


Abstract
To evaluate the extent of drug-drug interaction concerning metabolic inhibition in the liver quantitatively, we tried to predict the plasma concentration increasing ratio of midazolam (MDZ) by itraconazole (ITZ) or ketoconazole (KTZ) in rats. MDZ was administered at a dose of 10 mg/kg through the portal vein at 60 min after bolus administration of 20 mg/kg ITZ or during 0.33 mg/h/body of KTZ infusion. The ratio values in the area under the plasma concentration curve of MDZ in the presence of ITZ and KTZ was 2.14 and 1.67, respectively. The liver-unbound concentration to plasma-unbound concentration ratios of ITZ and KTZ were 11∼14 and 1.3, respectively, suggesting a concentrative uptake of both drugs into the liver. ITZ and KTZ competitively inhibited the oxidative metabolism of MDZ in rat liver microsomes, and Ki values of ITZ and KTZ were 0.23 μM and 0.16 μM, respectively. We predicted the ratio values of MDZ in the presence of ITZ and KTZ, usingKi values and unbound concentrations of both drugs in the plasma or liver. The predicted ratio values in the presence of ITZ or KTZ calculated by using unbound concentration in the plasma were 1.03∼1.05 and 1.39, whereas those calculated using unbound concentration in the liver were 1.73∼1.97 and 1.51, respectively, which were very close to the observed ratio values. These findings indicated the necessity to consider the concentrative uptake of inhibitors into the liver for the quantitative prediction of the drug-drug interactions concerning metabolic inhibition in the liver.
The hepatic metabolic inhibition of one drug by another is one of the most important events among pharmacokinetic drug-drug interactions. Such an interaction may induce adverse effects by elevating the plasma concentration of the interacted drug. In clinical cases, it has been reported that azole antifungal agents, macrolide antibiotics, and histamine H2-receptor antagonists inhibited the oxidative metabolism of various drugs in the liver (Fee et al., 1987; Olkkola et al., 1993,1994, 1996; Beckman et al., 1994; Ahonen et al., 1995; Baldwin et al., 1995). Most of the enzymes concerning drug-drug interactions are cytochrome P-450 (CYP).1 Isoforms of metabolic enzymes can be identified using anti-P-450 antibodies or specific inhibitors to CYP (Ghosal et al., 1996), which make it possible to predict the possibilities of drug-drug interactions qualitatively. Moreover, we can estimate the degree of interactions quantitatively to some extent if the metabolic Ki are obtained by using liver microsomes, primary cultured hepatocytes, and/or CYP-expressed cells (Pichard et al., 1990; Gascon and Dayer, 1991; Wrighton and Ring, 1994;Ghosal et al., 1996). However, it still remains difficult to predict precisely the increasing ratio of the concentrations of the interacted drug in plasma, because we must take into account such points as: 1) disposition of inhibitors in the liver, 2) concentrations of inhibitors in the portal vein or hepatic vein, 3) inhibition of metabolism in the gastrointestinal tract, and 4) drug-drug interaction in the absorption process (Sawada et al., 1996; Sugiyama and Iwatsubo, 1996). In this study, we focused on point 1 and tried to predict the increase of plasma concentration in rats. To exclude points 2, 3, and 4, inhibitors were administrated i.v. and the interacted drug was administrated through the portal vein. Midazolam (MDZ), a hypnotic, as an interacted drug, and itraconazole (ITZ) and ketoconazole (KTZ), azole antifungal agents, as inhibitors, were used in this study. Recently, we reported the quantitative prediction of histamine H2 receptor antagonists (cimetidine and nizatidine) and MDZ by using the same procedure (Takedomi et al., 1998).
Experimental Procedures
Materials
ITZ, hydroxy itraconazole (ITZ-OH), and KTZ were obtained from Janssen-Kyowa Co. (Tokyo, Japan). MDZ was purchased as MDZ injection (Dormicam Inj.) from Yamanouchi Pharmaceutical Co. (Tokyo, Japan). All other chemicals used as reagents were of reagent grade and reagents for HPLC.
Animals
Sprague-Dawley male rats (7 weeks) weighing 220 to 250 g were purchased from Nippon Bio-Supply Center (Tokyo, Japan). The rats were allowed access to water and food pellets ad libitum.
In Vivo Studies
Preparation of Drug Solutions.
Twenty milligrams of ITZ was dissolved with 0.1 ml of 12 N HCl, and 1.75 ml of polyethylene glycol 400 and 0.15 ml of 8 N NaOH were added to prepare 10 mg/ml of ITZ solution. The solution of KTZ for bolus administration was prepared by dissolving KTZ in a similar way. To this solution, saline was added to prepare the solution for constant infusion. MDZ injection (Dormicam Inj.; 10 mg/2 ml) was used as the solution of MDZ for intraportal administration.
Plasma and Liver Concentration Profiles of ITZ and KTZ.
Under light ether anesthesia, rats were cannulated through the femoral vein. After recovery from anesthesia, ITZ was administrated through the femoral vein at bolus doses of 5, 10, and 20 mg/kg. At 0.083, 0.25, 1, 4, 8, and 24 h after administration of ITZ (only at 1 h after doses of 10 and 20 mg/kg), blood was collected from the femoral artery and the liver was removed. Blood was centrifuged at 12,000 rpm for 2 min to obtain the plasma. Plasma and liver were stored at −20°C until analyzed.
For KTZ, the loading dose and subsequent maintenance dose were calculated by the pharmacokinetic parameters of KTZ obtained in a preliminary study. Briefly, 10 mg/kg of KTZ was bolusly administrated through the femoral vein to rats and blood was collected at 2, 5, 10, 15, 30, 60, 90, 120, and 180 min after administration of KTZ.
In the infusion study, KTZ was administered through the femoral vein at a bolus dose of 5 mg/kg and infused at a constant rate of 0.33 mg/h/body using a syringe infusion pump (Terufusion, STC-525; Terumo Co., Tokyo, Japan) for 6 h. The blood was collected at 0.25, 0.5, 1, 2, 3, 4, 5, and 6 h after the start of infusion. At 6 h, the liver was removed. To investigate the concentration dependency of liver/plasma concentration ratio (KpH) of KTZ, KTZ was administrated through the femoral vein at a bolus dose of 10 mg/kg. At 0.5, 1, 2, and 3 h after administration of KTZ, the blood was collected from the abdominal aorta and the liver was removed. Blood was centrifuged at 12,000 rpm for 2 min to obtain the plasma. Plasma and liver were stored at −20°C until analyzed.
Effect of ITZ and KTZ on Plasma Concentration Profiles of MDZ.
To investigate the effect of ITZ and KTZ on the plasma concentration profiles of MDZ, MDZ was administered through the portal vein at 60 min after i.v. bolus administration of ITZ or at the steady state of KTZ. Because elimination half-lives of ITZ and KTZ after i.v. administration were 8.8 h and 1.2 h, respectively, the plasma concentration of KTZ reached a steady state within several hours. However, it takes a long time for ITZ to reach a steady state; therefore, we used the ITZ concentrations at 1 and 4 h after i.v. bolus administration as the maximum and minimum concentrations of ITZ, respectively.
Rats were cannulated in the femoral vein, artery, and portal vein under light ether anesthesia. After recovery from anesthesia, ITZ was administered through the femoral vein at a dose of 20 mg/kg. At 60 min after ITZ administration, MDZ was administered through the portal vein at a dose of 10 mg/kg. KTZ was administered through the femoral vein at a dose of 5 mg/kg and infused at a constant rate of 0.33 mg/h/body using a syringe infusion pump. At 120 min after the beginning of KTZ infusion, MDZ was administered through the portal vein. The blood was collected at 2, 5, 10, 15, 30, 60, 90, 120, and 180 min after administration of MDZ. Blood was centrifuged at 12,000 rpm for 2 min to obtain the plasma. At 180 min, the liver was removed. The plasma and liver were stored at −20°C until analyzed.
In Vitro Studies
Unbound Fractions of ITZ and KTZ in the Plasma and Liver Tissue.Plasma protein binding of ITZ.
The protein unbound fraction (fp) of ITZ in rat plasma was measured by the ultrafiltration method with a Centrifree (MPS-3; Amicon, Beverly, MA). ITZ was mixed with the rat plasma at a concentration of 5 μg/ml, and then the mixture was incubated at 37°C for 30 min. One milliliter of the mixture was placed in the sample reservoir of Centrifree in triplicate. After 0.1 ml of methanol was added to the filtrate cup to prevent adsorption of ITZ, each Centrifree was centrifuged at 1600g for 5 min. After filtration, the filtrate cup was weighed to estimate the volume of filtrate. A 0.2-ml aliquot of each filtrate and 0.1 ml of rat plasma remaining in the sample reservoir were used to determine the concentrations of ITZ. This procedure was repeated using the same membrane more than four times until the unbound fraction became steady.
Liver tissue binding of ITZ.
It was assumed that the unbound fraction of drug in liver homogenates was the same as the unbound fraction in the intact liver under flow conditions. The unbound fraction of inhibitors in rat liver homogenates was calculated as follows. The liver tissue binding of ITZ was calculated using the distribution fraction to liver tissue cytosol and the unbound fraction to liver cytosol protein.
For determination of the distribution fraction to liver tissue cytosol, liver tissues were homogenized with 0.1 M phosphate buffer (pH 7.4) to prepare 20, 30, 40, 50, 60, and 75% tissue homogenates. Blood was not removed from the liver before homogenization. ITZ was mixed with the tissue homogenates at a concentration of 50 μg/ml. The mixtures were incubated at 37°C for 30 min, and were ultracentrifuged at 105,000g for 60 min to obtain cytosol fractions. The concentrations of ITZ in the cytosol fractions and tissue homogenates were determined for calculation of the distribution fraction to liver tissue cytosol.
The unbound fraction of ITZ in the liver tissue cytosol was measured by the charcoal adsorption method (Yuan, 1995). Twenty, 30, 40, 50, 60, and 75% liver tissue homogenates were ultracentrifuged at 105,000g for 60 min to obtain the cytosol fractions. A 0.3-ml aliquot of the cytosol fractions was preincubated at 37°C and ITZ was mixed with the liver tissue cytosols at a concentration of 100 ng/ml. At 5, 10, 15, and 30 s after adding ITZ, 0.2 ml of 50 mM Tris-HCl (pH 7.4) containing charcoal (final concentration, 0.05%) was added to the mixtures. The mixtures were centrifuged at 12,000 rpm to obtain the supernatant, and the concentration of ITZ in the supernatant was determined. The remaining ratio at time 0 was extrapolated from those at 5, 10, 15, and 30 s and was regarded as the bound fraction in the liver tissue cytosol of ITZ.
Total amount of drug in liver homogenate is equal to the sum of the amount of unbound and bound drug in cytosol and the amount of drug bound to liver organelle. Therefore, total amount of drug in liver homogenate is represented by the following equation:
The liver tissue unbound fraction (fh) was calculated according to the following equations:
Plasma protein and liver tissue binding of KTZ.
The plasma protein and liver tissue binding of KTZ were evaluated by the equilibrium dialysis method.
Dialysis was performed using an apparatus made of clear acrylic resin and consisting of two 1.5-ml chambers separated by a cellulose dialysis membrane (SC-101-M10H; DIACHEMA, München, Germany). KTZ was added to the rat plasma at a concentration of 5 μg/ml and applied to one chamber, and 0.1 M phosphate buffer (pH 7.4) was applied to the other chamber. After incubation at 37°C for 6 h, 0.1 ml of sample was collected from both chambers for assay.
For determination of the liver tissue binding of KTZ, liver tissues were homogenized with 0.1 M phosphate buffer (pH 7.4) to prepare 10, 20, and 40% tissue homogenates. Tissue homogenates were dialyzed twice with 100 volumes of 0.1 M phosphate buffer (pH 7.4) for 12 h to remove coenzymes. KTZ was mixed with the tissue homogenates at a concentration of 20 μg/ml. The mixture and 0.1 M phosphate buffer (pH 7.4) were added to the dialysis chamber and incubated at 37°C for 6 h. After the incubation, 0.5 ml of sample was collected from both sides for assay. The liver tissue unbound fraction was calculated according to the following equation:
Inhibitory Effect of ITZ, ITZ-OH, and KTZ on the Metabolism of MDZ in Rat Hepatic Microsomes.
From the preliminary experiment we confirmed that substrate depletion increased proportionally up to 5 min and in the range of 0.2 to 2 mg/ml of protein concentration on the condition of metabolic inhibition experiment. The kinetic and inhibition studies for MDZ in rat liver microsomes were performed on the incubation condition described as follows: 0.32 ml of incubation mixture containing rat liver microsome (protein concentration, 0.4 mg/ml) and a NADPH-regenerating system [100 mM phosphate buffer (pH 7.4), 2 mM NADP, 10 mM glucose-6-phosphate, 1 U G-6-P dehydrogenase, 0.1 mM EDTA, 5 mM MgCl2] were preincubated at 37°C for 2 min. The concentrations of MDZ were 1, 2, 5, 10, and 20 μM. The reactions were initiated by adding 0.04 ml of inhibitor solutions (ITZ, KTZ, ITZ-OH) and 0.04 ml of MDZ solution. The concentrations of inhibitors were as follows: ITZ, 0.1, 0.2, 0.5, and 1 μM; KTZ, 0.1, 0.2, 0.5, and 1 μM; and ITZ-OH, 10, 20, and 50 μM. After incubation at 37°C for 5 min, the enzyme reactions were terminated by adding 0.4 ml of cold acetonitrile and the reaction mixture was centrifuged at 3000 rpm for 2 min. Four-tenths milliliter of the supernatant was removed to determine the concentration of MDZ. The reaction velocity was estimated from the decrease of MDZ. The following equation was fitted to the observed data using the nonlinear iterative least-squares method (Yamaoka et al., 1981) to estimate kinetic parameters for the metabolism of MDZ and inhibition by ITZ, KTZ, and ITZ-OH in rat liver microsomes.
Ki value must be calculated based on unbound concentration of inhibitor. The unbound fraction of inhibitors in rat liver microsomes were calculated as follows. The unbound concentrations of ITZ and ITZ-OH in the incubation mixtures were measured by the charcoal adsorption method (Yuan, 1995), and that of KTZ was evaluated by the equilibrium dialysis method in the same way as described above.
For the determination of the unbound fractions of ITZ and ITZ-OH, 0.3 ml of the reaction solution without NADP was preincubated at 37°C. ITZ and ITZ-OH were mixed with the reaction solution at a concentration of 1 and 10 μM, respectively. At 10, 20, and 30 s after adding inhibitors, 0.2 ml of 50 mM Tris-HCl (pH 7.4) containing charcoal (final concentration, 0.05%) was added to the mixtures. The mixtures were centrifuged at 12,000 rpm to obtain the supernatant, and the concentrations of ITZ and ITZ-OH in the supernatant were determined. The remaining ratio at time 0 was extrapolated by those at 10, 20, and 30 s and was regarded as the bound fraction in the reaction solution of ITZ and ITZ-OH.
For the determination of the unbound fractions of KTZ, KTZ was mixed with the reaction solution without NADP at a concentration of 1 μM and applied to one chamber, and 0.1 M phosphate buffer (pH 7.4) was applied to the other chamber. After incubation at 37°C for 6 h, 0.1 ml of sample was collected from both chambers for determining the concentration.
Uptake of ITZ and KTZ by Isolated Rat Hepatocytes.
Rat hepatocytes were isolated by the procedure of Baur et al. (1975). Cell viability for each experiment was checked by the trypan blue exclusion test and was in the range of 85 to 95%. Protein concentration was determined by the colorimetric method of Lowry et al. (1951). All experiments were completed within 2 h after cell preparation, at which time the viability had not changed appreciably.
The uptake of ITZ and KTZ into isolated rat hepatocytes was investigated as follows. Isolated rat hepatocytes (protein concentration, 20 mg/ml) were suspended in Krebs-Henseleit buffer. Three-tenths milliliter of hepatocyte suspension and 2.7 ml of Krebs-Henseleit buffer (albumin-free) were mixed and preincubated at 37°C for 5 min. Thirty microliters of standard solution of drugs was added to each hepatocyte suspension at a concentration of 1 μM and incubated at 37°C. At 20 s after addition of drugs, 0.4 ml of the cell suspensions were placed in 1.5-ml polyethylene tubes previously layered with 0.5 ml of silicone-oil (specific gravity, 1.050) and 0.2 ml 3 N KOH. The samples were then centrifuged for 10 s in a tabletop microfuge capable of extremely rapid acceleration to separate the cells from the medium. The samples were frozen at −20°C, and then the sample tubes were cut at the middle of the oil layer. The concentrations in upper layers (medium) and lower layers (hepatocytes) were measured to investigate the initial uptake rate into isolated rat hepatocytes. The uptake of drugs was corrected for the adherent fluid volume and then converted to true intracellular concentration. The values of adherent fluid (2.2 μl/mg protein) and intracellular space (5.2 μl/mg protein) were obtained from the literature (Miyauchi et al., 1993).
Hepatocyte suspension was incubated at 27°C or 0°C to investigate the effect of temperature on the initial uptake rates of ITZ and KTZ into isolated rat hepatocytes. The effect of metabolic inhibitor on the initial uptake rates of ITZ and KTZ was investigated as follows. The cellular ATP content was reduced rapidly by adding 30 μM rotenone (Nakamura et al., 1994). Rotenone was added to hepatocyte suspension and then preincubated at 37°C for 10 min. The standard solution of drugs was added to each hepatocyte suspension and the initial uptake rates of drugs were measured.
Measurement of the Concentrations of ITZ, ITZ-OH, KTZ, and MDZ
For the determination of ITZ concentration in plasma, 0.1 ml of plasma, 0.5 ml 1 N NaOH, and 2.5 ml of n-hexane/isoamyl alcohol (98:2, v/v) were mixed and shaken for 5 min and then centrifuged at 3000 rpm for 5 min. Two milliliters of the organic phase was transferred to another tube and evaporated under nitrogen gas. The residue was dissolved with 0.2 ml of the mobile phase and 75 μl was injected into HPLC. The chromatographic system consisted of an autosampler 717 (Waters, Tokyo, Japan), a LC-10AD pump, and a SPD-10A variable wavelength UV detector operated at 263 nm (Shimadzu, Kyoto, Japan). The column was a reversed-phase Inertosil ODS, 4.6- × 250-mm (GL Science, Osaka, Japan) and was maintained at 40°C. The mobile phase was acetonitrile-10 mM phosphate buffer (pH 6.5; 8:2, v/v), and was pumped isocratically at a flow rate of 1 ml/min. The calibration curve was linear within the range of 50 to 5000 ng/ml (r = 0.999).
For the determination of ITZ concentration in the liver, 0.5 ml of 20% liver homogenate, 0.5 ml 1 N NaOH, and 5 ml ofn-hexane/isoamyl alcohol (98:2, v/v) were mixed and shaken for 5 min and then centrifuged at 3000 rpm for 5 min. Four milliliters of the organic phase was transferred to another tube and back-extracted with 3 ml 0.1 N HCl. To 2 ml of the aqueous phase, 0.5 ml of 1 N NaOH was added and extracted with 2.5 ml of n-hexane/isoamyl alcohol (98:2, v/v). Two milliliters of the organic phase was transferred to another tube and evaporated under nitrogen gas. The residue was dissolved with 0.2 ml of the mobile phase and 75 μl was injected into HPLC. The HPLC condition was the same as the plasma concentration of ITZ. The calibration curve was linear within the range of 500 to 50,000 ng/g liver (r = 0.999).
For ITZ-OH, the same method as the quantitative analysis of ITZ was carried out except for the extract solvent and mobile phase, where isopropylether as extract solvent and acetonitrile-10 mM phosphate buffer (pH 6.5; 7:3, v/v) as mobile phase were used. The calibration curves were linear within the range of 100 to 10,000 ng/ml (r = 0.999) and 200 to 20,000 ng/g liver (r = 0.999), for plasma and liver, respectively.
For the determination of KTZ concentration in plasma, the same method as the quantitative analysis of ITZ was carried out except for detected wavelength and mobile phase. KTZ was detected at 225 nm. The mobile phase was acetonitrile-10 mM phosphate buffer (pH 6.5; 7:3, v/v). The calibration curve was linear within the range of 100 to 10,000 ng/ml (r = 0.999).
For KTZ in the liver, the same extraction method as for ITZ was carried out except for the extract solvent, where isopropylether was used. The HPLC condition was the same as for the determination of the plasma concentration of ITZ. The calibration curve was linear within the range of 500 to 50,000 ng/g liver (r = 0.999).
For the determination of MDZ concentration in plasma, 0.1 ml of plasma, 0.5 ml of 1 N NaOH, and 3 ml of n-hexane were mixed and shaken for 5 min and then centrifuged at 3000 rpm for 5 min. Two milliliters of the organic phase was transferred to another tube and evaporated under nitrogen gas. The residue was dissolved with 0.2 ml of the mobile phase and 75 μl was injected into HPLC. The HPLC condition was the same as the plasma concentration of ITZ. MDZ was detected by measuring the absorption at 245 nm. The calibration curve was linear within the range of 50 to 10,000 ng/ml (r = 0.999).
In all measurements, coefficients of variation were less than 10%, and within-run accuracies were less than ±10%. When the concentrations in the samples were below the limit of quantitation, levels were determined by increasing the amount of sample.
Analysis of Data
Determination of Kinetic Parameters.
Plasma concentration profiles were fitted to the following biexponential equation using the nonlinear least squares method (Yamaoka et al., 1981).
Prediction of Increasing Ratio of Plasma Concentration of MDZ.
In the absence of inhibitors, metabolic velocities were expressed as the following equation (eq. 11). Assuming that the interaction of drug metabolism is of a competitive inhibition type, in the presence of inhibitors, metabolic velocities were expressed as eq. 6. Reaction velocities at varying concentrations of the substrate, MDZ were analyzed by nonlinear least-squares regression.
Statistical Analysis.
Statistical analysis was performed using Student’s ttest. Differences were regarded as statistically significant whenp values were below .05.
Results
Plasma and Liver Concentration Profiles of ITZ and KTZ.
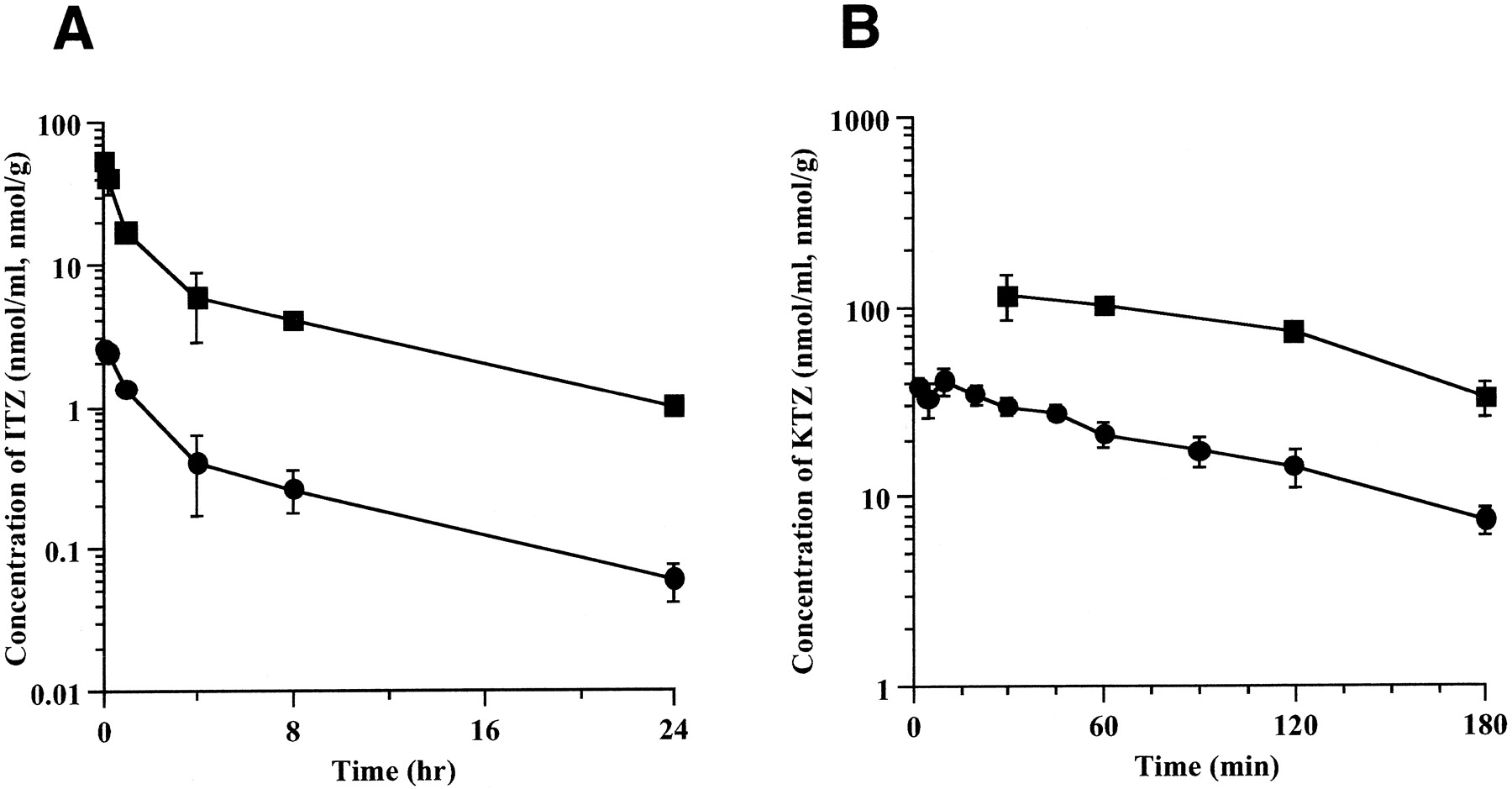
Figure 1 shows the plasma and liver concentration profiles of ITZ and KTZ after bolus injection of each drug. The liver concentrations of ITZ and KTZ were in parallel with the plasma concentration at 15 and 30 min or later after administration. Table 1 shows the pharmacokinetic parameters of ITZ and KTZ after i.v. administration. ITZ-OH concentration at 1 h after i.v. administration of ITZ was 0.104 ± 0.055 nmol/ml (8.1% of ITZ) in plasma and 2.46 ± 0.72 nmol/g (15.0% of ITZ) in the liver, respectively (mean ± S.D., n = 3). At 4 h, the ITZ-OH concentration was 0.144 ± 0.103 nmol/ml (36.5% of ITZ) in plasma and 3.00 ± 1.08 nmol/g (52.5% of ITZ), respectively (mean ± S.D.,n = 3). ITZ-OH concentrations were lower than those of ITZ in both plasma and liver. Figure 2shows the concentration dependency of the liver/plasma concentration ratio (KpH) of ITZ and KTZ. KpH was substantially constant within the range studied.

Plasma and liver concentration profiles of ITZ (A) and KTZ (B) after bolus injection of ITZ or KTZ to rats.
ITZ was administered through the femoral vein at a dose of 5 mg/kg. KTZ was administered through the femoral vein at a dose of 10 mg/kg. Each point represents the mean ± S.D. (n = 3). ●, plasma concentration; ■, liver concentration.
Pharmacokinetic parameters of ITZ and KTZ in rats

Concentration dependency of liver/plasma concentration ratio (KpH) of ITZ (A) and KTZ (B).
Horizontal line and shaded area represent the mean ± S.D.
Plasma Protein and Liver Tissue-Binding Assay of ITZ and KTZ.
The plasma unbound fraction of ITZ was 0.0034 ± 0.0002 (mean ± S.D., n = 3). The distribution fractions to liver tissue cytosol were 0.0112 ± 0.0007, 0.0078 ± 0.0009, and 0.0076 ± 0.0007 (mean ± S.D., n = 3), and the unbound fractions in liver cytosol were 0.39 ± 0.03, 0.49 ± 0.09, and 0.42 ± 0.13 (mean ± S.D.,n = 3) for 50, 60, and 75% homogenate of liver tissue, respectively. Consequently, the liver tissue unbound fractions calculated from eq. 3 were 0.0022 ± 0.0002, 0.0024 ± 0.0002, and 0.0024 ± 0.0004 (mean ± S.D., n= 3) for 50, 60, and 75% homogenate of liver tissue, and were substantially constant within the above ranges.
The plasma unbound fraction of KTZ was 0.0095 ± 0.0003 (mean ± S.D., n = 3). The liver tissue unbound fractions calculated from eq. 5 were 0.0039 ± 0.0008, 0.0031 ± 0.0005, and 0.0035 ± 0.0006 (mean ± S.D., n= 3) for 10, 20, and 40% homogenates of liver tissue, and were also constant regardless of the homogenate concentrations.
Table 2 shows the mean concentrations in plasma and liver and the liver concentration/plasma concentration ratios of ITZ and KTZ. The liver-unbound concentration to plasma-unbound concentration ratios (CHf/Cpf) of ITZ and KTZ were more than 1, suggesting concentrated uptake of both drugs into the liver.
Liver and plasma concentrations of ITZ and KTZ in rats
Effect of ITZ and KTZ on Plasma Concentration Profiles of MDZ.
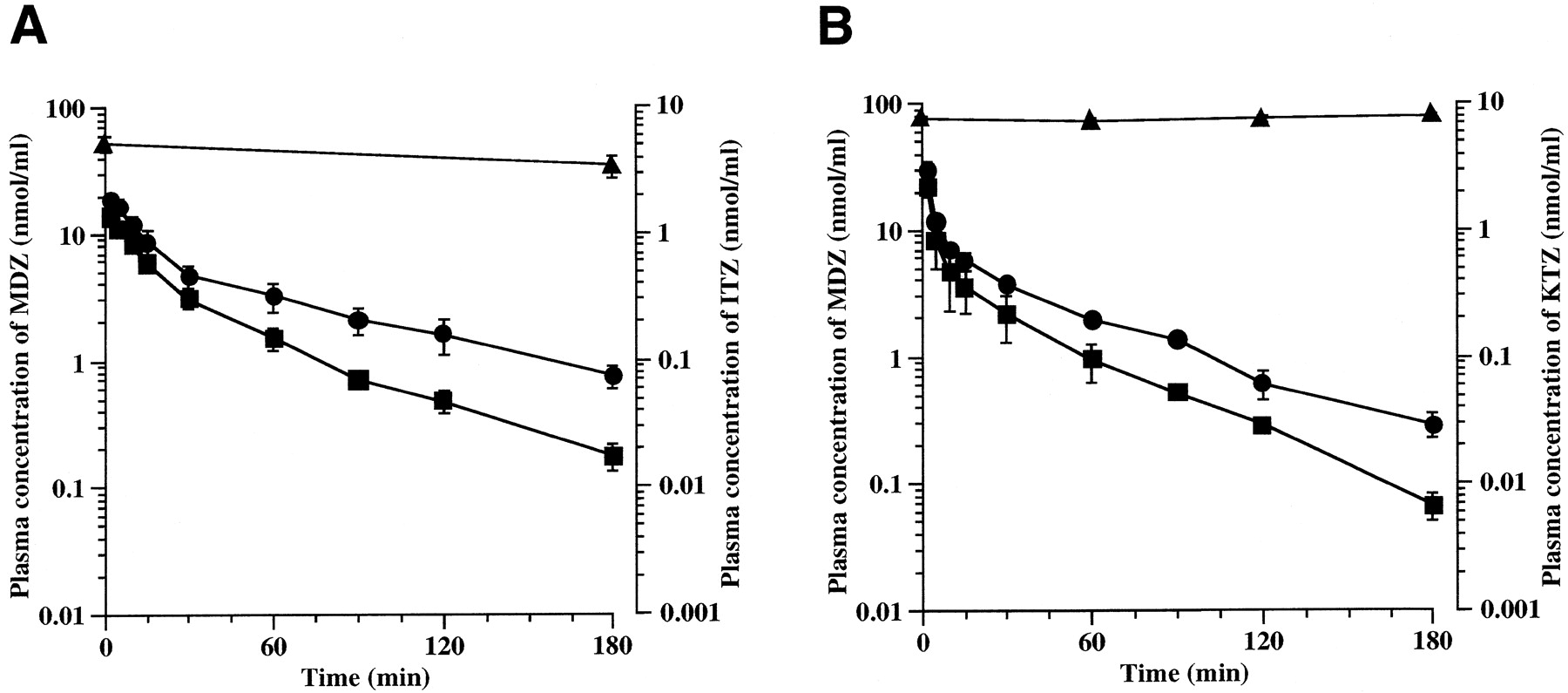
Figure 3 and Table3 show the plasma concentration profiles and pharmacokinetic parameters of MDZ after intraportal administration in the presence or absence of ITZ or KTZ. ITZ and KTZ increased the AUC of MDZ by 2.14-fold (p < .01) and 1.67-fold (p < .01).
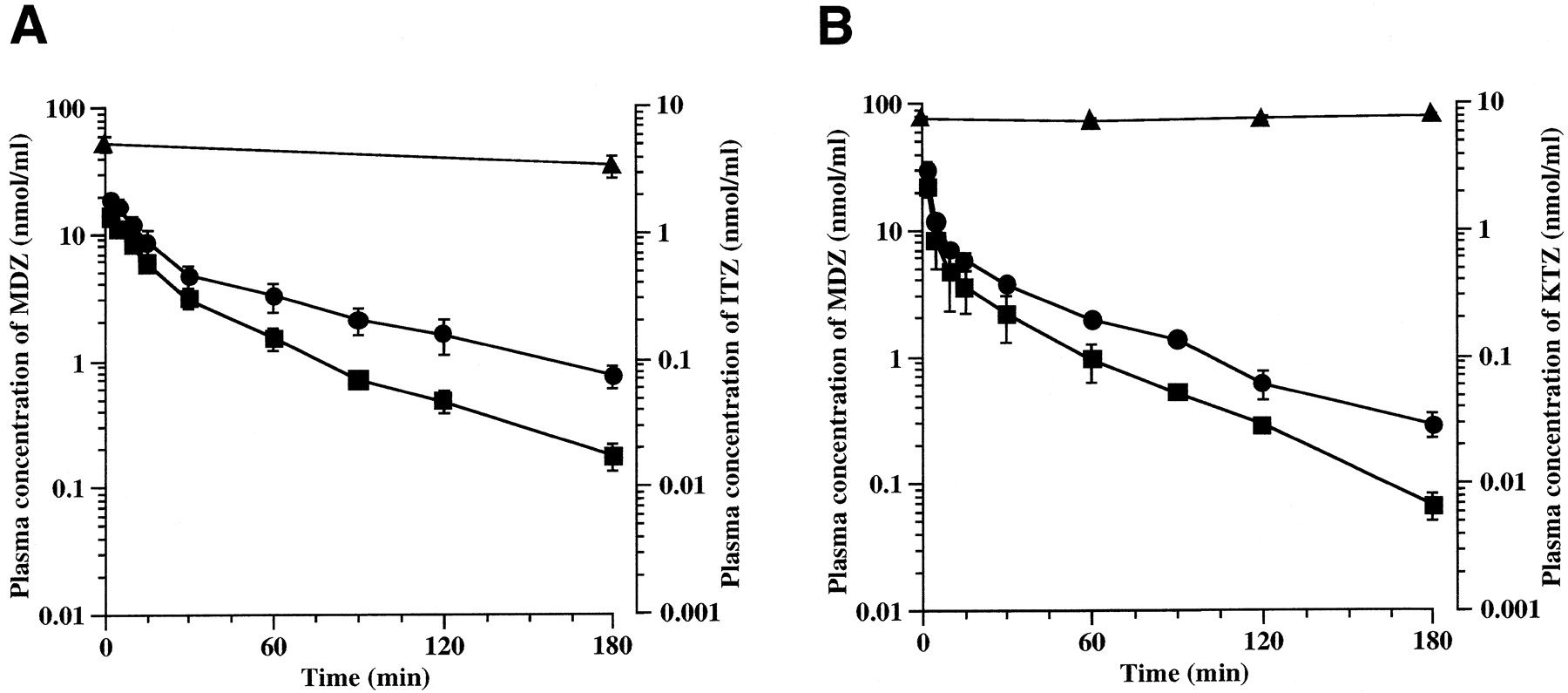
Effect of ITZ (A) and KTZ (B) on plasma concentration of MDZ.
A, ITZ was administered through the femoral vein at a dose of 20 mg/kg. MDZ was administered through the portal vein at a dose of 10 mg/kg at 60 min after administration of ITZ. Each point represents the mean ± S.D. (n = 4). ■, plasma concentration of MDZ alone; ●, plasma concentration of MDZ in the presence of ITZ; ▴, plasma concentration of ITZ. B, KTZ was administered through the femoral vein at a dose of 5 mg/kg and then infused into the rat at constant rate of 0.33 mg/h/body using a syringe infusion pump. MDZ was administered through the portal vein at a dose of 10 mg/kg at 120 min after the beginning of infusion. Each point represents the mean ± S.D. (n = 4). ■, plasma concentration of MDZ alone; ●, plasma concentration of MDZ in the presence of KTZ; ▴, plasma concentration of KTZ.
Pharmacokinetic parameters of MDZ in the absence or the presence of ITZ and KTZ
Inhibitory Effect of ITZ, ITZ-OH, and KTZ on the Metabolism of MDZ in Rat Hepatic Microsomes.
Figure 4 shows the inhibitory effect of ITZ, ITZ-OH, and KTZ on the metabolism of MDZ in rat hepatic microsomes based on the Michaelis-Menten equation. The metabolism of MDZ was competitively inhibited by ITZ, ITZ-OH, and KTZ. Table4 shows the kinetic parameters for the metabolism of MDZ and inhibition by ITZ, ITZ-OH, and KTZ in rat hepatic microsome. The averaged calculated values of Kmand Vmax were 7.01 μM and 3.34 nmol/mg protein/min, respectively. Ki values based on unbound concentration of inhibitors were 0.23 μM, 18.2 μM, and 0.16 μM, for ITZ, ITZ-OH, and KTZ, respectively.

The inhibitory effect of ITZ (A), KTZ (B), and ITZ-OH (C) on the metabolism of MDZ by rat liver microsomes based on a Lineweaver-Burk plot.
The concentrations of MDZ in inhibition of metabolism were 1, 2, 5, 10, and 20 μM, and the concentrations of inhibitors were ITZ: 0.1, 0.2, 0.5, and 1 μM; KTZ: 0.1, 0.2, 0.5, and 1 μM; ITZ-OH: 10, 20, and 50 μM. A and B: ○, MDZ alone; ●, with 0.1 μM ITZ, KTZ; ■, with 0.2 μM ITZ, KTZ; ▴, with 0.5 μM ITZ, KTZ; ⧫, with 1 μM ITZ, KTZ. C: ○, MDZ alone; ●, with 10 μM ITZ-OH; ■, with 20 μM ITZ-OH; ▴, with 50 μM ITZ-OH.
Kinetic parameters for metabolism of MDZ and inhibition by ITZ, ITZ-OH, and KTZ in rat microsomes
Uptakes of ITZ and KTZ by Isolated Rat Hepatocytes.
Figure 5 shows the effects of temperature and metabolic inhibitor on the initial uptake rates of ITZ and KTZ into isolated rat hepatocytes. The initial uptake rate of ITZ decreased to 43% at 27°C and 7% at 0°C. The initial uptake rate of KTZ decreased to 72% at 27°C and 35% at 0°C. Uptakes of ITZ and KTZ showed marked a temperature dependency and were reduced by adding 30 μM rotenone.

The effect of temperature and metabolic inhibitor on the initial uptake rates of ITZ (A) and KTZ (B) into rat hepatocytes.
Each point represents the mean ± S.D. (n = 4).
Comparison between Predicted Value and Observed Value of Increase of MDZ in Plasma Concentration.
Table 5 shows the predicted increase rate of plasma concentration of MDZ in the presence of ITZ or KTZ, according to eqs. 17 and 19 and the observed value. The increase rate (Rp) predicted from Cpf using eq. 19 was much underestimated, whereas the increase rate (RH) predicted from CHf using eq. 17 was very close to the observed increase value.
Comparison between predicted value and observed value of increase in ratio of plasma concentration of MDZ
Discussion
There have been few studies to predict the increase of blood concentration of interacted drugs on drug-drug interaction (Takedomi et al., 1998). To develop a methodology to predict the risk of drug-drug interaction quantitatively, it is necessary to solve several problems. Prediction of the disposition of inhibitors in the liver is very important because many drugs are transported into the liver by carrier-mediated hepatic uptake systems (Meijer et al., 1990; Yamazaki et al., 1996). We were successful in predicting the interaction between histamine H2 receptor antagonists (cimetidine and nizatidine) and MDZ, considering the concentrative uptake of both inhibitors in the liver (Takedomi et al., 1998; Table 5). Ervine et al. (1996) determined the ability of two azole antifungal agents, KTZ and fluconazole, to inhibit hepatic CYP activity in vivo in rats. The difference in activity was two orders of magnitude greater when Ki values were expressed in terms of unbound concentration in blood, which were more representative of hepatic tissue concentrations. These data confirm the conclusions based on in vitro findings that KTZ is a more inhibitory of CYP than fluconazole. Von Moltke et al. (1996) determined liver/plasma concentration ratio using liver homogenated with the plasma, and predicted the increase of plasma concentration of triazolam on the inhibition effect of KTZ. However, they did not take into account the concentrated uptake of the drug into the liver by carrier-mediated transport.
It was assumed that the unbound fraction of drug in liver homogenates was the same as the unbound fraction in the intact liver under flow conditions. The unbound fractions of inhibitors in rat liver homogenates were calculated from equations (3) and (5). The liver tissue unbound fractions of ITZ were 0.0022 ± 0.0002, 0.0024 ± 0.0002, and 0.0024 ± 0.0004 (mean ± S.D.,n = 3) for 50, 60, and 75% homogenate of liver tissue, and were substantially constant within the above ranges. As for KTZ, the liver tissue unbound fractions were 0.0039 ± 0.0008, 0.0031 ± 0.0005, and 0.0035 ± 0.0006 (mean ± S.D.,n = 3) for 10, 20, and 40% homogenates of liver tissue, and were also constant regardless of the homogenate concentrations. Table 2 shows that the liver-unbound concentration/plasma-unbound concentration ratios (CHf/Cpf) of ITZ and KTZ were more than 1, suggesting the concentrative uptake of both drugs into the liver.
To clarify the concentrative uptake into the liver, the effects of temperature and metabolic inhibitor on the initial uptake rates of ITZ and KTZ into isolated rat hepatocytes were investigated. Uptakes of ITZ and KTZ showed marked temperature dependency and were reduced by adding rotenone. The temperature dependency and the effect of metabolic inhibitor on the initial uptake rates of ITZ and KTZ suggested that concentrative uptakes of both drugs were due to an energy-dependent active transport system.
The Michaelis-Menten equation in the presence of a competitive inhibitor can be written as eq. 13, which assumes that the inhibitor concentration remains constant as the substrate concentration decreases. From a preliminary experiment, we confirmed that the inhibitor concentration did not decrease.
Figure 4 shows the inhibitory effect of ITZ, ITZ-OH, and KTZ on the metabolism of MDZ in rat hepatic microsomes based on a Lineweaver-Burk plot. The metabolism of MDZ was competitively inhibited by ITZ, ITZ-OH, and KTZ. Ki values based on unbound concentrations were 0.23 μM, 18.2 μM, and 0.16 μM, for ITZ, ITZ-OH, and KTZ, respectively. Table 5 summarizes the correspondence between the predicted and the observed values of the increase of MDZ concentrations in the plasma in the presence of ITZ and KTZ based on the in vivo and in vitro data. The predicted values were considerably underestimated, using plasma-unbound concentrations as the concentration of ITZ or KTZ near the metabolic enzymes, whereas the predicted values using unbound concentrations in the liver were very close to the observed value. It may be possible to use the unbound concentrations in the blood as the concentration of inhibitor only if they are equal to the unbound concentrations in the liver, the metabolic tissue. However, when the inhibitors are actively transported into the liver as ITZ and KTZ appear to be, the predicted increase of interacted drug concentration using the unbound concentrations of inhibitors in the plasma were underestimated, and the unbound concentrations in the liver may be more appropriate as the concentrations of inhibitor. We must take into account the concentrative uptake of inhibitors into the liver because the metabolic enzymes are localized on the endoplasmic reticulum in the hepatocyte and are physically separated from the blood by the plasma membrane, the Space of Disse, and capillary endothelium.
The concentrations in the plasma and the liver and theKi value of ITZ-OH were measured to examine the contribution of ITZ-OH to the inhibition of the metabolism of MDZ. The plasma and liver concentrations of ITZ-OH were lower than those of ITZ.Ki values of ITZ-OH and ITZ were 18.2 and 0.23 μM, respectively. The inhibition effect of ITZ exceeded that of ITZ-OH 80-fold. Accordingly, it is considered that ITZ-OH hardly inhibited the metabolism of MDZ.
There have been many reports on the interaction between MDZ and inhibitors including ITZ, KTZ, verapamil, and erythromycin in clinical cases and clinical studies (Olkkola et al., 1993, 1994, 1996; Beckman et al., 1994; Ahonen et al., 1995). The inhibition study on the metabolism of MDZ by the human liver microsome was also performed (Gascon and Dayer, 1991). To predict the extent of interaction, the concentrations of inhibitors in the portal or hepatic vein, inhibition of metabolism in the gastrointestine, and drug-drug interaction in the absorption process play an important role because these inhibitors are administered orally in clinical cases. In the effects of erythromycin and grapefruit juice influencing the plasma concentrations of MDZ after oral administration and i.v. injection, it was suggested that the contributions of metabolic inhibition and inhibition of secretion in the small intestine were larger than those of metabolic inhibition in the liver (Sawada et al., 1996). Recently, it was reported that pharmacokinetically estimated extraction ratio (0.43) in the gastrointestine was similar to that of the liver (0.44) (Paine et al., 1996; Thummel et al., 1996). Therefore, the gastrointestine can be a major site for presystemic metabolism after oral administration of MDZ. In clinical fields, the increase of AUC of MDZ after oral administration of inhibitors was much larger than the predicted value using the unbound concentrations in the plasma as the concentrations of inhibitors around the metabolic enzyme, or using the unbound drug in the liver estimated on the assumption that there were no species differences in the distribution of unbound drugs in the liver between human and rat (Sawada et al., 1996). This underestimation may be due to the metabolism in the intestinal wall or the change of absorption process. However, the contribution of these factors remains unclear, and further studies are required. It is also necessary to take into account that the drug concentration in the portal vein is higher than that in the systemic circulation after oral administration (Tabata et al., 1995; Hoffman et al., 1995; Fujieda et al., 1996). The degree of the inhibition will be underestimated if the concentration of inhibitor in the systemic circulation is used. Therefore, it is necessary to estimate the concentration in the portal vein from the absorption rate in the gastrointestine, and the concentration in the systemic circulation to estimate the unbound concentrations in the liver as the concentrations of inhibitors around the metabolic enzymes.
In conclusion, the increase of the plasma concentrations of MDZ by the azole antifungal agents, ITZ and KTZ, could be quantitatively predicted using the unbound concentrations of inhibitors in the liver and the inhibition constant on the metabolism of MDZ. When the inhibitors, ITZ and KTZ, were actively transported into the liver, the inhibition effects were underestimated using the unbound concentrations of inhibitors in the plasma. At the present time, most of the drugs causing problems in a clinical situation are administered orally. Consequently, in the future, a pharmacokinetic model to estimate the drug-drug interaction on the metabolism and the absorption in the gastrointestine must be developed to predict the increase of plasma concentrations of the inhibited drugs that are orally administered.
Footnotes
-
Send reprint requests to: Katsuhiro Yamano, Biopharmaceutical and Pharmacokinetic Research Laboratories, Fujisawa Pharmaceutical Co., Ltd., 1-6, Kashima 2-chome, Yodogawa-ku, Osaka 532-8514, Japan.
- Abbreviations used are::
- MDZ
- midazolam
- ITZ
- itraconazole
- ITZ-OH
- hydroxy itraconazole
- KTZ
- ketoconazole
- CYP
- cytochrome P-450
- AUC
- area under the plasma concentration curve
- Kph
- liver/plasma concentration ratio
- Chf
- unbound concentration in the liver
- Cpf
- unbound concentration in the plasma
- fh
- liver tissue-unbound fraction
- fp
- protein unbound fraction
- Received June 15, 1998.
- Accepted December 7, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}