


1996 ASPET N-Glucuronidation of Xenobiotics Symposium
Abstract
Glucuronidation of amines has been shown to exhibit species differences in vitro and in vivo. Substrates for N-glucuronidation can be classified according to the chemical structures of the resulting glucuronides into two groups: compounds that form non-quaternary N-conjugates, and those that form the quaternary counterparts. For compounds of the former class—such as sulfonamides, arylamines, and alicyclic, cyclic, and heterocyclic amines—species differences appear to be less striking and are of a quantitative nature. No one common laboratory animal species used routinely in metabolism research (e.g. rat, mouse, dog, non-human primate, rabbit, and guinea pig) has been shown to be deficient in N-glucuronidation when all of the substrates studied and reported are taken into consideration. The ability of a species to form N-glucuronides is compound-dependent, although rabbit and guinea pig appear to exhibit the highest capacity for this bioconjugation among preclinical species. For tertiary amines, most notably the tricyclic antidepressant and antihistamine drugs,N-glucuronidation is commonly observed in non-human primates and man. There are examples, however, of quaternary glucuronidation occurring in lower animal species. In exploring species differences in amine conjugation in vivo, it is noted that the apparent absence of N-glucuronides in animal urine may not reflect the inability of that species to form such conjugates, since the N-glucuronides may be excreted in bile. Problems such as degradation or low recoveries commonly encountered in isolation and identification of in vivo metabolites further complicate the interpretation of data. Because of the wide range ofpKa values exhibited by various classes of amines, caution also should be exercised for in vitrostudies since incubation conditions for N-glucuronidation often are substrate- and species-dependent. Explanations for the species differences observed in N-glucuronidation appear to be emerging as rapid advances are made in the understanding of the glucuronosyltransferases at the molecular level. More information, however, remains to be gathered from the glucuronosyltransferase genes of animal species other than humans before a better understanding of species differences in N-glucuronidation can be achieved.
Glucuronidation of amines represents a less-common metabolic pathway among phase II conjugation reactions of a wide variety of endo- and xenobiotics. Aryl- and alkylamines, sulfonamides, and heterocyclic amines have been reported to undergo glucuronidation in vitro and in vivo in a large number of animal species and in humans. The primary function of N-glucuronidation, similar to that of O-glucuronidation, is believed to be detoxification (Caldwell, 1979), although in some cases (e.g. arylamines), N-glucuronidation is postulated to mediate the toxic effect of the parent compound.N-glucuronidation is catalyzed by UGT,1 which has been established to consist of a large family of isozymes that are present in hepatic and extrahepatic tissues in all animal species (Dutton and Burchell, 1977; Dutton, 1980; Mulder et al., 1990; Burchellet al., 1991; Burchell et al., 1995). Identification of the isozymes responsible forN-glucuronidation, however, has only become possible recently because of the progress made in the cloning and expression of UGTs and the availability of the purified and expressed enzymes. For several classes of amines, especially the clinically important tertiary amines and aromatic primary amines that have been shown to be carcinogenic,N-glucuronidation appears to be catalyzed by UGT1*4 in humans (Green et al., 1995; Green and Tephly, 1996, 1998), other compounds, such as heterocyclic amines, tetrazole (e.g. losartan), and triazines (e.g.lamotrigine), have been shown to be substrates of human UGT1*6, rat UGT2B1 (Huskey et al., 1994), and human UGT1*4, respectively.
One striking feature of N-glucuronidation is that distinct species differences have been observed among certain substrates; for example, the preferential glucuronidation of tertiary amines to form quaternary glucuronides by humans and higher primates (chimpanzees). For other substrates, however, the reaction appears to be less species-selective but remains an important issue that has captured the attention of many investigators.
The objective of this review is to highlight the literature with respect to differences in N-glucuronidation observed among various species, with substrates classified according to their chemical structures. It should be noted that most of the in vivo data available from the literature are from earlier studies wherein detection and quantitation of glucuronides presented a dual problem because of the chemical instability of certain classes ofN-glucuronides and the lack of sensitivity in quantitative assays utilized for biological fluid (bile, feces, and urine) in an era before the availability of LC-MS/MS. Therefore, reports included in this review are limited to those in which more thorough studies have been described in comparing and confirming the species differences of glucuronidation and in which the results are less ambiguous.
Non-Quaternary N-Glucuronides
Sulfonamides.
Studies on the species differences of N-glucuronidation of sulfonamide drugs were among the earliest reports devoted to this subject matter available in literature. The non-quaternaryN-glucuronide of sulphadimethoxine (2,4-dimethoxy-6-sulphanilamidopyrimidine) (fig.1) was detected initially in human urine; subsequent studies conducted by Bridges and Adamson et al.(Bridges et al., 1968; Adamson et al., 1970) compared the urinary metabolites of this drug in man, eight species of non-human primates (rhesus monkey, baboon, squirrel monkey, green monkey, capuchin, bushbaby, slow loris, and tree shrew), and nine species of non-primates (rat, mouse, Indian fruit bat, hen, cat, ferret, dog, guinea pig, and rabbit). The N1-glucuronide was shown to be the major metabolite present in urine, accounting for 4%–27% of the dose of all primate species, whereas in the non-primates, this metabolite was a minor component (1%–6% of dose) in urine. Three species, cat, ferret, and rabbit, did not produce any detectable amounts of the N1-glucuronide. TheN4-glucuronide, however, was found in small amounts in the urine of all species studied. Thorough studies also were carried out by these investigators to examine bile for the presence ofN-glucuronides. Thus it was confirmed that theN1-glucuronide of sulphadimethoxine was absent in rabbit bile or urine, but in rat bile it accounted for 7% of the dose. TheN4-glucuronide also was detected in trace amounts (<1%) in bile of rat and rabbit.

Chemical structures of compounds that form non-quaternary N-glucuronides.
Arrows indicate the positions of conjugation. Glucuronidation of FANFT occurs after N-deformylation and U-93385E after despropylation; pinacidil glucuronide exists in the non-quaternary, pyridonimine structure, not shown in this figure. Two positions of N-OH-PhIP glucuronidation are shown and both resulting conjugates are non-quaternary:N-OH-PhIP-N2- andN3-glucuronides.
It is of interest to note that the formation ofN-glucuronides in this class of compounds was associated with 2,4-disubstitution of the pyrimidine ring. Thus, when the positions of the two methoxy groups were altered, as in 2-, 4-, or 5-methoxy- and 2,5- or 4,5-dimethoxy-6-sulphanilamidopyrimidines, the compounds did not form appreciable amounts of N1-glucuronide and resulted in no distinct species difference in their disposition. This structure-metabolism relationship was best illustrated in two compounds, sulphamethomidine (4-methoxy-2-methyl-6-sulphanilamidopyrimidine) and sulphasomidine (2,4-dimethyl-6-methyl-6-sulphanilamidopyrimidine) (fig. 1), which differed structurally only in the replacement of one (sulphamethomidine) or both pyrimidylmethoxy groups (sulphasomidine) of sulphadimethoxine with a methyl group (Bridges et al., 1969). Sulphamethomidine exhibited a marked species difference in metabolism: in man and monkeys, the N1-glucuronide was excreted as a major metabolite (40% and 20% of dose, respectively), whereas in rats and rabbits, it was not detected (Bridges et al., 1969). This is in contrast to sulphasomidine, which did not form the N1-glucuronide.
Arylamines.
N-glucuronidation is an important pathway for metabolism of aromatic amines. Among the earliest studies on this class of compounds, including aniline, p-phenetidine, o-,m-, and p-anisidine, and 4,4′-diaminodiphenylsulfone, N-glucuronides of the parent compounds were detected in the urine of rats and/or rabbits (Smith and Williams, 1949). Investigators of these early studies were cautious about the possibility that N-glucuronidation might be an artifact because of the reaction between free amines and glucuronic acid in urine. Nevertheless, Boyland et al. confirmed that the N-glucuronide of 2-naphthylamine (2-NA) was present in the urine of rats and rabbits (Boyland et al., 1957). Subsequently, many arylamines, such as 4-aminobiphenyl (ABP), benzidine, dapsone, 1- and 2-NA, were studied, among which benzidine has been studied extensively.
Hepatic N1-glucuronidation of benzidine (fig. 1) in dogs was proposed as an important intermediate metabolic step responsible for bladder carcinogenesis in this species (Babu et al., 1992,1993a, 1993b). It was postulated that in dogs, afterN1-glucuronidation (detoxification) of benzidine in the liver, the acid-labile conjugate was transported in plasma while bound to proteins, filtered by the kidney, and accumulated in urine, wherein acid hydrolysis regenerated the amine. The amine was then activated by bladder enzymes to initiate the carcinogenic process. This postulate was supported by results from in vitro metabolism studies of benzidine in liver microsomes prepared from rats, dogs, and humans. The rate of N1-glucuronidation was shown to follow the rank order human > dog ≫ rat. In all three species, benzidine underwent N-acetylation in the liver, giving rise to an arylamide that was susceptible to activation by oxidation, leading to binding to hepatic DNA. The low capacity of rat liver UGT for conjugation of benzidine thus was hypothesized to be partly responsible for the observed liver cancer, rather than bladder cancer, of this compound in this species. Recently, benzidineN1-glucuronidation was shown to involve UGT1*4 expressed in HK293 cells (Green et al., 1995; Green and Tephly, 1998).
Recently, it was reported that expressed human UGT1*6 and UGT1*7 proteins catalyze the glucuronidation of 1- and 2-NA and, to a lesser extent, 4-ABP (Lilienblum and Bock, 1984).N1-Glucuronidation of 2-NA has been shown to involve human UGT1*4 (Green and Tephly, 1996). In the rat,N-glucuronidation of 1- and 2-NA involved UGT2B1 (expressed protein) (Orzechowski et al., 1994), UGT2B2 and UGT2B3 (purified enzymes) (Chowdhury et al., 1986; Green and Tephly, 1987; Kadlubar et al., 1992), and that of 4-ABP (“bulky amine”) involved only UGT2B2 (Pritchard et al., 1994). Interested readers are referred to a review by Green and Tephly in this issue (Green and Tephly, 1998).
For another class of arylamines represented byN-[4-(5-nitro-2-furyl)2-thiazolyl]formamide (FANFT) and its deformylated metabolite, 2-amino-4-(5-nitro-2-furyl)thiazole (ANFT) (fig. 1), N-glucuronidation of the primary amine (after deformylation of FANFT) was a predominant pathway that accounted for 18% and 80% of radioactivity excreted in urine, respectively, corresponding to 3.4% and 24% of the respective oral dose to guinea pigs (Dawley et al., 1991). These metabolites can also be produced in vitro using liver and kidney microsomes prepared from guinea pigs but not rats, although the UGT activity as assayed using p-nitrophenol in the two species was not very different. The facile conjugation by guinea pig was believed to be responsible for the much-reduced free ANFT levels observed in this species and may partially explain the resistance of this species to FANFT-induced bladder cancer.
Arylamine N-OH.
One of the earliest studies comparing in vitro N-glucuronidation activity in liver microsomes prepared from various species (rat, dog, and human) was conducted by Kadlubar and co-workers on N-hydroxy arylamines of 1- and 2-NA, 4-ABP, 2-aminofluorene (AAF), 4-aminoazobenzene, andN-acetyl-2-aminofluorene (Kadlubar et al., 1977). The relationship of glucuronidation to urinary bladder carcinogenesis also was explored. These studies led to the conclusion that hepatic glucuronidation of the N-OH arylamines proceeded largely, if not exclusively, by conjugation with the nitrogen, rather than the oxygen, atom of the substrate. The stability of these metabolites was in marked contrast to the lability of the O-glucuronides ofN-OH-AAF. In microsomes fortified with UDPGA, rats, dogs, and humans appear to have similar rates of glucuronide formation withN-OH-2-NA and N-OH-1-NA. TheN-glucuronides were believed to be the metabolic intermediates that upon acid hydrolysis in urine, produced the ultimate electrophilic carcinogen in the bladder lumen via a reactive arylnitrenium ion (Dawley et al., 1991).
Another group of compounds of the N-OH arylamine class that has been studied thoroughly with respect to their glucuronidation potential is phenylimidazole derivatives of pyridine, quinoline, and quinoxaline (Kaderlik et al., 1994). These compounds include 2-amino-3-methylimidazo[4,5-f]-quinoline (IQ), 2-amino-1-methyl-6-phenylimidazo[4,5-b]-pyridine (PhIP), 2-amino-6-methyl-dipyrido-[1,2-a:3′,2′-d]imidazo[4,5-f](Glu-P-1), and 2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline. In vitro experiments were conducted in the presence of liver microsomes for the formation of N-OH derivatives of the above compounds. Interesting results were obtained forN-OH-PhIP (fig. 1), which formed twoN-glucuronides: the imidazole ring N3-glucuronide and the exocyclic N-glucuronide. The rat preferentially produced the former conjugate (ratio, 15:1), whereas the dog and human formed the latter (ratios, 1:2.5 and 1:3.1). These results were in excellent agreement with in vivo observations in that theN3-glucuronide was the major (40% of metabolites) biliary metabolite in PhIP-treated rats and the exocyclicN-glucuronide was the metabolite present in PhIP-treated dogs. These results suggest that the UGT enzymes involved in the formation of the two conjugates are different and that species differences may reflect the species-related differences in substrate specificity of the particular UGT responsible for formation of the conjugate.
Cyclic Amines.
There are only a small number of nonaromatic cyclic amines that have been reported to undergo N-glucuronidation. An interesting example of species-specific N-glucuronidation was described recently for a cyclic secondary amine by Martin et al.(Martin et al., 1993). After an oral dose of U93385E (fig.1), an aminotetralin and a 5HT1A agonist, rats and cynomolgus monkeys excreted 85.4% and 65.8% of the dose in urine, respectively. The major metabolite in monkey urine, accounting for 33.5% of the oral dose, was identified as the N-glucuronide of the despropylated parent compound. This metabolite was not detected in rat urine.
Another secondary cyclic amine, 1,2,3,4-tetrahydroisoquinoline-7-sulfonamide, has been shown to form anN-glucuronide in the rat exclusively at the ring nitrogen instead of the sulfonamide nitrogen. There are no data available for the presence of this conjugate in species other than the rat (Kuoet al., 1986).
Heterocyclic Aromatic Amines.
Pimobendan (fig. 1), a pyridazinone derivative of benzimidazole, is a cardiotonic vasodilator. In man, pimobendan was reported to undergo demethylation, followed by O- andN-glucuronidation (Pahernik et al., 1995). It was also N-glucuronidated directly. In vitro, the major metabolite formed was the O-glucuronide, whereasin vivo the N-glucuronide was the major metabolite in urine. These observations led to the postulate thatin vivo glucuronidation was taking place in extrahepatic organs, such as the kidneys.
Pinacidil (fig. 1), a pyridine derivative, was shown to undergoN-glucuronidation at the pyridine ring, and the product was detected in rabbit urine after oral dosing (Sakamoto and Nakamura, 1993). The hydroxy metabolite O-glucuronide and theN-oxide glucuronide were detected in mouse urine. The five species studied could be classified into two groups based on metabolic pathways: rats, dogs, and humans as a group vs. rabbits and monkeys. Species in each group share similar pathways. It is of interest to note that for pinacidil N-glucuronide, theNH group at the 4-position of the pyridine assumed the pyridonimine structure. Therefore, the N-glucuronide is not a quaternary conjugate.
Glucuronidation of a hydantoin has been observed for nirvanol (5-ethyl-phenylhydantoin) (Maguire et al., 1982). The species difference in N-glucuronidation reported appeared to be insignificant. The major urinary metabolite in the dog was theN-glucuronide (>80%), whereas in man it represented 50% of the dose.
Acidic Aromatic Amines.
The Tetrazoles. Tetrazole is used frequently as a substructure in drug molecules to impart acidity equivalent to a carboxylic acid. The first tetrazole N-glucuronide was reported for AA-344, 6-ethyl-3-(1H-tetrazole-5-yl)-chromone (fig.2), an orally active antiallergic agent (Nohara, 1980). After oral dosing of the radiolabeled drug, the maximum levels of AA-344 and half-lives of the drug were highest and longest in dogs, followed by monkeys, guinea pigs, rats, and rabbits. The major route of excretion was urine in all species studied with the exception of the guinea pig (which was equally divided between urine and feces). Major urinary metabolites were oxidative derivatives.N1-glucuronide was detected in rabbits and dogs. In these two species, total radioactivity excreted in urine accounted for 85% and 71% of the dose, of which ∼6% and 13% were the glucuronide, respectively. The dog also excreted 20% of the dose in bile, of which 33% was the glucuronide. Monkey excreted the glucuronide as a very minor metabolite, ∼1%–2% of total dose.

Chemical structures of compounds containing aromatic heterocyclic amines (tetrazole, triazole, and imidazole) that form non-quaternary N-glucuronides.
Arrows indicate the positions of conjugation. For tetrazole-containing compounds, conjugation occurred on N2, except for the chromone derivative, AA-344, which conjugates atN1.
Several other tetrazole glucuronides have been discovered more recently. Losartan (fig. 2), a nonpeptidyl angiotensin II (AII) receptor antagonist, has been shown to form a N2-glucuronide at the tetrazole moiety in vitro after incubation of the compound with liver slices from rats, monkeys, and humans (Stearnset al., 1992). While this biotransformation occurred in all three species, the primary route of metabolism was oxidative in the rat, leading to either hydroxylated or oxidized (carboxylic acid) metabolites, whereas in monkeys, glucuronidation of the tetrazole moiety predominated. An equal mixture of both oxidized and glucuronic conjugated metabolites was formed in incubations with human liver slices. N-glucuronidation of losartan gave rise to an inactive AII antagonist and was expected to shorten the duration of antihypertensive activity of the drug. The in vitroglucuronidation of losartan was studied further in liver microsomes from rats, dogs, monkeys, and humans fortified with UDPGA (Huskeyet al., 1993). For this reaction, the optimal pH was determined to be 5.0 with liver microsomes from monkeys and humans and 6.2 with those from rats and dogs. The rate of glucuronidation (nmol/min/mg protein) in these four species followed the rank order monkey (2.1 ± 0.2) > dog (0.6) > rat (0.05) ∼ human (0.02). The rate of glucuronidation in rats was shown to be higher in liver microsomes prepared from animals induced with dexamethasone (DEX). It is of interest to note that for losartan, a molecule that possesses a primary OH group in the molecule which also can be glucuronidated, the two reactions were shown to be equally facile in the rat when the compound was incubated at neutral pH (7.7). Dogs and humans produced only the N2-glucuronide, whereas monkeys generated both glucuronides, with the N-2-glucuronide being the predominant metabolite (ratio, 10:1). At lower pH (5.0 or 6.2), all four species formed only the N-glucuronide. Two other tetrazole-containing AII antagonists, L-158,809 and L-158,338, and a model compound, methylbiphenyl (MB)-tetrazole (fig. 2), were studied similarly. The rate of glucuronidation followed the same rank order in the four species for these two AII antagonists. The model compound, on the other hand, was glucuronidated at nearly equal rates in all species studied. Furthermore, liver microsomes from DEX-induced male rats and PB-induced female rats showed a significant increase inN-glucuronidation and a slight increase inO-glucuronidation of these compounds. These results suggested that there was probably more than one UGT isozyme involved in conjugation reactions of the tetrazole. This postulate has been substantiated in subsequent studies using recombinant UGT2B1 and UGT1*6 expressed stably in V79 cells (Huskey et al., 1994).
Studies reported for losartan and the AII compounds emphasized the importance of optimization of pH in in vitro studies. These reports are among the few describing the regioselectivity ofN-glucuronidation. Other tetrazole-containing AII compounds studied were SR47436 (BMS186295) and GR117289 (fig. 2). For GR117289, a compound containing a tetrazole and a carboxylic acid, glucuronidation occurred at both moieties. Both the N2 and acylglucuronide were detected in dog bile (Bowers et al., 1994). However, acylglucuronidation was shown to be the major route of the two in the rat. SR47436 (BMS186295), unlike losartan and GR117289, has only one site for glucuronidation (Perrier et al., 1994). It underwent glucuronidation in cynomolgus monkey liver microsomes with a relative catalytic efficiency (Vmax/Km ) of 11- and 2.6-fold higher than that observed in rat and human liver microsomes, respectively. SR47436 (BMS186295) glucuronidation was postulated to involve a DEX-inducible UGT that was also responsible for glucuronidation of monodigitoxigenin-monodigitoxoside.
Other Nitrogen-Containing Aromatic Heterocycles.
Glucuronidation of other nitrogen-containing heterocycles has been studied in triazoles (1,2,3- and 1,2,4-substituted) and imidazoles (C2- and C4-substituted) (fig. 2) (Huskey et al., 1994). As in other N-glucuronides of heterocyclic amines, determination of the exact position of conjugation was difficult and the assignments were based solely on NOE difference NMR spectroscopy. In general, relatively low reactivity was found at nitrogens located next to a substituted carbon in heterocycles such as N3 in methyl biphenyl-C4-imidazole. When the rates of the reactions were compared under optimal reaction conditions, most compounds showed higher reactivity with liver microsomes from monkeys than those from rats, except for N2-glucuronidation of MB-tetrazole and MB-1,2,3-triazole. The trend for relative N-glucuronidation reactivity of these compounds in humans, however, was quite different from that in rats and monkeys. It appeared that the human UGT responsible for N-glucuronidation preferentially conjugated compounds with higher pKa (1,2,4-triazole andC4-imidazole), whereas the more acidic substrates (1,2,3-triazole and tetrazole) were preferred substrates in the rat. Regioselectivity among species also was observed:N1-glucuronidation of MB-1,2,3-triazole was favored over that at N2 in monkeys, whereas the opposite was true for rats and humans. When these compounds were studied with recombinant UGTs from humans (UGT1*6) and rats (UGT2B1), only one glucuronide (N2) was detected in incubates with the former enzyme but both (N1 and N2) were detected with the latter (Green and Tephly, 1996). UGT1*6, UGT2B1 and UGT2B4 did not produce any glucuronides when imidazole and 1,2,4-triazole were used as substrates.
Quaternary N-Glucuronides
Cyclic Tertiary Amines—Piperidines and Piperazines.
Species differences exhibited in N-glucuronidation by this class of compounds are the most striking and involve a larger number of clinically important drugs. The identification by Porter et al. (Porter et al., 1975) of cyproheptadineN-glucuronide in urine of humans after an oral dose of cyproheptadine represents the first report recognizing a quaternaryN-glucuronide as a metabolite (fig.3). Subsequently, Fischer et al. compared urinary excretion of cyproheptadineN-glucuronide in monkeys, chimpanzees, and humans after a single oral dose (Fischer et al., 1980). Over a 48-hr period, the amount of N-glucuronide excreted accounted for 12.4% and 8.6% of the total dose in humans and chimpanzees, respectively, compared with a trace amount (0.5%) in the urine of monkeys, including various Old World monkeys and a New World monkey (cebus). In lower laboratory species such as dogs, cats, and rats,N-glucuronide was not detected in urine; however, as the authors pointed out, since approximately half of the cyproheptadine dose given to the lower laboratory species was excreted in fecesvia bile, it could not be concluded that these species did not form N-glucuronides until the bile was studied. Results from these studies also suggested that unlike rats, dogs, and cats, rabbits might share the same pathway as humans since theN-glucuronide was formed when cyproheptadine was incubated with immobilized rabbit hepatic microsomal system (Lehman et al., 1982). It is worth noting, however, that in separate experiments using liver microsomes and in vivo, the formation of cyproheptadine N-glucuronide has not been observed in this species. Cyproheptadine N-glucuronidation has been shown to involve UGT1*4 in human liver microsomes (Green and Tephly, 1998).

Chemical structures of drugs containing tricyclic tertiary amines (piperidines or piperazines) that form quaternary ammonium–linked glucuronides.
The positions of conjugation are the tertiary nitrogens of the piperidine and piperazine rings, indicated by arrows.
The species-specific glucuronidation observed for cyproheptadine is exhibited by another tricyclic piperidine–containing drug, ketotifen (fig. 3). In rat hepatocyte culture, ketotifen underwentN-demethylation (forming norketotifen) andN-oxidation; these were the same metabolic pathways as those observed in vivo (Le Bigot et al., 1987). In rabbit hepatocyte cultures, the major metabolites detected were theN-sulfate of norketotifen and the N-glucuronide of the parent drug. In human hepatocytes, the major metabolites were those resulting from ketoreduction and N-glucuronidation of the parent. Trace amounts of norketotifen and N-oxide also were detected. These results were in excellent agreement with those from in vivo studies: 46% and 37% of the urinary metabolites were confirmed to be the N-glucuronide in rabbit and man, respectively. No N-glucuronide was detected in rat urine; however, these results from rats may require confirmation as the rat might have excreted the conjugate in bile. KetotifenN-glucuronidation has been shown recently to also involve UGT1*4 in human liver microsomes (Green and Tephly, 1998).
Other piperazine-containing drugs that undergoN-glucuronidation in humans are clozapine, loxapine, and olanzapine (fig. 3); all are tricyclic antipsychotic agents. For olanzapine administered to humans, N-glucuronidation, one of the three major metabolic pathways identified for this compound, occurred at both the piperazinyl (N4′) and diazepine nitrogen (N10) atoms, giving rise to quaternary and tertiaryN-glucuronides, respectively (Kassahun et al., 1997). In preclinical species such as rats, dogs, and rhesus monkeys, however, the major metabolic pathways involve the oxidation of the benzene moiety as shown by metabolites identified in rat urine and bile (Mattliuz et al., 1994). N-glucuronidation of clozapine and loxapine have been shown to involve UGT1*4 (Greenet al., 1995).
Another piperazine-containing drug that undergoesN-glucuronidation is mianserin (fig. 3), a tetracyclic piperazinoazepine antidepressant. The piperazine moiety in this compound is methyl-substituted. Metabolic pathways of mianserin include hydroxylation, demethylation, N-oxidation, andN-glucuronidation (Delbressine et al., 1992). Metabolic disposition using the radiolabeled drug was studied in rats, mice, guinea pigs, and humans; the N-glucuronide was only found in man.
Alicyclic Tertiary Amines.
A large number of compounds from this class have been reported to formN-glucuronides. A series of antihistamines, chlorpheniramine, pheniramine, diphenhydramine, doxylamine, pyrilamine, triplennamine, promethazine, have been shown to produceN-glucuronides by humans after an oral dose of the drug (Luoet al., 1991). The conjugates were detected primarily in urine. The structures of all of these compounds contain a terminal dimethyl-substituted amino group, and the N-glucuronides have not been found in lower laboratory species. For a group of tricyclic antidepressants, imipramine, amitriptyline, cyclobenzaprine, clomipramine, trimipramine, and chlorpromazine (fig.4), the quaternaryN-glucuronides were shown to be the major metabolites in human urine (Luo et al., 1995). Consistent with the in vivo results, amitriptyline was shown to undergoN-glucuronidation in vitro in the presence of human liver microsomes (Dahl-Puustinen and Bertilsson, 1987). The conjugation activity in humans varies among subjects (sevenfold in 13 subjects) and was inhibited by p-nitrophenol but not by morphine. Amitriptyline, imipramine, and chlorpromazine were shown to undergo N-glucuronidation in immobilized rabbit liver microsomal systems (Lehman et al., 1983), indicating that this non-primate lower laboratory species had the potential of forming the quaternary N-glucuronides. Recent studies using recombinant UGT1*4 confirmed that all three of the above tertiary amines were substrates of this isozyme. Published reports are not available yet on the involvement of rabbit UGT with these compounds.

Chemical structures of tricyclic antidepressant drugs that form quaternary N-glucuronides.
The positions of conjugation are the alicyclic tertiary nitrogens indicated by arrows.
The most thorough investigations on species differences in tertiary amine N-glucuronidation were conducted with cyclobenzaprine (fig. 4) by Hucker et al. (Hucker et al., 1978a,1978b), using the C-14–labeled drug. In rats, dogs, rhesus monkeys, and humans cyclobenzaprine was well absorbed after oral administration. Rats eliminate the drug primarily in the feces, whereas urinary excretion is predominant in dogs, monkeys, and man. In man, theN-glucuronide metabolite represents 48.5% of urinary radioactivity in 24 hr and ∼24% in 120 hr. This metabolite was not detected in rat urine and was present only in trace amounts in dog urine.
Aromatic Heterocyclic Amines. Imidazoles.
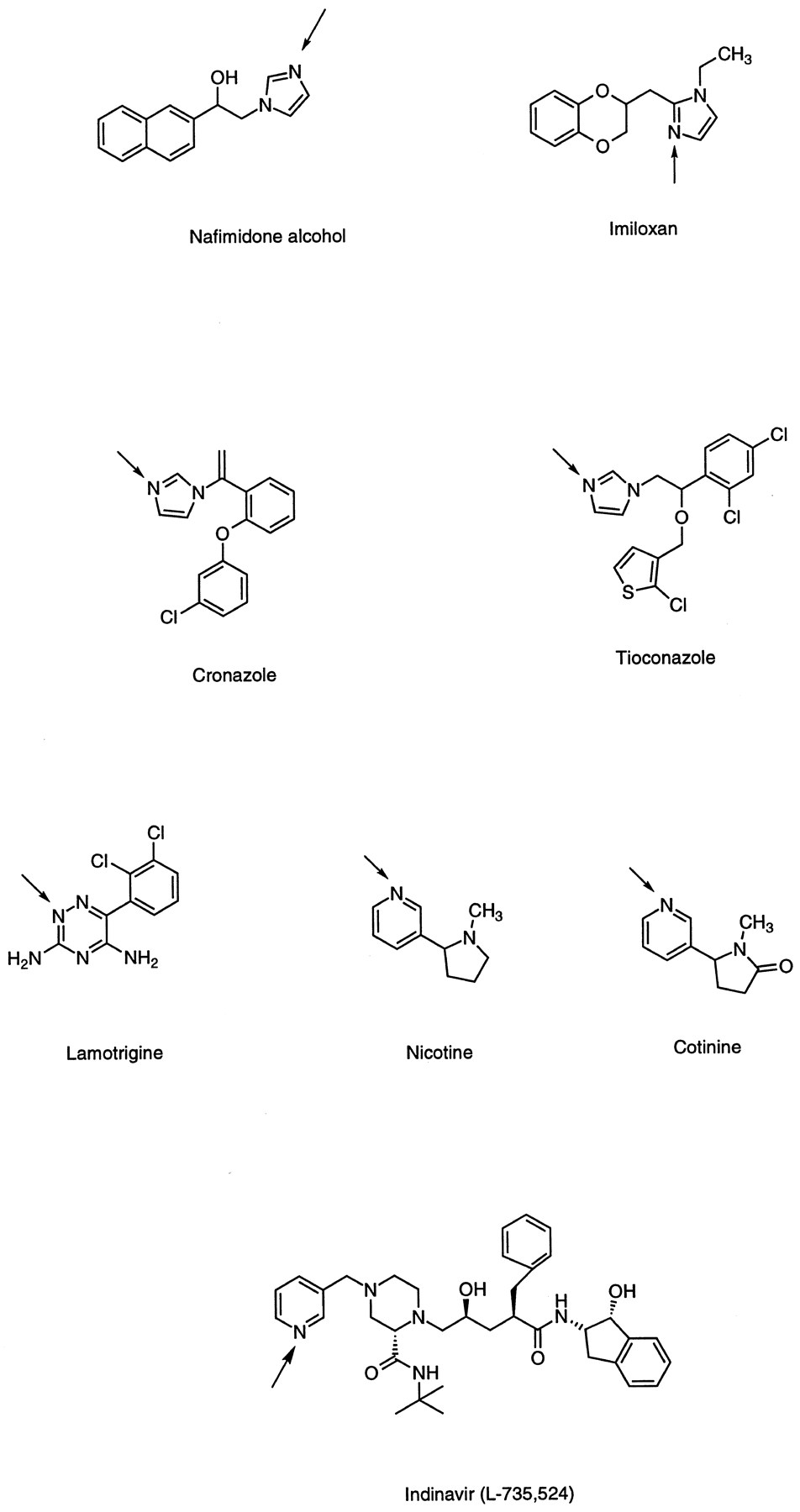
The imidazole is a common pharmacophore in drug molecules. Several imidazole-containing drugs have been shown to form interesting glucuronides. Nafimidone, an anticonvulsant, was well absorbed in rats, dogs, primates (cynomolgus monkeys and baboons), and man. Urinary excretion consisted essentially only of metabolites (Graham et al., 1987; Rush et al., 1990). TheN-glucuronide of nafimidone alcohol (fig.5), the primary metabolite resulting from ketoreduction, was excreted in the urine of humans, baboons, and cynomolgus monkeys, accounting for ∼24%, 11%, and 22% of the oral dose in these species. This conjugate was not detected in dog urine and rat bile. It is of interest to note that the major metabolite (80%) found in dog urine was the O-glucuronide, whereas for the other primate species, including man, both O- andN-glucuronides were present. Two other imidazole-containing drugs, imiloxan (fig. 5) and idazoxan, alpha-adrenoceptor antagonists, have also been studied (Rush et al., 1992).N-glucuronide formation, linked through the 2′ nitrogen of the imidazole ring, was reported in man (Rush et al., 1992).N-glucuronides of the imidazole moiety also were observed for tiaconazole and croconazole (fig. 5), two anti-fungal agents (Takeuchi et al., 1989; Macrae et al., 1990). When given to humans, 25% of the tiaconazole dose was recovered in urine, of which 42% was a quaternary N-glucuronide. No conjugate was detected in feces. In rats, the tiaconazole glucuronide was a major component, as in cynomolgus monkey bile. This glucuronide was similar to that in human urine and to the major circulating metabolite in humans. The tiaconazole N-glucuronide was cited as the first reported example for a quaternaryN-glucuronide as a major metabolite in rats. The quaternaryN-glucuronide metabolite of croconazole was detected in the urine of rabbits and humans (Takeuchi et al., 1989). In rabbits, it accounted for 2.8% of an iv dose of C-14–labeled croconazole and was cited as the first example reported for the formation of a quaternary N-glucuronide in this species.

Chemical structures of drugs containing aromatic heterocycles (imidazole, pyridine, and triazine) that form quaternary glucuronides.
The positions of conjugation are the ring nitrogens indicated byarrows.
Pyridines.
The most important N-glucuronide known for pyridine-containing compounds is that of nicotine and cotinine (fig.5). The metabolism of nicotine in humans has been studied extensively. However, it was not known until recently that this compound undergoesN-glucuronidation in animals (rats and monkeys) and humans. Seaton et al. reported that humans (smokers and nonsmokers) excreted glucuronides of nicotine, (S)-(−)-cotinine and 3′-hydroxycotinine in urine after receiving an iv dose of radiolabeled racemic nicotine, and these conjugates accounted for a major portion (29%) of total nicotine metabolites excreted in urine (80%) (Seatonet al., 1993). Both N-glucuronides were present as metabolites in rat bile after the animals were given C-14–racemic nicotine (Caldwell et al., 1992).
Indinavir (L-735,524), a potent HIV-1 protease inhibitor approved recently for the treatment of AIDS, has been reported (Balani et al., 1996a, 1996b) to form a quaternary N-glucuronide at the pyridyl nitrogen as a metabolite in human and monkey. Analysis by LC-MS/MS indicated that this metabolite was absent in rat and dog urine and rat bile. After a 400-mg oral dose, the pyridineN-glucuronide accounted for ∼0.5% of the radiolabeled dose administered to humans and monkeys. It also was reported that human liver slices converted indinavir to the pyridineN-glucuronide. Data are not yet available for other species.
Triazine.
The metabolism of lamotrigine (fig. 5), a 1,2,4-triazine–containing anticonvulsant, was studied in detail in rats, dogs, monkeys, guinea pigs, and humans. In human urine, the N-glucuronide accounted for 90% of the recovered dose; a total of 85% of the dose was recovered in urine (Sinz and Remmel, 1991). The compound was glucuronidated extensively in monkeys, but the glucuronide was only a minor metabolite in rats and dogs. In guinea pigs, the species reported to have high concentrations of UGT activity in liver microsomes, compared with other species, lamotrigine underwent metabolism to form the 2N-glucuronide, which accounted for 60% of an iv dose when excreted in urine. Lamotrigine thus represents the second amine that has been shown to undergo extensive N-glucuronidation in a lower laboratory species (Sinz and Remmel, 1991; Remmel and Sinz, 1991). Pretreatment of rats with BNF, a known inducer of UGT in rats, failed to induce the enzymatic activity toward lamotrigine orp-nitrophenol. The formation of the N-glucuronide also was observed in guinea pig microsomes and freshly isolated hepatocytes (Remmel and Sinz, 1991). An interspecies comparison of lamotrigine glucuronidation (humans, rabbits, rats, and monkeys) revealed that the rate of glucuronidation was low. Of all of the species considered, humans glucuronidated the drug to the greatest extent, with a specific activity twofold higher than that observed in rabbit liver microsomes (Magdalou et al., 1992). In contrast, the activity was >20 times lower in rat liver microsomes. However, in this species, PB pretreatment enhanced lamotrigine glucuronidation slightly. In vitro, glucuronidation of lamotrigine was inhibited by chlorpromazine, but not by imiprimine, amitrityline, or cyproheptadine, although all four drugs were known to undergo quaternary N-glucuronidation. In both male and female human liver microsomes, testosterone and ethinyl estradiol competitively inhibited lamotrigine glucuronidation with similar apparent Ki values, thus suggesting that the drug and the hormones were substrates of the same molecular forms of UGT. In contrast, testosterone glucuronidation was not affected by lamotrigine but was decreased to various extents by structurally different tertiary amines. These results highlight the strict specificity of the UGT isozyme toward this endogenous substrate. Lamotrigine N-glucuronidation was shown to involve human UGT1*4 (Green and Tephly, 1998).
Recent Progress
A survey of the literature on species differences in amine glucuronidation leads to the following conclusions: (1) Species differences observed for various amines are substrate-dependent. For primary and secondary amines or other nitrogen-containing compounds that are substrates for glucuronidation, giving rise to non-quaternary glucuronides, species differences appear to be less striking and are of a quantitative nature. No one species among the common laboratory animal species used in metabolism research (rats, mice, dogs, primates, rabbits, and guinea pigs) has been shown to be deficient inN-glucuronidation of all of the known substrates reported in literature; (2) For tertiary amines that are substrates for quaternaryN-glucuronidation, species differences are striking, and these reactions are commonly observed in non-human primates and man. There are examples, however, for the same reactions occurring in lower animal species; (3) The apparent absence of N-glucuronides in animal urine may not reflect the actual disposition of the compounds but may be due to instability of certain classes of these conjugates, the excretion route and problems commonly encountered in isolation, and identification of in vivo metabolites; and (4) In vitro N-glucuronidation conditions for each substrate are species-dependent; thus the absence of glucuronides in in vitro incubates with liver microsomes could be the result of suboptimal conditions utilized in the studies. Explanations for species differences observed in N-glucuronidation, however, appear to be emerging as rapid advances are made with the glucuronosyltransferases at the molecular level. In the last decade, molecular-biological approaches have enabled rapid progresses in characterization of UGTs with respect to their structures and their genomic expression. cDNAs of UGT messengers have been made and sequenced and have provided probes to identify related cDNAs coding for other forms of the enzyme. Subsequently, these cDNAs have been transfected into various cells (cos cells, V79, etc.) in which UGT activity can be expressed. This has allowed studies to be carried out on the substrate specificity of the UGT corresponding to a particular cDNA (Green et al., 1995). Of the more than 30 UGT isozymes that have been purified or cloned and expressed, many have been shown to catalyze N-glucuronide formation for various amines. It is also known now that the structure of the UGT1 gene complex is highly conserved across species, and it is possible, as postulated by Green and Tephly (Green and Tephly, 1996), that for the formation of glucuronides, such as the N-glucuronides, a mutation in the first exon encoding UGT1*4 has resulted in a pseudo-gene that is responsible for the inability of some species to form this type of glucuronide. This postulate remains to be verified when more information is available on the UGT genes from species other than man. Among them, rabbits and guinea pigs may be of particular interest because of the accumulated data on their relatively higher activity in N-glucuronidation.
Acknowledgments
The authors wish to thank Drs. Anthony Y. H. Lu and Thomas A. Baillie for helpful discussions in the preparation of this manuscript.
Footnotes
-
Send reprint requests to: Dr. Shuet-Hing Lee Chiu, Department of Drug Metabolism, Merck Research Laboratories, Rahway, New Jersey 07065.
- Abbreviations used are::
- UGT
- UDP-glucuronosyltransferase
- LC-MS/MS
- liquid chromatography-mass spectroscopy/mass spectrometry
- 2-NA
- 2-naphthylamine
- ABP
- aminobiphenyl
- FANFT
- N-[4-(5-nitro-2-furyl)2-thiazolyl]formamide
- ANFT
- 2-amino-4-(5-nitro-2-furyl)thiazole
- AAF
- 2-aminofluorene
- UDPGA
- uridinediphosphoglucose
- IQ
- 2-amino-3-methylimidazo[4,5-f]-quinoline
- PhIP
- 2-amino-1-methyl-6-phenylimidazo[4,5-b]-pyridine
- DEX
- dexamethasone
- MB
- methylbiphenyl
- PB
- phenobarbital
- The American Society for Pharmacology and Experimental Therapeutics



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}