYP 3A and 2B, respectively.

Induction of CYP 2B1/2 (A) and CYP 3A1/2 (B) in rat hepatocytes vs. Sprague-Dawley rats. Rat hepatocytes and Sprague-Dawley rats were treated with 13 drug candidates at a dose of 50 μM and at 400 mg/kg, respectively, as described in Materials and Methods. Results for CYP 3A1/2 induction in vitro vs. in vivo are expressed as percentage induction relative to dexamethasone (Dex) treatment, whereas CYP 2B1/2 induction is defined as percentage induction relative to phenobarbital (PB) treatment as described inMaterials and Methods. Values are expressed as means.
Induction and Basal Expression of CYP 3A4 in Human Hepatocytes.
As found with rat hepatocytes, human hepatocytes cultured on Matrigel retained a rounded morphology, whereas those cultured on collagen rapidly flattened out into monolayers. Addition of 10 μM rifampicin, a well known CYP 3A4 inducer, caused a marked increase in CYP 3A4 immunoreactive protein in cells maintained on Matrigel or collagen (fig. 6). Variability in CYP 3A -fold induction as a result of rifampicin treatment in four different hepatocyte preparations ranged from a 4–35-fold increase (results not shown). Addition of 50 μM phenytoin, another well known CYP 3A4 inducer, also caused induction of CYP 3A4 in cells on Matrigel that was indistinguishable from that of cells on collagen (results not shown).

Immunoblot analysis of CYP 3A4 protein in human hepatocytes cultured on Matrigel and collagen. Cells cultured on Matrigel or collagen were incubated in the absence and presence of 50 μM rifampicin for 48 hr. CYP 3A4 was analyzed as described inMaterials and Methods.
Fig. 7 illustrates basal CYP 3A4 expression in human hepatocytes over a 5-day culture period. CYP 3A4 protein levels decreased steadily in cells cultured on either Matrigel or collagen. Dexamethasone (1 μM) was included in the media at all times.

Maintenance of CYP 3A4 protein in human hepatocytes cultured on Matrigel and collagen. Cells were cultured for 96 hr on Matrigel (closed bar) or on collagen (open bar) dishes, and immunochemical quantitation of CYP 3A4 protein was analyzed every 24 hr as described inMaterials and Methods. Band densities were then expressed as percentages relative to a sample at time zero. Values shown are the means of three separate experiments (±SD).
Induction of CYP 3A1/2 in Rat vs. CYP 3A4 in Human.
As demonstrated in fig. 8A, rifampicin caused a dose-dependent increase in CYP 3A4 induction in human hepatocytes but did not induce CYP 3A1/2 in rat cells (fig.8B) when cultured on Matrigel. In contrast, dexamethasone was a potent inducer of CYP 3A1/2 in the rat (fig. 8B) but a weak inducer of CYP 3A4 in human hepatocytes (fig. 8A). Furthermore, incubation of human hepatocytes with two drug candidates caused a much higher induction in CYP 3A expression in rat compared with human hepatocytes. Interestingly, when induction of CYP 3A by drug X (50 μM) was calculated as a fold increase, we observed a range from 2- to 8-fold increase in hepatocytes from four different donors. However, when the same induction for drug X was expressed as a percentage of a classic inducer (10 μM rifampicin), the range was from 16 to 34%. On average, maximum -fold induction of CYP 3A1/2 by dexamethasone and CYP 3A4 induction by rifampicin were similar (approximately 10-fold increase).

Induction of CYP 3A4 in human hepatocytesvs. induction of CYP 3A1/2 in rat hepatocytes cultured on Matrigel. Human (A) and rat (B) hepatocytes were incubated for 48 hr in the presence of rifampicin (Rif) at 10, 2, 0.4, and 0.08 μM; dexamethasone (Dex) at 10 μM; drug X at 50, 10, and 2 μM; and drug Y at 50 μM, as indicated. Induction of CYP 3A4 and 3A1/2 expression was defined as per cent of 10 μM rifampicin induction and per cent of 10 μM dexamethasone induction, respectively. Values are expressed as means of three separate experiments (± SD).
Discussion
The objective of this study was 2-fold: first, to contrast basal and inducible P450 expression in rat (CYP 3A1 and 2B1) and human (CYP 3A4) hepatocytes cultured on collagen vs. Matrigel; second, to validate the hepatocyte model for P450 induction by directly comparing results obtained in vitro with those from in vivo in the rat.
In this study, we found that basal CYP 3A expression in rat hepatocytes was affected by the substratum used. Cells cultured on Matrigel were better able to maintain CYP 3A expression compared with those cultured on collagen. In contrast, Kocarek et al. (1992) reported that CYP 3A mRNA in rat hepatocytes cultured on either Matrigel or collagen disappears almost completely during the first 2 days in culture. A possible explanation for this discrepancy may be due to the difference in media used. In the latter study, the culture media used were devoid of dexamethasone whereas the media used in this study were supplemented with 100 nM dexamethasone. We did find that if dexamethasone was absent from the media, basal CYP 3A expression was not retained. Other beneficial characteristics reported to be afforded by low doses of dexamethasone include prevention of cell dedifferentiation (Sidhu and Omiecinski, 1995), stimulation of phenobarbital-mediated induction of CYP 2B1 (Sinclair et al., 1990; Waxman et al., 1990), and maintenance of cell shape and viability (Castellino et al., 1992;Leroux-Nicollet et al., 1983).
In contrast with maintenance of basal CYP 3A expression, inducibility of CYP 3A in cells cultured on either collagen or Matrigel was retained. This confirms previous studies indicating that Matrigel is not necessary for retaining CYP 3A inducibility by rat hepatocytes in culture (Schuetz et al., 1988).
Constitutive expression of CYP 2B in rat hepatocytes was poorly detected in all the culture conditions studied. This may be due to a combination of low levels of CYP 2B protein in rat hepatocytes as well as the limitation of the detection assay. Therefore, culture conditions best suited for basal CYP 2B expression were not determined. However, induction of CYP 2B expression in response to phenobarbital was dependent on the substratum. Induction of CYP 2B expression was approximately 50-fold in hepatocytes cultured on Matrigel but only approximately 5-fold in cells cultured on collagen. Previous studies have reported that hepatocytes cultured on collagen either failed to respond to phenobarbital or required highly sensitive radiometric assays to be detected (Bissell and Guzelian, 1980; Newman et al., 1982). Our studies show that even though cells cultured on Matrigel had a much stronger response, hepatocytes cultured on collagen did respond to phenobarbital, albeit poorly. More recently, others have also demonstrated that hepatocytes cultured on collagen can respond to phenobarbital induction of CYP 2B when a low dose of dexamethasone is included in the media (Sinclair et al., 1990; Waxmanet al., 1990). Interestingly, Kocarek et al.(1994a) and Sidhu and Omiecinski (1995) reported that phenobarbital induction of CYP 2B is markedly enhanced by the presence of a low dose of dexamethasone (100 nM) but is dramatically down-regulated in the presence of higher dexamethasone concentrations.
Northern blot analysis showed that accumulation of CYP 3A and 2B protein in induced rat hepatocytes was preceded by a rise in CYP 3A1 and 2B1 mRNA. Increase in CYP 3A and 2B protein in response to inducers was first observed 8 hr after inducer was added (results not shown). This suggested that the induction occurred via transcription. This is in agreement with in vivo experiments demonstrating that induction of CYP 3A by dexamethasone required both protein and mRNA synthesis (Castellino et al., 1992).
To further validate the use of primary hepatocytes to predict for P450 induction in vivo, a series of compounds varying in their abilities to induce CYP 3A and 2B in rat hepatocytes was also examinedin vivo in rats. The treatment doses for both experiments were assumed to cause a maximum response. However, it is possible that some compounds may have not reached sufficiently high blood levelsin vivo because of poor bioavailability. Even so, results demonstrated an excellent correlation for CYP 3A and 2B expression obtained in vitro with those from in vivoexperiments. These data strongly support the use of primary hepatocytes to predict for CYP 3A and 2B induction in the rat.
In these latter experiments, we defined P450 induction by drug candidates as a percentage of a classic inducer rather than as -fold protein increase. In the case of CYP 3A, defining induction as -fold increase resulted in a high degree of variability between different hepatocyte preparations (fig. 2). This may be due to a number of reasons, including inter-individual variations in basal level of CYP 3A and general state of the cells. Because this assay is not only used to study P450 induction by individual compounds but also to rank order compounds according to their induction potentials, an internal control was deemed necessary to address variability between different hepatocyte preparations. In the case of CYP 2B, basal levels were already at or below the level of detection, hence expressing induction as a -fold increase over basal levels was impossible with our current detection methods. Therefore, induction of CYP 3A and 2B was defined as a percentage of dexamethasone and phenobarbital, respectively.
Ultimately, the reason for using animal models is to provide us with insight into the possible response in humans. However, differences in P450 induction between species are known to exist, and therefore rat hepatocytes may not necessarily be predictive for human. Results from the rat hepatocyte experiments in this study suggest that hepatocytes are predictive of P450 induction in vivo, and therefore human hepatocytes should be predictive for human. Recent studies with human hepatocytes cultured on collagen have reported that these cells also retain inducible P450s, including 3A4 (Donato et al., 1995; Pichard et al., 1990). However, to our knowledge, comparison of induction of CYP 3A4 in human hepatocytes cultured on collagen vs. Matrigel has not been reported. As clear differences were observed with the rat hepatocyte model, inducibility of CYP 3A4 in human hepatocytes cultured on collagen vs.Matrigel was investigated. In contrast with rat hepatocytes, basal CYP 3A4 expression in primary cultured human hepatocytes decreased rapidly in cells cultured on either collagen or Matrigel. Inducibility of 3A4 upon incubation with high doses of rifampicin was also similar in cells cultured on either matrix. Whereas previous studies have shown rifampicin to cause induction of CYP 3A4 in human hepatocytes, high doses of 50 μM are most often used. In this study, a dose of 2 μM rifampicin was found to elicit a near maximum response. This may be of particular relevance when induction potentials of drug candidates are being directly compared with well known inducers like rifampicin.
As reported previously for rat, rifampicin did not cause induction of CYP 3A in rat hepatocytes (Kocarek et al., 1994b; Wrighton et al., 1985). Interestingly, dexamethasone was found to be a weak inducer of CYP 3A4 in all of the human cells obtained to date. Furthermore, induction of CYP 3A in the rat model by two drug candidates did not translate into similar induction potency of CYP 3A4 in the human hepatocyte model. Therefore, as previous studies have shown, CYP 3A induction in the rat may be of questionable relevance to human, and this further stresses the importance of developing in vitro models from human tissue.
Induction potentials of compounds studied in the human hepatocyte model were also defined as a percentage induction relative to rifampicin, which was included as a standard inducer in every experiment. This enabled us to compare induction potentials of compounds in cell cultures from different individuals. Our findings are in agreement with previous reports, which indicate induction of CYP 3A4 expression in human primary hepatocytes shows a large inter-individual variability (Donato et al., 1995; Kocarek et al., 1994b; Pichard et al., 1990). For example, Pichard et al. (1990) reported CYP 3A4 induction by rifampicin and dexamethasone to range from 3.7- to 24-fold and from 3.4- to 11.7-fold, respectively, in a study with human hepatocytes from several donors. Results from our own study indicated that variability was greatly minimized by expressing induction as a percentage of a standard inducer.
In summary, these studies suggest that compared with collagen, Matrigel is a better matrix for the maintenance of basal CYP 3A expression in rat hepatocytes. Also, as previously concluded, CYP 2B induction is markedly greater in hepatocytes cultured on Matrigel than collagen. With human hepatocytes, both basal and inducible CYP 3A4 expression were similar in cells cultured on either matrix. The use of percentage induction relative to a standard inducer is presented as an approach that gives less variable data than -fold induction. This allows for rank ordering of compounds and provides a strategy for circumventing inter-individual variability. This in vitroassay and calculation approach correlated well with in vivoinduction. In conclusion, this assay allows for numerous compounds to be evaluated at the drug discovery stage using minimal quantities of drug and markedly fewer animals.
Acknowledgments
We acknowledge Dr. Marc Bilodeau from the Hopital Saint-Luc, Montreal, and Dr. Emile Levy from the St. Justine Hospital, Montreal, for providing human liver tissue. The excellent technical assistance from the Laboratory Animal Resource group at Merck Frosst is greatly appreciated.
Footnotes
-
Send reprint requests to: José M. Silva, Ph.D., Merck Frosst Centre for Therapeutic Research, P.O. Box 1005, Pointe-Claire-Dorval, Quebec H9R 4P8, Canada.
-
↵1 Current address: Astra Research Centre Montreal, 7171 Frederick-Banting, Saint-Laurent, Quebec, Canada.
- Abbreviations used are::
- P450
- cytochrome P450
- G3PDH
- glyceraldehyde-3-phosphate dehydrogenase
- Received June 26, 1997.
- Accepted January 23, 1998.
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}