


Abstract
A marked difference in hepatic activity of aldehyde oxidase between rats and monkeys was found to be responsible for the previously reported marked species difference in the metabolism of Zaleplon in vivo. In the postmitochondrial fractions,S-9s, from liver homogenates of these animals, Zaleplon was transformed in the presence of NADPH into the side chain oxidation product, N-desethyl-Zaleplon, and the aromatic ring oxidation product, 5-oxo-Zaleplon. In the rat S-9,N-desethyl-Zaleplon and 5-oxo-Zaleplon were a major and a very minor metabolites, respectively. However, in the monkeyS-9, Zaleplon was transformed into 5-oxo-Zaleplon at a much higher rate than that for N-desethyl-Zaleplon formation. N-Desethyl-Zaleplon was formed in the monkeyS-9 at a rate almost equal to that in the ratS-9. N-Desethyl-5-oxo-Zaleplon was formed at a minor rate only in the monkey S-9 through N-desethyl-Zaleplon as an obligatory intermediate. The hepatic activity for the formation of 5-oxo-Zaleplon in the monkey and rat was localized in cytosol and did not require NADPH. Sensitivity to various inhibitors and requirement of water as oxygen source, using H218O, strongly suggested that the hepatic cytosolic formation of 5-oxo-Zaleplon was mediated by aldehyde oxidase.N-Desethyl-Zaleplon was formed in the presence of NADPH by microsomes from the liver of rats and monkeys, and its formation was strongly suggested using various cytochrome P-450 inhibitors to be mediated by a number of cytochrome P-450 isoforms, such as 3A, 2C, and 2D subfamilies.
Zaleplon, CL 284,846 (N-[3-(3-cyanopyrazolo[1,5-a]pyrimidine-7-yl)phenyl]-N-ethyl-acetamide), is a nonbenzodiazepine compound with a preclinical profile indicative of sedative-hypnotic properties (Vanover et al., 1994). Zaleplon is currently being developed as an ultra-short-acting sleep inducer with a prompt onset of action, and it has been submitted for a new drug application to the Food and Drug Administration in the United States, and late phase II studies have been completed in Japan. A marked species difference was found in metabolism of Zaleplon administered orally, i.e., the major metabolite in the plasma of the rat, mouse, and dog is N-desethyl-Zaleplon, a side chain oxidation product, and in that of the monkey and human is5-oxo-Zaleplon, an oxidation product at a position adjacent to the nitrogen atom of the pyrimidine ring (Chaudhary et al., 1994). The in vivo study also demonstrated that the N-deethylation and ring oxidation products were found as minor metabolites in the plasma of monkey and of the rat, mouse, and dog, respectively, and that N-desethyl-5-oxo-Zaleplon existed as a minor metabolite in the plasma of the human and monkey.
It may be presumed that the N-deethylation reaction from Zaleplon to N-desethyl-Zaleplon proceeds by microsomal cytochrome P-450 (CYP; EC1.14.14.1) in the presence of NADPH and molecular oxygen (Wislocki et al., 1980). On the other hand, the aromatic ring oxidation of Zaleplon to 5-oxo-Zaleplon may be mediated by the molybdenum hydroxylase(s), aldehyde oxidase (AO1; EC 1.2.3.1), xanthine oxidase (XO; EC 1.1.3.22), and/or xanthine dehydrogenase (XD; EC 1.1.1.204), because Zaleplon, a pyrimidine derivative, seems to be a structurally typical substrate for these enzymes (Johns et al., 1969). Pyridine, quinoline, pyrimidine, and their derivatives, including various drugs, are known to be oxidized at the α-carbon to the nitrogen atom of the heterocycles by AO, XO, and/or XD in mammalian liver cytosol (Krenitsky et al., 1972;Beedham, 1987). Many of the N-heterocycles for AO are oxidized by XO and XD, e.g., pyridoxal, 4-hydroxypyrimidine, 6-mercaptopurine, and pyrazolo[3.4-d]pyrimidine (Johns et al., 1969; Krenitsky et al., 1972).
The hepatic oxidases, AO and XD, have been demonstrated to be products from the corresponding genes in the human (Ichida et al., 1993; Wright et al., 1993; Xu et al., 1994). XO is a post-translational modification product derived by an oxidative process from XD as a direct translation product from the gene (Stirpe and Corte, 1968; Corte and Stirpe, 1972;Waud and Rajagopalan, 1976). Both XO and XD play an important role in catabolism of hypoxanthine to uric acid via xanthine. Of these three molybdenum hydroxylases, only XD requires NAD+ as an electron acceptor for the maximal activity in the oxidation ofN-heterocycles, but the others do not require any cofactor for their enzyme function (Rajagopalan, 1980). These cytosolic enzymes contain a common electron transfer system in each subunit, i.e., one molybdenum atom, two Fe/S clusters, and one flavin adenine dinucleotide molecule (Rajagopalan et al., 1962; Rajagopalan, 1980; Beedham, 1987). Water is used by these enzymes as a source of the oxygen atom incorporated into the N-heterocycles with concomitant reduction of molecular oxygen to superoxide (Beedham, 1987).
This article provides evidence that Zaleplon is transformed to5-oxo-Zaleplon selectively by cytosolic AO and to N-desethyl-Zaleplon by microsomal CYPs in the liver of monkeys and rats. Evidence will also be provided that the previously reported marked species difference in Zaleplon metabolism in the monkey and rat in vivo is due to the marked difference in the hepatic AO activity between these animals.
Materials and Methods
Chemicals.
Zaleplon, its metabolites, and CL 218,872 as an internal standard for HPLC were supplied by Wyeth-Ayerst Research Drug Metabolism Division (Princeton, NJ). NAD+ and NADPH were purchased from Oriental Yeast Co., Ltd. (Tokyo, Japan), and allopurinol, methotrexate, oxipurinol, α-naphthoflavone, quinidine, and quinine were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Quinacrine, menadione, troleandomycin, and chlorozoxazone were obtained from Sigma Chemical Co. (St. Louis, MO) and SKF 525-A (proadifen hydrochloride) was obtained from Funakoshi (Tokyo, Japan). Furafylline, sulfaphenazole, and (S)-(+)-mephenytoin were purchased from Salford Ultrafine Chemicals and Research Ltd. (Manchester, UK), H218O (97 atom%) was purchased from Isotec Inc. (Miamisburg, OH). All other chemicals were of analytical grade.
Preparation of Subcellular Fractions.
Male Sprague-Dawley rats (6 weeks old) were obtained from Charles River Japan Laboratories, Inc. After starvation for 18 h, the rats were sacrificed by exsanguination under ether anesthesia. The livers were perfused with ice-cold 1.15% (w/v) potassium chloride (KCl), minced, and homogenized to 30% (w/v) in the ice-cold 1.15% KCl/50 mM sodium-potassium phosphate buffer solution (pH 7.4) with a Teflon-glass homogenizer. The homogenate was centrifuged to obtain a postmitochondrial fraction for 30 min at 9000g and 4°C and an aliquot of the resultant supernatant was frozen rapidly using liquid nitrogen and stored as S-9 at −80°C before use. All remaining supernatant was centrifuged for 60 min at 105,000gand 0°C and the supernatant was dialyzed against approximately 1000-fold volume of 10 mM phosphate buffer (pH 7.4) for 18 h, then frozen and stored as described above. The precipitate was resuspended in an appropriate volume of ice-cold 1.15% KCl/50 mM phosphate buffer solution (pH 7.4). The suspension was recentrifuged at 105,000g for 1 h to remove cytosol. The washed precipitate was resuspended with 1 mM EDTA/50 mM phosphate buffer solution (pH 7.4) to gram liver equivalent per milliliter and then stored at −80°C. One milliliter of S-9 or the cytosolic fraction was equivalent to 333 mg of the liver and 1 ml of the microsomal suspension to 1 g of the liver. The male cynomolgus monkey liver was supplied under frozen conditions in dry ice from Shin Nippon Biomedical Laboratories, Ltd. (Kagoshima, Japan) and prepared to obtain S-9, cytosol, and microsomes by the same procedure as described above. Protein concentration of each microsomal fraction was determined by the Lowry method (Lowry et al., 1951) using BSA as a standard and the others were by the Lowry-trichloroacetic acid method. The protein concentrations of the S-9, cytosolic, and microsomal fractions were 43.03, 30.25, and 20.12 mg/ml for monkey and 21.39, 20.89, and 8.14 mg/ml for rat, respectively.
Enzyme Assays.
The incubation mixture consisted of 20- to 50-mg liver equivalents per ml of rat and monkey subcellular fractions, 0.1 mM EDTA and 0.1 M potassium phosphate, pH 7.4, in the presence or absence of 5 mM NADPH. After preincubation at 37°C for 1 min, the reaction was initiated by adding DMSO solutions of Zaleplon to make a final concentration of 50 or 500 μM and followed by incubation at 37°C for 10 min in a shaking water bath. DMSO had no effect on the metabolic formation at a final concentration of less than 0.2% (v/v). Under the incubation conditions chosen, metabolite formations were linear with respect to time of incubation, protein concentration, and substrate concentration. Reactions were terminated by adding 2-fold volumes of chilled acetonitrile that included an internal standard. After vigorous stirring, the mixtures were centrifuged (1500g for 15 min) and 500 μl of clear supernatant was removed and evaporated at 40°C under a nitrogen stream. The residues were dissolved in 200 to 400 μl of 20% (v/v) acetonitrile-50 mM potassium phosphate, pH 6.8, and 50 μl of these solutions were injected into HPLC. For the CO inhibition study in cytosol and microsomes, incubation mixtures were pretreated by bubbling with 80% (v/v) CO/O2 obtained with a gas divider apparatus (SGD-XC51, STEC Inc.) for 2 min. Zaleplon (50 μM) was added to the reaction mixture, the gas bubbling was continued for 1 more min, and then the reaction system was quickly sealed with an airtight cap and incubated for 20 min. The inhibition of5-oxo-Zaleplon formation was studied in cytosol in the presence of potassium cyanide (Coughlan et al., 1980), allopurinol (Elion et al., 1966; Massey et al., 1970), oxipurinol (Massey et al., 1970), methotrexate (Lewis et al., 1984), quinacrine (Rajagopalan and Handler, 1964; Johns, 1967), menadione (Johns, 1967), and SKF 525-A (Yoshihara and Tatsumi, 1985; Robertson and Bland, 1993). The inhibitors were added in distilled water for potassium cyanide and quinacrine or in DMSO solution [final concentration of 0.1% (v/v)] for allopurinol, oxipurinol, methotrexate, menadione, and SKF 525-A. For the inhibition study on N-desethyl-Zaleplonformation in microsomes, the incubations were performed in the presence of specific inhibitors/substrates for CYP such as furafylline (Rodrigues, 1994; Newton et al., 1995), α-naphthoflavone (Rodrigues, 1994; Newton et al., 1995), sulfaphenazole (Rodrigues, 1994; Newton et al., 1995), tolubutamide (Loft et al., 1991; Masimirembwa and Hasler, 1994), (S)-(+)-mephenytoin (Loft et al., 1991), quinidine (Masimirembwa and Hasler, 1994), quinine (Masimirembwa and Hasler, 1994), chlorozoxazone (Peter et al., 1990), and troleandomycin (Rodrigues, 1994; Newton et al., 1995). The inhibitors were added in distilled water for quinidine, quinine, sulfaphenazole, and chlorozoxazone or in DMSO solution [final concentration of 0.1% (v/v)] for furafylline, α-naphthoflavone, tolubutamide, (S)-(+)-mephenytoin, and troleandomycin. After preincubation for 1 min in the presence of inhibitor and NADPH (5 mM), the reaction was initiated by addition of Zaleplon and followed incubation at 37°C for 10 min.
HPLC Analysis.
In all cases, chromatographic analysis was performed using a Develosil ODS-UG-5 column (4.6 × 150 mm; Nomura Chemical Co., Ltd., Aichi, Japan), with a mobile phase composed of acetonitrile and 50 mM phosphate buffer (pH 6.8) at ambient temperature. The column was eluted with acetonitrile gradient from 20% to 60% over 18 min by Waters LC Module-1 and a flow rate of 1.0 ml/min; the eluate was monitored continuously at 245 nm. No significant interference peaks appeared from the samples of the drug-free incubation mixture. The retention time for Zaleplon, N-desethyl-Zaleplon,5-oxo-Zaleplon, N-desethyl-5-oxo-Zaleplon, and the internal standard was 17.6, 14.5, 8.5, 3.4, and 20.8 min, respectively. The amounts of the metabolites were determined from the standard curve based on the peak area ratio between the metabolites and the internal standard (calculated automatically by a weighted linear regression least squares method using Waters 820J Workstation). The standard curves were produced by measurement of the authentic standard samples through the same preparation procedures as those for the reaction samples. Calibration lines for all compounds were liner in the range 0.05 to 25 μM.
Incorporation of 18O into 5-Oxo-Zaleplon.
Partially purified rat liver AO was prepared by the method of Stell et al. (1989) and underwent lyophilizing. The reaction system contained the lyophilized preparation (6 g wet liver equivalent), 5 mM Zaleplon, and 0.1 M phosphate buffer (pH 7.4) consisting of 2.71 mg KH2PO4, 28.67 mg Na2HPO4 and 1 ml of H218O or H2O. In each measurement, the reaction system was incubated at 37°C for 5 h in a shaking water bath, and then centrifuged at 15,000g for 10 min. The supernatant was applied to Sep-Pack C-18 (Waters) and eluted with a step gradient of aqueous methanol (0–50%). The fractions (10–30% methanol) were gathered and evaporated to concentrate the5-oxo-Zaleplon (determined by HPLC) and analyzed for incorporation of 18O or 16O into5-oxo-Zaleplon by a JEOL JMS-SX102A mass spectrometer under the following condition: instrument, fast atom bombardment (positive); gun high voltage, 3 KV; accelerating voltage, 10 KV; filament current, 2 A; emission current, 5 mA, and gas, Xe.
Results
Marked Species Difference in Metabolism of Zaleplon by Rat and Monkey Liver S-9s.
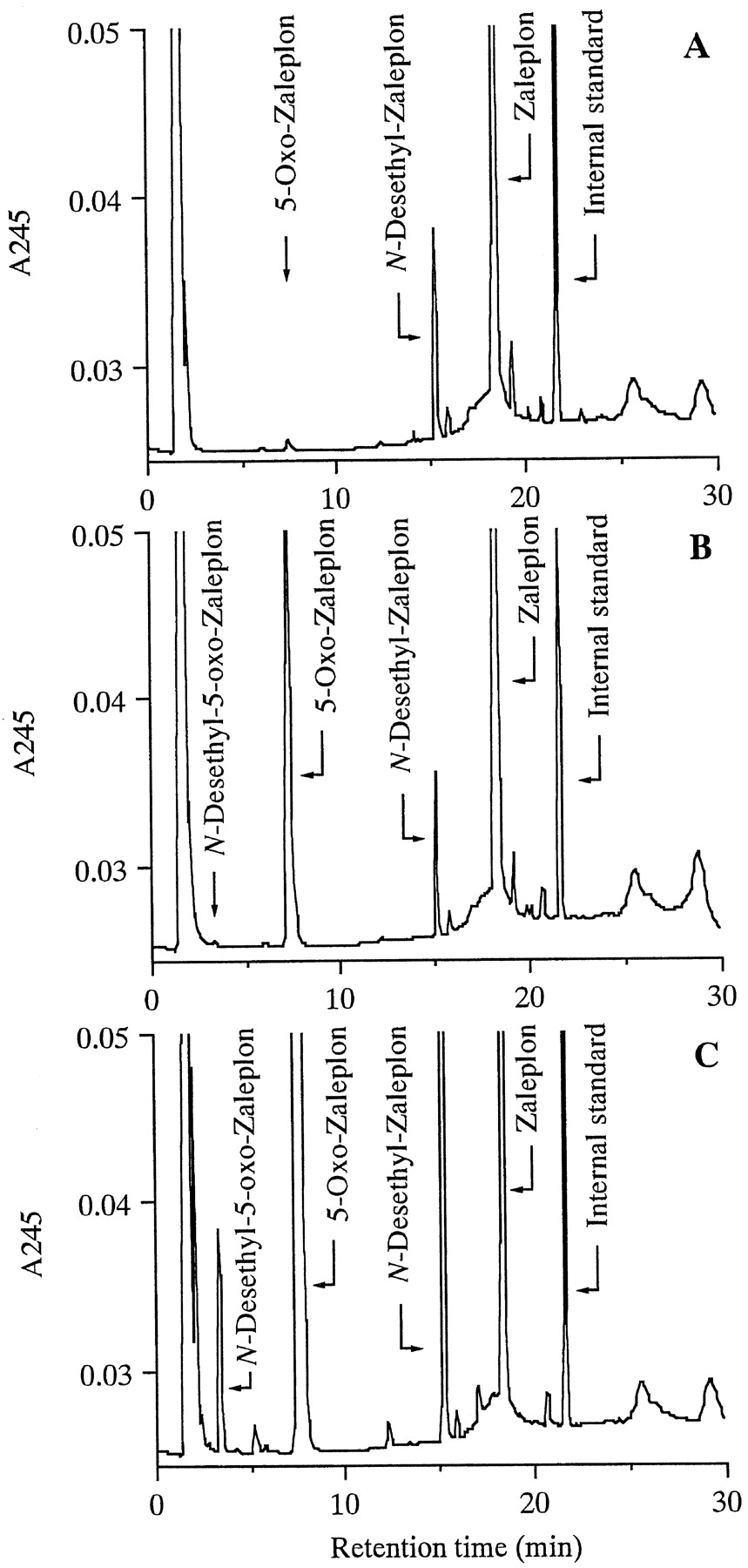
In the presence of NADPH, the postmitochondrial fraction,S-9, from rat liver homogenate transformed Zaleplon to N-desethyl-Zaleplon and 5-oxo-Zaleplonas a major and a very minor metabolite with higher polarities, respectively (Fig. 1A). In contrast, monkey liver S-9 transformed the sedative-hypnotic to5-oxo- and N-desethyl-Zaleplons as a major and a minor metabolite, respectively, under the same incubation conditions as used for rat liver S-9 (Fig. 1B).5-Oxo-N-desethyl-Zaleplon with higher polarity than the above two was also isolated by HPLC as a very minor metabolite from the reaction medium with the monkey S-9 when incubation was prolonged over 30 min (Fig. 1C), but not from that with the rat S-9. None of the metabolites were formed when theS-9s were heated (100°C, 10 min) before incubation (data not shown). These metabolites were further identified with the corresponding synthetic specimens by mass spectroscopy (MS) and UV absorption spectroscopy after being eluted from the HPLC column (data not shown).

HPLC of metabolites formed by incubations with rat (A) and monkey (B) liver S-9s for 10 min and with monkey liver S-9 for 120 min (C).
Zaleplon (500 μM) was incubated at 37°C with rat liverS-9 (20 mg liver/ml) in the presence of EDTA (0.1 mM) and NADPH (5 mM) in 0.1 M phosphate buffer (pH7.4) for 10 min (A), and with monkey liver S-9 for 10 min (B) and for 120 min (C) under the same conditions. HPLC was performed with acetonitrile extracts from incubation mixtures as described in Materials and Methods.
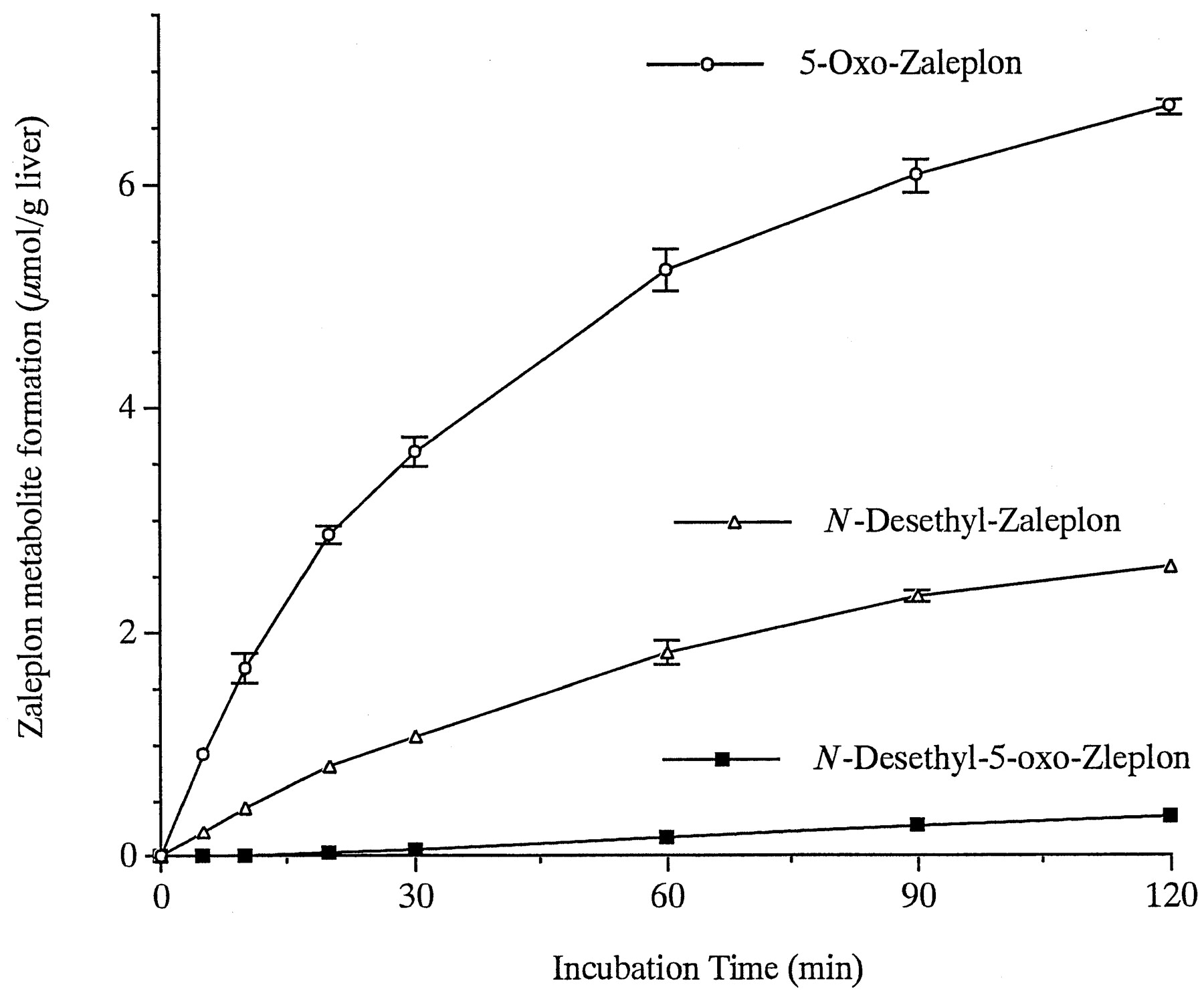
Under the HPLC conditions used, 5-oxo-,N-desethyl-, N-desethyl-5-oxo-,and unreacted Zaleplon showed approximately equal molecular absorbance at a wavelength of 245 nm. Therefore, direct comparison of the peak areas of the metabolites in the chromatograms indicated that the major metabolite, N-desethyl-Zaleplon, in rat liverS-9 was formed at an early stage (10 min) of incubation at an amount almost equal to that identified as a minor metabolite in monkey liver S-9. On the contrary, in the monkey liverS-9, 5-oxo-Zaleplon was formed at a significantly high rate compared with that for the N-desethyl-Zaleplon formation; the ratio of the ring oxidation to the oxidative N-deethylation was 4:1. The HPLC analysis also indicated the apparent rates of formation of5-oxo- and N-desethyl-Zaleplons to be 1.95 ± 0.06 and 41.53 ± 1.71 nmol/g liver/min, respectively, in the rat liver S-9 and to be 167.28 ± 1.93 and 42.28 ± 0.74 nmol/g liver/min, respectively, in the monkey liver S-9.
There was a time lag of about 10 min for the formation of N-desethyl-5-oxo-Zaleplon in the monkey liverS-9 until N-desethyl-Zaleplon formed increased to a concentration higher than about 7 μM. N-Desethyl-5-oxo-Zaleplon was found to be formed in the monkey liver S-9 from N-desethyl-Zaleplon at a rate of 103.33 ± 1.33 nmol/g liver/min and to a much lesser extent (9.65 ± 0.47 nmol/g liver/min) from the more polar metabolite, 5-oxo-Zaleplon, when the latter two metabolites were separately used as substrates (500 μM) for the metabolic reaction.
Characterization of Enzyme Catalyzing 5-oxo- and N-Desethyl-5-oxo-Zaleplon Formations in Monkey Liver Cytosol.
Zaleplon was incubated with subcellular fractions from monkey liverS-9, which was reconstituted from washed microsomes and dialyzed cytosol, for the characterization of the type of enzyme catalyzing the 5-oxo-Zaleplon formation. In the presence of NADPH, the reconstituted S-9 and dialyzed cytosol had almost equal activities of 167.28 ± 13.60 and 176.08 ± 2.93 nmol/g liver/min, respectively (Fig. 2a). The activity for 5-oxo-Zaleplon formation from Zaleplon in the monkey S-9 was localized in the cytosolic fraction, but not in microsomes (Fig. 3A). The cytosolic5-oxo-Zaleplon formation in the monkey liver was little affected in the absence of NADPH. Similar results were obtained for the5-oxo-Zaleplon formation as a very minor metabolic pathway by subcellular fractions of rat liver S-9 (data not shown).

Formation of 5-oxo-, N-desethyl-, andN-desethyl-5-oxo-Zaleplons from Zaleplon in monkey liver S-9.
Zaleplon (500 μM) was incubated at 37°C with monkey liver S-9 (20 mg liver/ml) in the presence of EDTA (0.1 mM) and NADPH (5 mM) in 0.1 M phosphate buffer (pH7.4), as described in Materials and Methods. Data are expressed as mean ± S.D. of triplicate determinations.

Effect of NADPH on formations of5-oxo-Zaleplon by subcellular fractions from monkey liver (A) and of N-desethyl-Zaleplon by those from rat and monkey livers (B).
Zaleplon (500 μM) was incubated at 37°C for 10 min with monkey and rat liver S-9s, microsomes (Ms.) and cytosols (Cy.) (20 mg liver/ml each) and EDTA (0.1 mM) in 0.1 M phosphate buffer (pH7.4) in the presence and absence of NADPH (5 mM), as described inMaterials and Methods. Data are expressed as mean ± S.D. of triplicate determinations.
Monkey liver cytosol had an activity oxidizing N-desethyl-Zaleplon to its 5-oxo-derivative at almost the same rate as did the S-9. NAD+ had no appreciable effect on the cytosolic 5-oxidation of Zaleplon and N-desethyl-Zaleplon(data not shown). Carbon monoxide (80%)/oxygen used as a gaseous phase for the incubation mixture had no effect on the5-oxo-Zaleplon formation in cytosol (Table1). MS of the metabolite,5-oxo-Zaleplon, indicated that an 18O atom was incorporated into Zaleplon from H218O (97 atom %) that had been used as an incubation medium in which lyophilized cytosolic proteins and buffer components were dissolved. The molecular ion peaks of the metabolite isolated by HPLC appeared at m/z 346 for [M + Na]+ and at m/z 324 for [M + H]+ (Fig. 4A), whereas those of 5-oxo-Zaleplon metabolically formed in H2O showed the molecular ion peaks atm/z 344 for [M + Na]+ and atm/z 322 for [M + H]+ (Fig. 4B).
Effect of various molybdenum hydroxylases inhibitors on formation of 5-oxo-Zaleplon in monkey liver cytosol
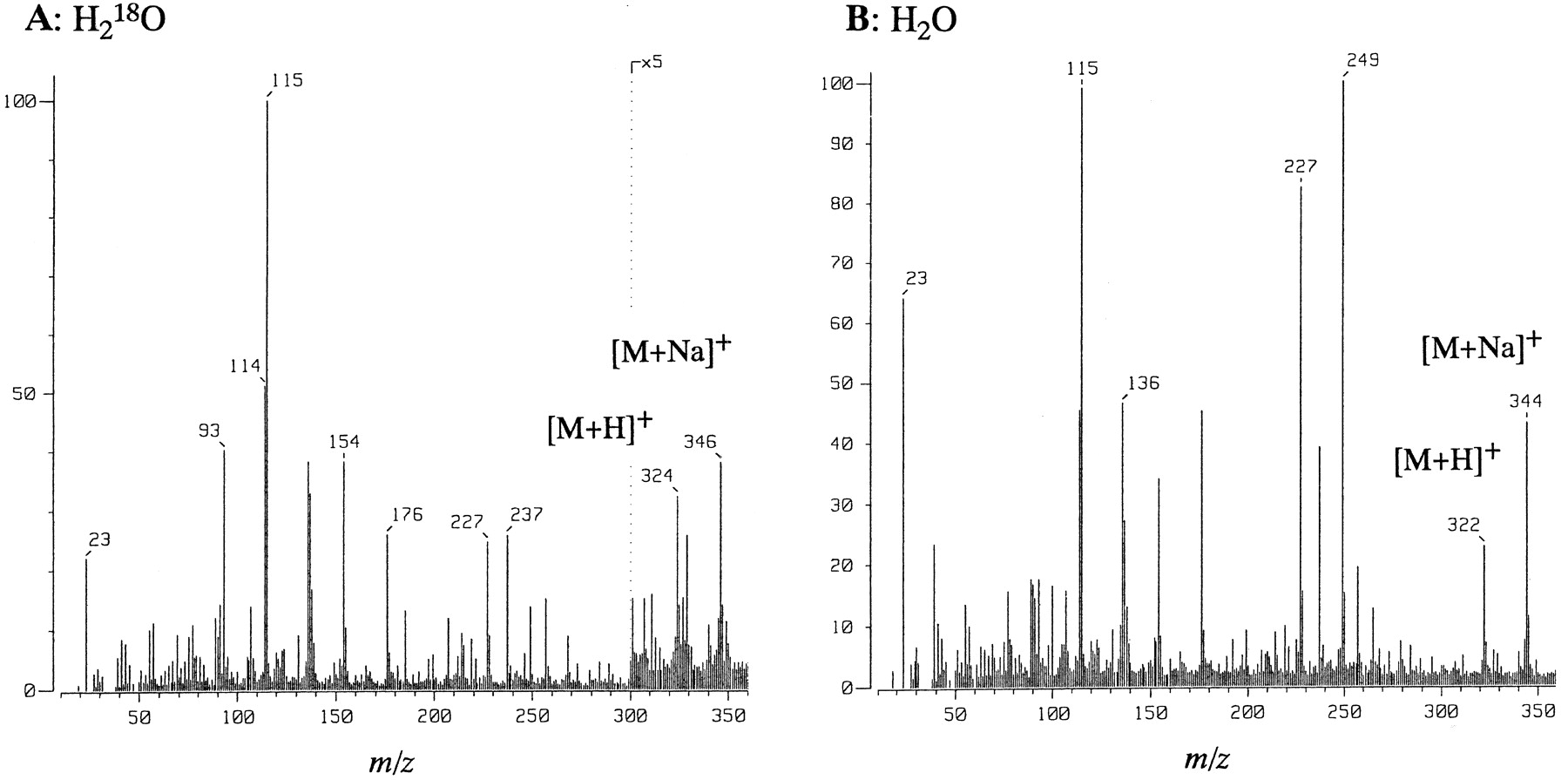
Fast atom bombardment MS of5-oxo-Zaleplon formed by incubation of Zaleplon with rat liver cytosol in H218O (A) and H2O (B).
5-Oxo-Zaleplon was isolated by solid extraction from incubation mixtures containing Zaleplon (0.5 mM) and lyophilized rat liver cytosol corresponding to 6 g of liver in H2O and H218O.
Zaleplon was incubated with dialyzed liver cytosol from monkeys in the presence of molybdenum hydroxylase inhibitors for further characterization and classification of the cytosolic enzyme involved in the 5-oxo-Zaleplon formation. The common irreversible inhibitor, KCN, for all molybdenum hydroxylases completely inhibited the hepatic cytosolic 5-oxo-Zaleplon formation (Table 1). Allopurinol, oxipurinol, and methotrexate had only a slight inhibitory effect on the 5-oxo-Zaleplon formation. However, the metabolic reaction was inhibited very strongly by menadione and strongly by quinacrine and SKF 525-A.
Characterization of CYPs Catalyzing MicrosomalN-desethyl-Zaleplon Formation in Rat and Monkey Livers.
Activity of the N-dealkylation of Zaleplon to N-desethyl-Zaleplon, a typical reaction mediated by CYP(s), was localized in microsomes but not in cytosols from rat and monkey livers (Fig. 3B). The microsomal N-dealkylation reaction required NADPH as a cofactor and was strongly inhibited by CO (Table 2). Sulfaphenazole, quinidine, quinine, and troleandomycin strongly inhibited the microsomalN-deethylation of Zaleplon at their concentration of 0.1 mM, suggesting the microsomal reaction to be mediated by multiple isoforms of CYPs such as 2C, 2D, and 3A subfamilies.
Effect of various CYP isoform inhibitors/substrates onN-desethyl-Zaleplon formation in rat and monkey liver microsomes
Discussion
The present study provides direct evidence that the previously demonstrated marked species difference in the in vivo metabolism of the oral sedative-hypnotic, Zaleplon, between the monkey and rat (Chaudhary et al., 1994) results from by AO activity in their hepatic postmitochondrial fractions, S-9s (Figs. 3 and5). The present study also provides the first evidence for the oxidation by AO of the pyrazolo[1,5-a]pyrimidine, a fundamental structure of Zaleplon. As demonstrated previously in the plasma of monkeys orally administered Zaleplon (Chaudhary et al., 1994), the major metabolite was 5-oxo-Zaleplon and the minor metabolites were N-desethyl-Zaleplon and N-desethyl-5-oxo-Zaleplon in the monkey liverS-9. Similarly, N-desethyl- and5-oxo-Zaleplons were a major and a very minor metabolite, respectively, in the rat liver S-9 as well as in the plasma after orally administered Zaleplon as reported previously (Chaudhary et al., 1994). However, there was only a little difference in hepatic activity of N-deethylation of Zaleplon in the S-9 and microsomal fractions between rats and monkeys. N-Desethyl-5-oxo-Zaleplon was formed at a very low rate from 5-oxo-Zaleplon used as a substrate in the monkey S-9 and microsomal fraction in the presence of NADPH, but not in the absence of the cofactor (data not shown).

Summary results of Zaleplon metabolism in rat and monkey liver.
In the monkey liver S-9, the major metabolite,5-oxo-Zaleplon, was formed in cytosol without any cofactor and the minor, N-desethyl-Zaleplon, by microsomes in the presence of NADPH. N-Desethyl-5-oxo-Zaleplon was formed only from N-desethyl-Zaleplon as an obligatory intermediate by the cytosolic 5-oxygenation of N-desethyl-Zaleplon in the absence of NADPH in the monkey liver.
The hepatic cytosolic 5-oxygenation of Zaleplon and N-desethyl-Zaleplon was attributable to a family of molybdenum hydroxylases consisting of AO, XO, and XD, which are widely distributed in cytosol of various tissues, especially the liver, in mammals (Krenitsky et al., 1974; Beedham et al., 1987a). H218O was found by MS to be the source of the oxygen atom incorporated into the substrate. This has been demonstrated directly using H218O with the oxidation of xanthine by XO (Murray et al., 1966; Hill and Sprecher, 1987) and indirectly under the anaerobic condition with N1-methylnicotinamide by AO (Quinn and Greengard, 1966). Of these enzymes, XD is excluded from possible participants in the aromatic oxidation of Zaleplon because NAD+had no effect on the 5-oxo-Zaleplon formation in monkey and rat liver cytosols (data not shown). AO and XO are known to be completely inhibited by cyanide anion acting as a potent ligand on the molybdenum ion located in the active site of the enzymes (Coughlan et al., 1980). The hepatic cytosolic Zaleplon 5-oxidation was completely inhibited by 1 mM KCN but not influenced by CO, the latter of which, however, strongly inhibited the microsomal N-deethylation of Zaleplon.
Representative inhibitors have been proposed for clearly differentiating the reaction mediated by AO from that by XO: quinacrine (Rajagopalan and Handler, 1964; Johns, 1967), menadione (Johns, 1967), and SKF 525-A (Yoshihara and Tatsumi, 1985; Robertson and Bland, 1993) for AO and allopurinol (Elion et al., 1966; Massey et al., 1970), oxipurinol (Massey et al., 1970), and methotrexate (Lewis et al., 1984) for XO. The Zaleplon 5-oxidation was inhibited very strongly by menadione and strongly by quinacrine and SKF 525-A, but slightly inhibited by allopurinol, oxipurinol and methotrexate (Table 1). Similar evidence was provided for the formation of5-oxo-Zaleplon as a very minor metabolite in rat liver cytosol (data not shown). In a similar manner using these inhibitors, the hepatic cytosolic oxidation of the α-carbon adjacent to aromatic nitrogen has been demonstrated with BRL 55792 (Harrell et al., 1994), famciclovir (Clarke et al., 1995), andO6-benzylguanine (Roy et al., 1995) to be mediated by AO in mammalian liver cytosol. Actually, oxidation of the N-containing aromatic heterocycles, 6-methylpurine (Krenitsky et al., 1974), bromonidine (Acheampong et al., 1996), and cinchona antimalarials (Beedham et al., 1987b) are known to proceed at much higher rates in hepatic cytosol from monkeys and humans than in that from rats as demonstrated in the present study on the 5-oxidation of Zaleplon. To our knowledge, except for the above drugs, nothing is known about the oxidation of N-containing heterocyclic aromatic compounds, especially pyrazolo[1,5-a]pyrimidine, a fundamental structure of Zaleplon, by AO in monkey liver.
The hepatic microsomal N-deethylation of the Zaleplon side chain in rats and monkeys was a reaction mediated by CYP, because of the requirement of NADPH as a cofactor and the strong inhibition by CO (Table 2). Use of various inhibitors at a concentration of 0.1 or 10 and 100 μM for characterizing the molecular species of CYPs in the rat strongly suggested that at least two CYPs were involved in the oxidative N-deethylation of Zaleplon (Table 2), because the microsomal N-dealkylation reaction was strongly inhibited by quinine and quinine, inhibitors for CYP2D subfamily in the rat and human, respectively. In addition, these alkaloids are good substrates for CYP3A (Guengrich et al., 1986), so that they may play a role as competitive inhibitors in the N-deethylation of Zaleplon. A possibility of the direct participation of CYP 3A in the microsomalN-deethylation was strongly supported by the potent inhibition with troleandomycin, an irreversible inhibitor for human CYP3A3/4. Sulfaphenazole, an inhibitor for human CYP 2C9/10, also strongly inhibited the microsomal N-dealkylation reaction. A further study is in progress in our laboratories to identify the molecular species of human CYPs involved in the ZaleplonN-deethylation using recombinant human enzymes and to provide direct evidence for the participation in the Zaleplon 5-oxidation of AO purified from monkey liver cytosol.
Acknowledgments
We thank K. Hayashi for generating mass spectral data for5-oxo-Zaleplon. We also thank the following personnel of Wyeth-Ayerst Research (Princeton, NJ): Dr. G. Fisher, Dr. S. E. Ball, Dr. J. A. Scatina, and Dr. V. V. Subrahmanyan for helpful discussions during the course of this work and K. V. Wimbert for proofreading the manuscript.
Footnotes
-
Send reprint requests to: Kosuke Kawashima, Department of Pharmacokinetics, Medical Research Laboratories, Lederle (Japan), Ltd., 1–6-34 Kashiwacho, Shikishi, Saitama 353-8511, Japan. E-mail:kosuke_k{at}kt.rim.or.jp
- Abbreviations used are::
- AO
- aldehyde oxidase
- XD
- xanthine dehydrogenase
- XO
- xanthine oxidase
- Received September 11, 1998.
- Accepted December 15, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}