


Abstract
The metabolism of retigabine in humans and dogs is dominated byN-glucuronidation (McNeilly et al., 1997), whereas in rats, a multitude of metabolites of this new anticonvulsant is observed (Hempel et al., 1999). The comparison of the in vivo and in vitro kinetics of retigabine N-glucuronidation in these species identified a constant ratio between retigabine and retigabineN-glucuronide in vivo in humans and dog. An enterohepatic circulation of retigabine in these species is likely to be the result of reversible glucuronidation-deglucuronidation reactions. Rats did not show such a phenomenon, indicating that enterohepatic circulation of retigabine via retigabineN-glucuronide does not occur in this species. In the rat, 90% of retigabine N-glucuronidation is catalyzed by UDP-glucuronosyltransferase (UGT)1A1 and UGT1A2, whereas family 2 UGT enzymes contribute also. Of ten recombinant human UGTs, only UGTs 1A1, 1A3, 1A4, and 1A9 catalyzed theN-glucuronidation of retigabine. From the known substrate specificities of UGT1A4 toward lamotrigine and bilirubin and our activity and inhibition data, we conclude that UGT1A4 is a major retigabine N-glucuronosyl transferase in vivo and significantly contributes to the enterohepatic cycling of the drug.
Retigabine [N-(2-amino-4-(4-fluorobenzylamino)phenyl)carbamic acid ethyl ester] (Fig. 1) is a new anticonvulsant that is now in clinical phase II development. It shows effects in many in vitro and in vivo models of epilepsy and has multiple modes of action (Kapetanovic et al., 1995; Rostock et al., 1996; Tober et al., 1996; Skeen et al., 1995).

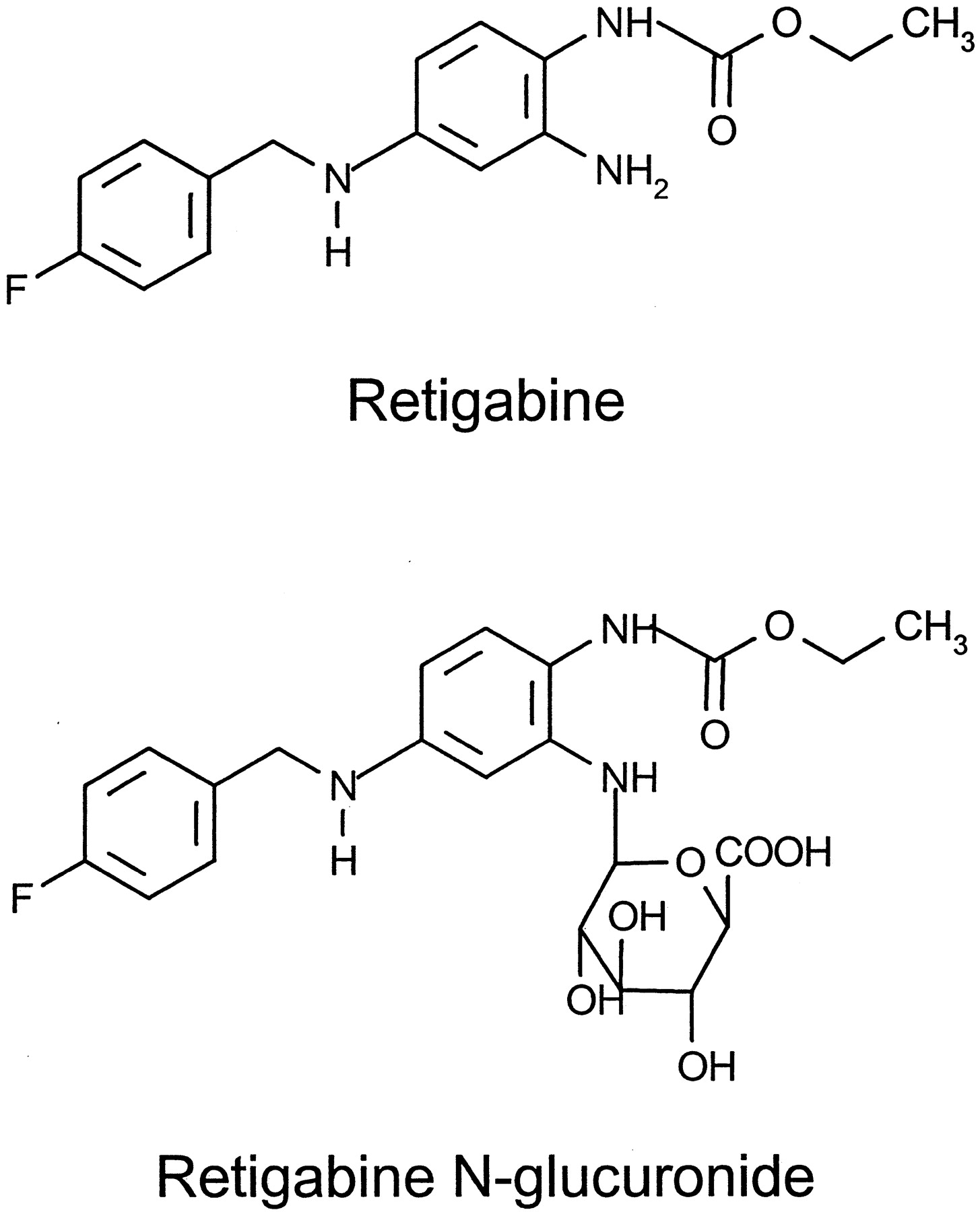
Formulae of retigabine and retigabine N-glucuronide.
The in vitro and in vivo metabolism of retigabine in humans and dog is dominated by glucuronidation reactions resulting in the formation of two distinct N-glucuronides (McNeilly et al., 1997). TheN2-glucuronide (retigabine N-glucuronide, Fig.1), where the primary amino group in the 2 position is the site of glucuronidation, exceeds by far the amount of theN4-glucuronide, where the secondary amino group in 4 position is glucuronidated. We report here only data on the formation or cleavage of the major retigabine N-glucuronide. Retigabine metabolism in rats is more complex. Although retigabineN-glucuronide is the major metabolite in rat plasma and bile, urine contains more than 20 metabolites that are mainly produced by acetylation, ring closure reactions, and N-dealkylation of the fluorobenzylic side chain (Hempel et al., 1999).
The importance of retigabine N-glucuronide in vivo relative to free retigabine is unclear. We therefore determined both compounds in human blood plasma from a phase I clinical study in volunteers and compared those to data from animals. Interestingly, we found that in contrast to rats, dogs and humans show a characteristic constant ratio between their retigabine and retigabine N-glucuronide plasma levels. To understand the mechanism by which the constant ratio is maintained and to understand the role of enzymes contributing to this mechanism, we: 1) characterized the enzyme kinetics of retigabineN-glucuronide formation in liver microsomes, 2) determined the interindividual variability of retigabine N-glucuronide formation in vitro in a panel of 16 human livers, and 3) characterized UDP-glucuronosyltransferases (UGTs)1contributing to retigabine N-glucuronidation by inhibition studies and by investigations with heterologously expressed enzymes. Our results suggest that the considerable interindividual variation of retigabineN-glucuronidation in vitro does not affect the ratio between retigabine and retigabine N-glucuronide in vivo and that UGT1A4 contributes significantly to the glucuronidation of retigabine.
Materials and Methods
Chemicals.
Retigabine was synthesized by ASTA Medica AG (Dresden, Germany). [14C]retigabine (9 mCi/mmol) was synthesized by Amersham (Buckinghamshire, UK). UDP-glucuronic acid and ascorbic acid were purchased from Fluka AG (Buchs, Switzerland). β-Glucuronidase (Escherichia coli), bilirubin, and alamethicin were purchased from Sigma (St. Louis, MO). Human UGT1A1 and UGT1A4 expressed in lymphoblasts were obtained from NatuTec (Frankfurt, Germany). Lamotrigine was kindly provided by Glaxo Wellcome (Research Triangle Park, NC). All other chemicals were of analytical grade and purchased from commercial suppliers.
Preparation of Microsomes from Animals and Humans,
Frozen livers from Wistar rats (Crl:(WJ)BR, Charles River GmbH, Sulzfeld, Germany) and Gunn Rats (HsdBlu:GUNN-j; Harlan Winkelmann GmbH, Detmold, Germany), which are genetically different in family I UGTs, were used for preparation of liver microsomes. Human liver samples were obtained from livers of kidney transplant donors (Meier et al., 1983) or from livers obtained from the University of Greifswald, Germany. Dog liver microsomes were prepared from a male Beagle dog (Versuchstierzucht Günter Halangk, Germany). Human, dog, and rat liver microsomes were prepared according to the method ofMeier et al. (1983) with modifications:
All of the following steps were performed on ice or at 4°C. Approximately 1 g of human or dog liver was thawed and homogenized in 5 ml of homogenization buffer (154 mM KCl, 1 mM EDTA, 50 mM Tris/HCl pH 7.4) using a Polytron PT3000 (Kinematica GmbH, Lucerne, Switzerland) set at 17,000 rpm for 45 s. A postmitochondrial supernatant was prepared by centrifugation of the homogenate at 10,000g for 20 min (SW 28 rotor; Beckman Instruments, Berkeley, CA). Microsomes were sedimented from this supernatant at 202,000g for 70 min (rotor 70lTl; Beckman), the pellet was washed once in ∼15 ml of 0.1 M Na2P2O7(pH 7.8) using a Dounce homogenizer. After centrifugation at 202,000g the pellet was resuspended in approximately 1 ml of 100 mM KH2PO4/K2HPO4 (pH 7.4), 1 mM EDTA (pH 8.0), 20% (v/v) glycerol. The microsomal suspension, which typically had a protein concentration of 10 mg/ml, was frozen as aliquots in liquid nitrogen and stored at −80°C. For the preparation of rat liver microsomes, about 3 g of liver was homogenized in 20 ml homogenization buffer with six strokes at 1200 rpm in a Potter-Elvehjem homogenizer. The remainder of the procedure was as described above.
The microsomal protein content was estimated using the bicinchoninic acid assay (Pierce, Rockford, IL) with BSA as standard.
Preparation of Human Recombinant UGTs.
Insect Sf9 cell extracts containing expressed UGT1A1, UGT1A7, and UGT1A10 were prepared as described previously (Strassburg et al., 1996,1998). The entire coding region of human UGT1A3, UGT1A4, and UGT2B7 were cloned from total liver cDNA using specific oligonucleotides to these RNAs. The cDNAs were cloned into Invitrogens BlueBac 4 transfer vector and the proteins expressed in Sf9 cells, as described previously (Nguyen and Tukey, 1997; C.P.S., A. Strassburg, N.N., Q.L., M.P. Manns and R.H.T., in press). Human UGT1A9 and UGT2B15 were cloned directly from a human liver cDNA and expressed in Sf9 cells as outlined (C.P.S., A. Strassburg, N.N., Q.L., M.P. Manns and R.H.T., in press). The cloning of the UGT2B4 cDNA was isolated from a human liver λ cDNA library with rabbit UGT2B13 cDNA as a probe. The insert was recovered by digestion of the plasmid with EcoRI andBamHI, followed by cloning of the insert into pBlueBac4. The UGT1A6 cDNA was cloned from human liver mRNA by reverse transcription followed by PCR using specific primers that span the coding region. The insert was cloned into pBlueBac4. The transfer vectors were then transfected into log phase Spodoptera frugiperdainsect cell (Sf9) monolayers as described (Nguyen and Tukey, 1997).
All of the expressed proteins were active based on catalytic activities toward octyl gallate (UGT1A1, UGT1A3, UGT1A7, UGT1A9, and UGT1A10), androsterone (UGT1A4), 4-nitrophenol (UGT1A6), hyodeoxycholic acid (UGT2B4 and UGT2B7), and 4-hydroxybiphenyl (UGT2B15). Analysis of catalytic activity for these substrates was carried out as described (Strassburg et al., 1996).
Glucuronidation Assays.
The final incubation mixture had a total volume of 200 μl and contained 80 to 400 μM retigabine added in 50 μl of 20% (v/v) aqueous methanol, 10 mM ascorbic acid, 7 mM MgCl2, 100 mM Tris-HCl (pH 8.0), 2.5 mM UDP-glucuronic acid (UDPGA) and 40 μg of total protein except for rat liver microsomes, where 100 μg total protein was used. The reaction was initiated at 37°C by addition of UDPGA. The mixture was then incubated for 40 min at 37°C with gentle shaking and the reaction was stopped by a 2-min incubation at 70°C. This stopping procedure resulted in an almost complete loss of retigabineN-glucuronidation activity (>99%). The amount of retigabine N-glucuronide was decreased during this incubation by about 3%. After sedimentation of the microsomal proteins at 100,000g for 20 min, the supernatant was analyzed by high-performance liquid chromatography (HPLC), as described below.
The methanol concentration of 5% (v/v) in the assay leads to a 8% loss of catalytic activity when compared with a methanol content of 1.25% (data not shown). Activation of retigabine glucuronidation by treatment with Triton X-100, Chaps, or Tween 20 could not be observed with detergent concentrations up to 0.04% (w/v) or up to a detergent to protein ratio of 2, therefore, we did not use detergents in the reaction mixture.
The biosynthesis of retigabine N-glucuronide catalyzed by human and dog liver microsomes was linear as a function of incubation time up to 40 min and as a function of protein content up to 40 μg per assay. Therefore an incubation period of 40 min and a protein content of 40 μg were chosen for subsequent experiments. Because of analogous examinations the incubations in rat liver microsomes yielded a linear range up to of 100 μg of microsomal protein and 40 min of incubation time. These conditions were used for the rat assay.
The synthesis of retigabine N-glucuronide by human lymphoblast-expressed UGT1A1 was linear over time up to 80 min and up to a total protein content of 70 μg. Hence an incubation period of 80 min and a protein content of 70 μg were chosen for subsequent experiments. Because of analogous examinations the incubations with human lymphoblast-expressed UGT1A4 yielded a linear range up to of 70 μg of microsomal protein and 60 min of incubation time. These conditions were used for the UGT1A4 assay. Thus, an incubation mixture had a total volume of 200 μl and consisted of 70 μg of microsomal protein, 100 mM Tris-HCl (pH 8.0), 80 to 400 μM retigabine dissolved in methanol (5% v/v final), 2.5 mM UDPGA, 10 mM ascorbic acid, 7 mM MgCl2, and 12.5 μg/ml alamethicin. Alamethicin is an antibiotic, presumably increasing the substrate availability through pore formation in the microsomal membrane. The remainder of the procedure was performed as described above.
For inhibition experiments bilirubin was added to reaction mixtures containing 360 μM retigabine as a complex with BSA (final concentration: 330 μM bilirubin, 111 μM BSA) to increase the solubility of bilirubin (limit of solubility without albumin ∼30 μM). Control experiments were performed in the absence of bilirubin.
Inhibition experiments with lamotrigine were carried out with reaction mixtures containing 40 μM retigabine and 4 mM lamotrigine, dissolved in dimethyl sulfoxide. The final concentration for dimethyl sulfoxide and methanol in the reaction mixture was 5% (v/v) and 0.5% (v/v), respectively.
Data Analysis.
Apparent Km andVmax values were determined with a nonlinear least-squares fitting program Kinetik 2.4 (Arzneimittelwerk Dresden, Germany). The analyses were done by fitting the unweighted data using an algorithm developed by Marquard (1963). Evaluation of the pharmacokinetic variables of the plasma concentration-time profiles was performed using the pharmacokinetic software TopFit 2.0 (Tanswell and Koup, 1997) by means of noncompartmental analysis. Areas under the plasma concentration-time curve from T0 up to T∞ (AUCs) were determined by the linear trapezoidal rule and the area from the last point to infinity was estimated by triangulation, with the terminal rate constant determined from the slope of the regression line.
HPLC Analysis.
HPLC analyses were carried out using a HPLC system 400 (Kontron Instruments, Neufahrn, Germany) equipped with data system 450-MT2, HPLC pumps 420 and 422, autosampler 465, and UV detector 432. The sample preparation was performed with column switching HPLC techniques (Roth et al., 1981) except for using a 10-port valve instead of two 6-port valves. Briefly, the supernatant, after centrifugation of the incubation mixture, was injected onto a precolumn (extraction column) which was integrated into the HPLC instrument. Here, the sample was concentrated and highly polar compounds, e.g., salts or proteins, were removed by flushing with 20 mM KH2PO4 (pH 7.2), whereas the less polar compounds were adsorbed. Three minutes after the injection the HPLC separation on the analytical column was started with 20 mM KH2PO4 (pH 7.2)/acetonitrile 77:23 (w/w) by switching the 10-port valve. The mobile phase and the washing buffer were gassed for 10 min with helium. Chromatographic conditions are summarized in Table1.
Chromatographic conditions for retigabine and retigabine N-glucuronide determination by HPLC
The identity of retigabine N-glucuronide was verified through the decrease of the 3.5-min peak after β-glucuronidase treatment (Fig. 2). No peak at this retention time could be detected in microsomal incubations where UDPGA was omitted.

Cleavage of retigabine N-glucuronide in human urine by E. coli β-glucuronidase.
The incubation was performed for 145 min at 37°C in 20 mM KH2PO4 (pH 6.8) in the presence of different amounts of β-glucuronidase. ▪: retigabine; ●: retigabineN-glucuronide; ▴: half of the total peak area.
Quantification was performed using calibration curves based on the peak area of separate chromatographed authentic retigabine. The determination of retigabine N-glucuronide was based on the extinction coefficient of retigabine because of the structural similarities of the chromophore of retigabine and itsN-glucuronide.
Human blood plasma retigabine concentrations could not be determined by this method; therefore, the drug was determined by an HPLC assay with fluorescence-detection using the above described column switching technique. Briefly, 500 μl of human blood plasma was concentrated for 6 min with Milli-Q water (flow rate: 1.5 ml/min) on the extraction column (Perisorb RP-8, 10 × 4.6 mm, 30–40 μm; Merck, Darmstadt, Germany). The HPLC separation on the analytical column (LiChrospher 60 select B1, 120 × 4.0 mm, 5 μm; Merck) was started with 1.2 ml/min 75 mM KH2PO4 (pH 3.2)/acetonitrile, 74:26 (v/v), by switching the 10-port valve. Retigabine was detected by fluorescence at an excitation wavelength of 254 nm and an emission wavelength of 372 nm. The HPLC assay was carried out at room temperature, whereas the plasma samples were stored in cooled racks at 15°C before examination. The quantification of D-23129 was performed with calibration curves from spiked control plasma. The lower limit of quantification was about 5 pmol retigabine.
For determination of retigabine concentrations in blood plasma of rats and dogs extraction columns packed with Lichroprep RP-2 of 25 to 40 μm particle size (Merck, Germany) were used. The HPLC separation on the analytical column was performed with 75 mM KH2PO4 (pH 3.2)/acetonitrile, 76:24 (v/v). The remainder of the procedure was as described for retigabine determination in human plasma.
Pharmacokinetic Studies in Humans.
Human blood plasma samples were obtained from a clinical phase I study performed at the Department of Clinical Pharmacology, ASTA Medica AG, Frankfurt. The study was conducted in healthy male Caucasian volunteers between 18 and 45 years old. Retigabine was given over 28 days as capsules in a dosage of 2 × 200 mg per day in the morning and in the evening at least 1/2 h before a meal. On the first day only the morning dose was given. On the first day blood samples were obtained predose and at 0.33, 0.66, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 16, and 24 h. On days 3, 4, 5, 6, 7, 14, 21, 26, 27, and 28 blood sampling was taken in the morning before the next drug intake.
Additionally the blood plasma of two male healthy volunteers who received a single dose of 600 mg retigabine in capsules was analyzed. Blood samples were taken predose as well as at 0.33, 0.66, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, and 72 h after drug administration.
Samples of blood were collected using lithium heparin in Sarstedt Monovette plastic tubes and mixed gently. The tubes were then centrifuged at 3400 rpm for 10 min at 4°C. Plasma was removed and stored in polypropylene tubes at −20°C until HPLC analysis. The results from the phase I study will be reported elsewhere.
Pharmacokinetic Studies in Animals.
The pharmacokinetic studies in Beagle dogs after oral dosing of 8.25 mg/kg [14C]retigabine were performed as described (Hempel et al., 1999). Plasma samples of rats were taken from a pharmacokinetic study in Wistar rats after a single oral dose of 8.25 mg/kg [14C]retigabine according to the protocol described by Hempel et al. (1999).
Total radioactivity in dog and rat blood plasma was determined by liquid scintillation counting.
Results
HPLC Analysis of Retigabine and RetigabineN-Glucuronide.
To identify the formation of retigabine N-glucuronide in vitro in microsomal incubates and in vivo in plasma and urine we developed an HPLC method for the simultaneous determination of retigabine and retigabine N-glucuronide (Fig.3). Sample preparation was integrated into the isocratic HPLC method using a precolumn enrichment/clean-up method that yields clean samples in combination with a >98% recovery for both retigabine and retigabine N-glucuronide. The detection was linear up to 1.6 nmol for retigabine and 1.2 nmol for retigabine N-glucuronide, respectively. The limit of detection was 0.15 nmol (retigabine) and 0.05 nmol (retigabineN-glucuronide) based on a signal-to-noise ratio of 1:3.

HPLC separation of retigabine N-glucuronide (peak 1) and retigabine (peak 2) after incubation of retigabine with human liver microsomes in the presence of UDPGA (top), human urine (middle), and human blood plasma (bottom).
UV absorption was monitored at 220 nm.
Figure 2 shows the cleavage of retigabine N-glucuronide by E. coli β-glucuronidase. The sum of the peak areas of retigabine and retigabine N-glucuronide were nearly constant over all points. This is evidence that the extinction coefficients of retigabine and its N-glucuronide are similar. This enabled us to determine the concentration of retigabine N-glucuronide using the response factor of retigabine as described in Materials and Methods.
Humans and Dogs, but Not Rats, Show a Constant Ratio between Retigabine and Its N-Glucuronide In Vivo.
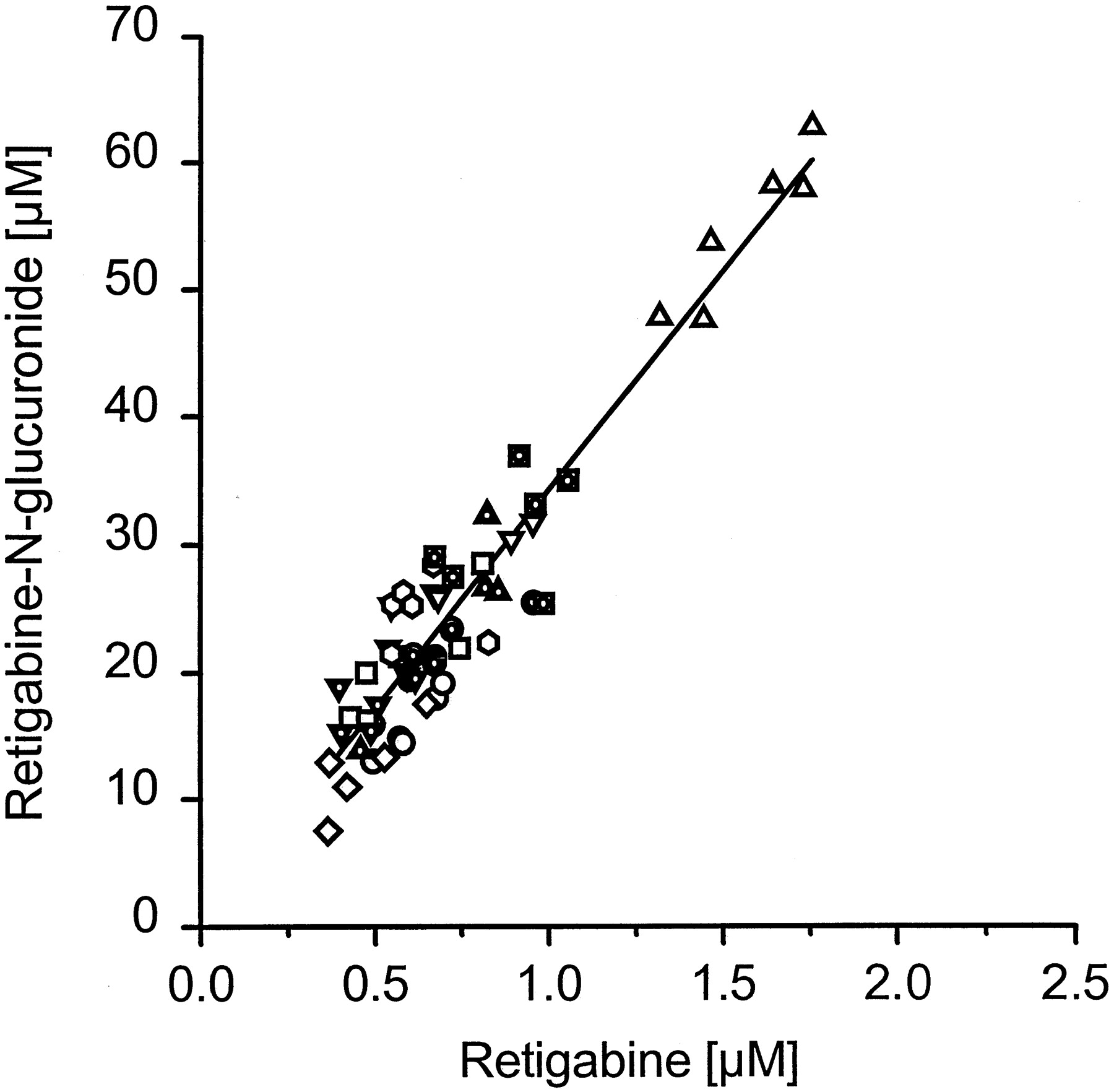
When we determined the concentration of retigabine and retigabineN-glucuronide in plasma of a human volunteer, we found rapid and extensive N-glucuronide formation as early as 20 min after dosing (Fig. 4). Retigabine and retigabine N-glucuronide reached the maximum plasma concentration at 40 min and 1.5 h, respectively.N-glucuronide concentrations exceeded those of retigabine by a factor of 24 ± 10-fold. In accord with this, the AUC was 24-fold larger for retigabine N-glucuronide than for retigabine. The parallel decline of plasma levels that we observed in this volunteer and two other volunteers at a higher dose (Table2) suggested to us that a constant ratio exists between retigabine and its N-glucuronide. This is also suggested by the very similar terminal half-lives of 11.4 and 12.3 h for retigabine and retigabine N-glucuronide, respectively. This factor was slightly higher (35 ± 5) when retigabine was given to the same volunteer in a multiple dose study over another 27 days. To investigate whether this ratio is invariant in different volunteers, we determined plasma concentrations of retigabine and retigabine N-glucuronide of ten volunteers after multiple dosing on six different days. As shown in Fig.5 the individual data points lie close to a straight line. This indicates that the ratio of retigabineN-glucuronide to retigabine is constant in different individuals (p < .001).
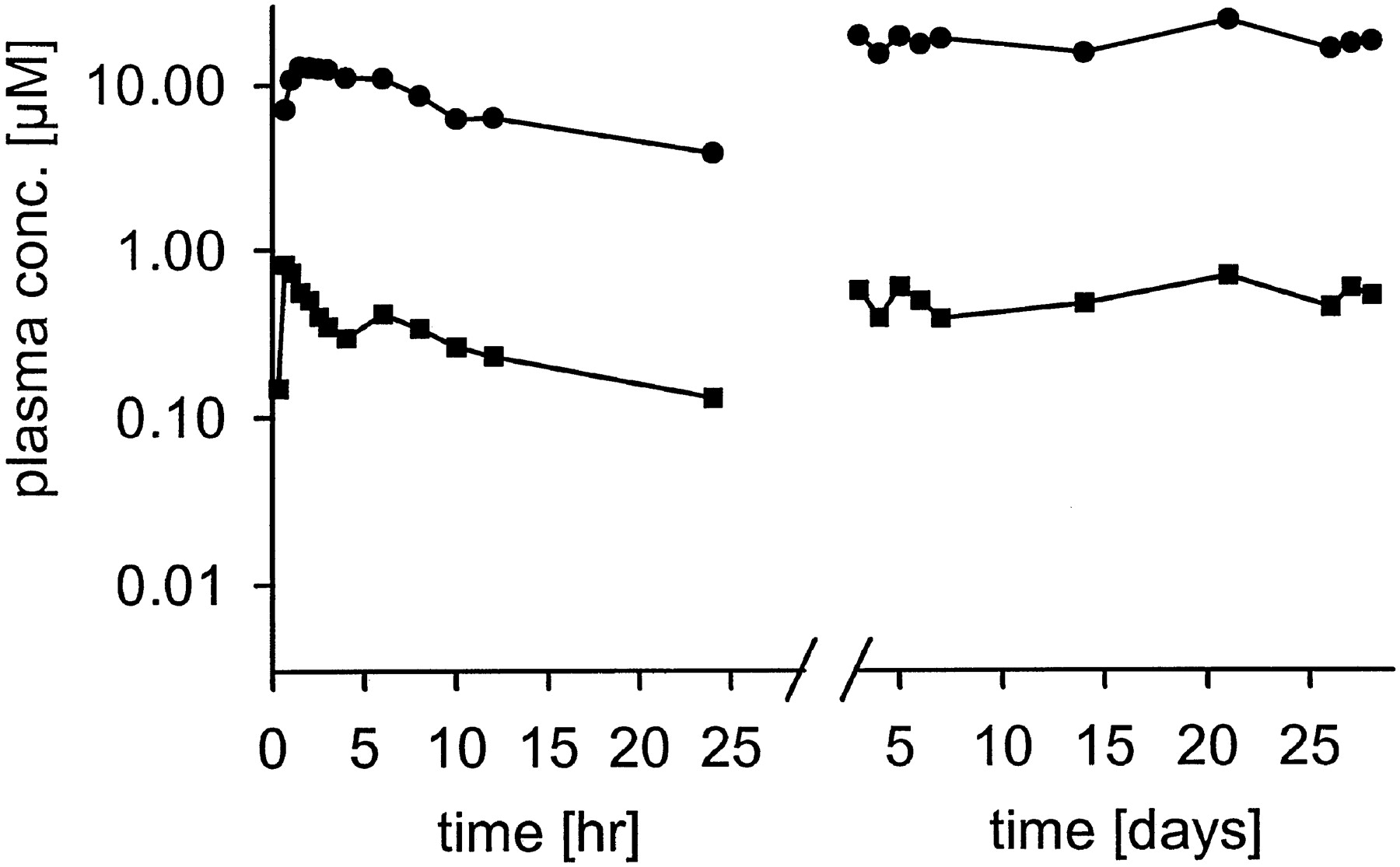
Pharmacokinetics of retigabine and retigabine N-glucuronide in a human volunteer after single (left) and multiple (right) oral administration of 200 mg retigabine.
Blood sampling was taken in the morning before the next drug intake. ●: retigabine N-glucuronide; ▪: retigabine.
Comparison of pharmacokinetic data for retigabine and retigabine N-glucuronide in healthy volunteers after the indicated single dose of retigabine

Retigabine and retigabine N-glucuronide plasma concentrations of ten volunteers at days 3 to 7 and 14 after multiple oral administration of 200 mg retigabine.
Blood sampling was taken in the morning before the next drug intake. Each symbol represents the values for one volunteer.
Similar to our findings in humans we found evidence for the existence of a constant ratio between retigabine and its N-glucuronide in dogs. After oral administration of 8.25 mg/kg [14C]retigabine we could observe a parallel decline of total plasma radioactivity and retigabine concentration (Fig. 6). As retigabineN-glucuronide is the major metabolite in the circulation system of dogs (Hempel et al., 1999), its concentration can be approximated as the difference between total radioactivity and retigabine concentration. The terminal half lives of retigabine as well as of retigabine N-glucuronide were determined to be approximately 5 h. The calculated AUC for retigabineN-glucuronide was 25 ± 12 (n = 12)-fold higher than the corresponding value for retigabine.
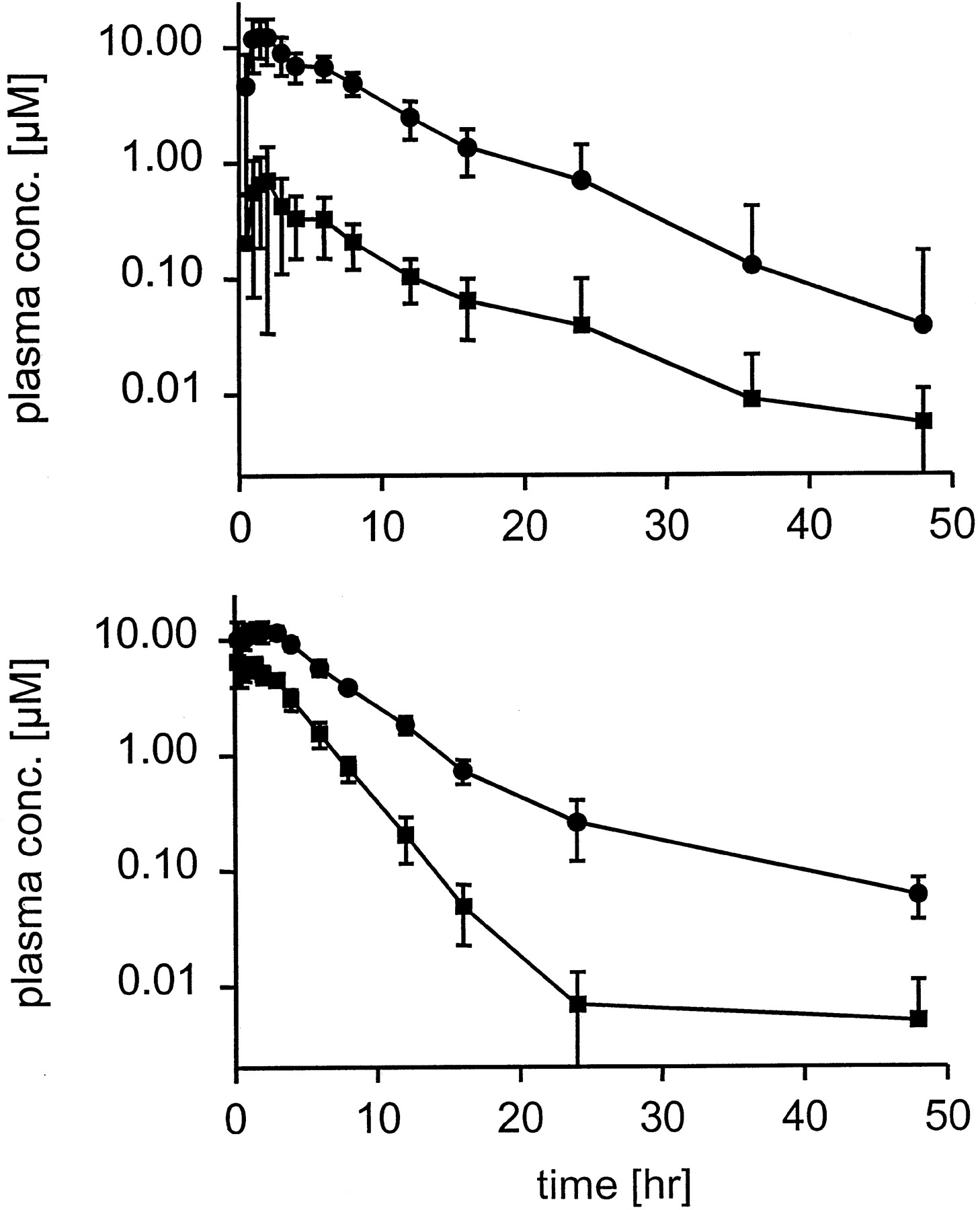
Pharmacokinetics of retigabine (▪) and total radioactivity (●) in dogs (top, n = 12) and rats (bottom, n = 12) after single oral administration of 8.25 mg/kg [14C]retigabine.
Data are represented as mean ± S.D.
Contrary to the findings in humans and dogs we could not observe this constant ratio in rats. After oral dosing of 8.25 mg/kg [14C]retigabine we found a pronounced difference in the decline of retigabine and total radioactivity (Fig.6). The terminal half-life of retigabine was determined to be in the range of 2 h, whereas the corresponding half-life of total radioactivity was found to be 3.3 h, i.e., 1.6-fold higher. The AUC of total radioactivity exceeded that of the unchanged drug by a factor of 3.
Michaelis-Menten Kinetic Analysis of RetigabineN-Glucuronidation.
To investigate whether the different findings in rats in vivo can be explained by the different kinetics of retigabineN-glucuronide formation in vitro we investigated retigabine glucuronidation in liver microsomal fractions from dog, rat, and humans.
In the presence of 5% (v/v) methanol the maximum solubility of retigabine was 400 μM as determined by visual inspection of the samples after a 30-min incubation at room temperature. Due to the limited water solubility of retigabine, it was not possible to achieve concentrations that approached Vmax. The apparent kinetic constants Km andVmax for the N-glucuronidation of retigabine in human liver microsomes were 145 ± 39 μM and 1.2 ± 0.3 nmol · min−1 · mg−1protein respectively (Fig. 7). The corresponding values for dog and rat liver microsomes are given in Table 3. Because of the limited solubility of retigabine the Km of 420 μM in rat liver microsomes is only a rough estimate. These data show that the decreased activity of rat liver microsomes is due to both decreasedVmax and increasedKm when compared to dogs or human liver microsomes. Although dog and human liver microsomes show similarVmax, apparentKm is higher in humans. The enzyme efficiency (Vmax/Km) is therefore decreasing from dog to human to rat (Table 3).

Michaelis-Menten kinetics of glucuronidation of retigabine by human liver microsomes.
Microsomes (40 μg total protein) were incubated 40 min in the presence of 2.5 mM UDPGA and increasing amounts of retigabine. Inset, respective Lineweaver-Burk plot where the graph was calculated using the values obtained from nonlinear regression. The curve represents a single experiment where each data point was determined in triplicate. Data are shown as mean ± S.D.
Michaelis-Menten parameters of retigabine glucuronidation in liver microsomal fractions of man, rat, and dog. Values are the mean ± S.D. (man: four liver samples (HGW10, HGW11, HGW12, and HGW13 microsomes of Fig. 8); rat: four animals; dog: 1 animal, 2 independent determinations).
Interindividual Variation of Retigabine Glucuronidation in Humans In Vitro.
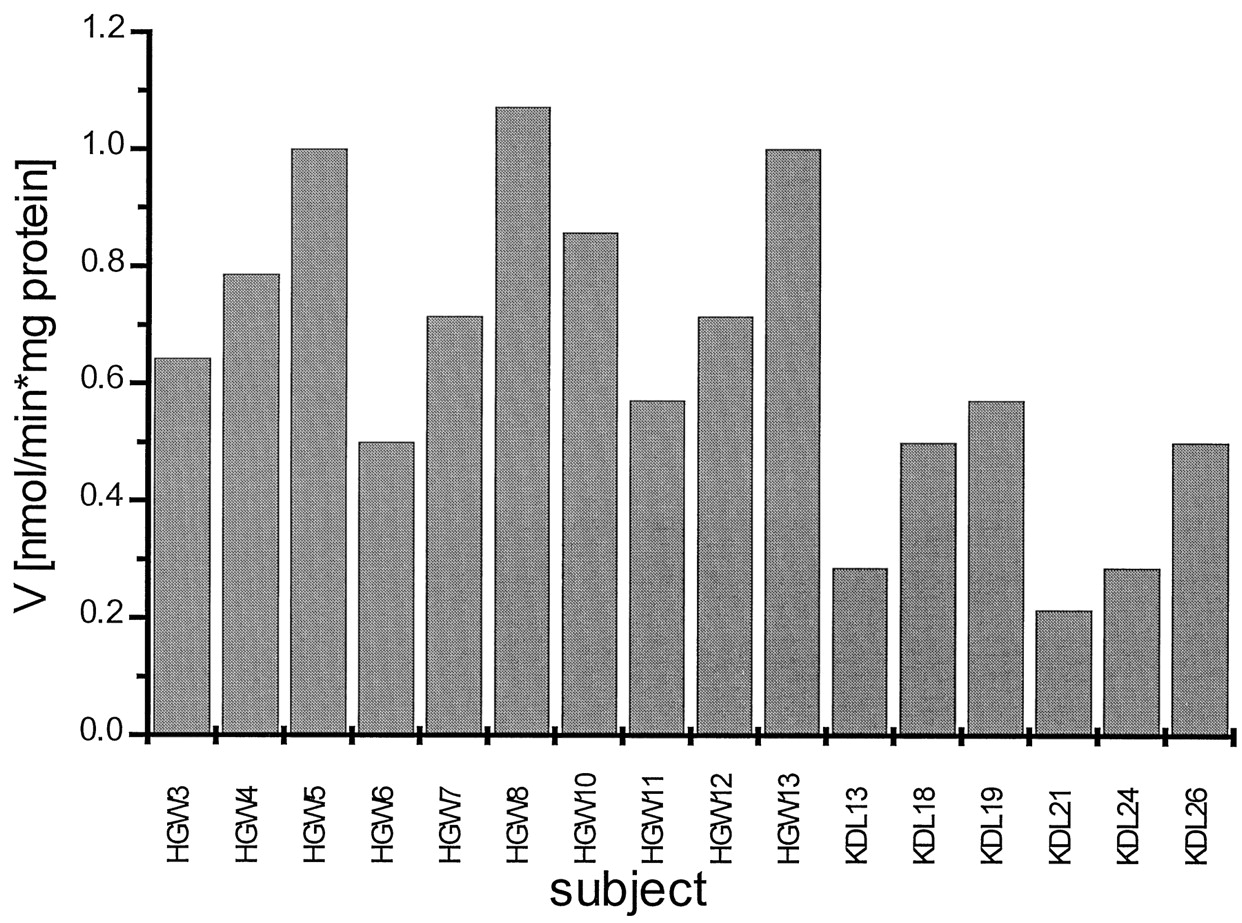
Figure 8 shows the interindividual variation of retigabine glucuronidation in 16 individual samples of human liver microsomes. Saturation conditions were not reached because concentrations of retigabine higher than 400 μM exceed the solubility in our incubations. We therefore used a retigabine concentration of 360 μM, which is 2.5-fold higher than the apparentKm. From the Michaelis-Menten equation, it follows that at this concentration a velocity of 70% ofVmax is obtained.

Interindividual variability of retigabine glucuronidation by human liver microsomes of different subjects.
Retigabine was used at a concentration of 360 μM. Each data point is represented as the mean of two independent determinations.
Large interindividual variations were observed in vitro, and differences up to 5-fold were detected. In this population the average specific activity was 0.64 nmol · min−1 · mg−1protein and the coefficient of variation was 0.41 (S.D./mean). It is interesting to note that in vivo we also found an ∼5-fold variation of plasma levels of retigabine N-glucuronide whereas the ratio between retigabine N-glucuronide and retigabine was unaffected. This suggests that the variableN-glucuronidation activity that we detected in vitro is not responsible for the variation of retigabine N-glucuronide levels in plasma in vivo.
Retigabine Is Glucuronidated by Multiple Isoenzymes.
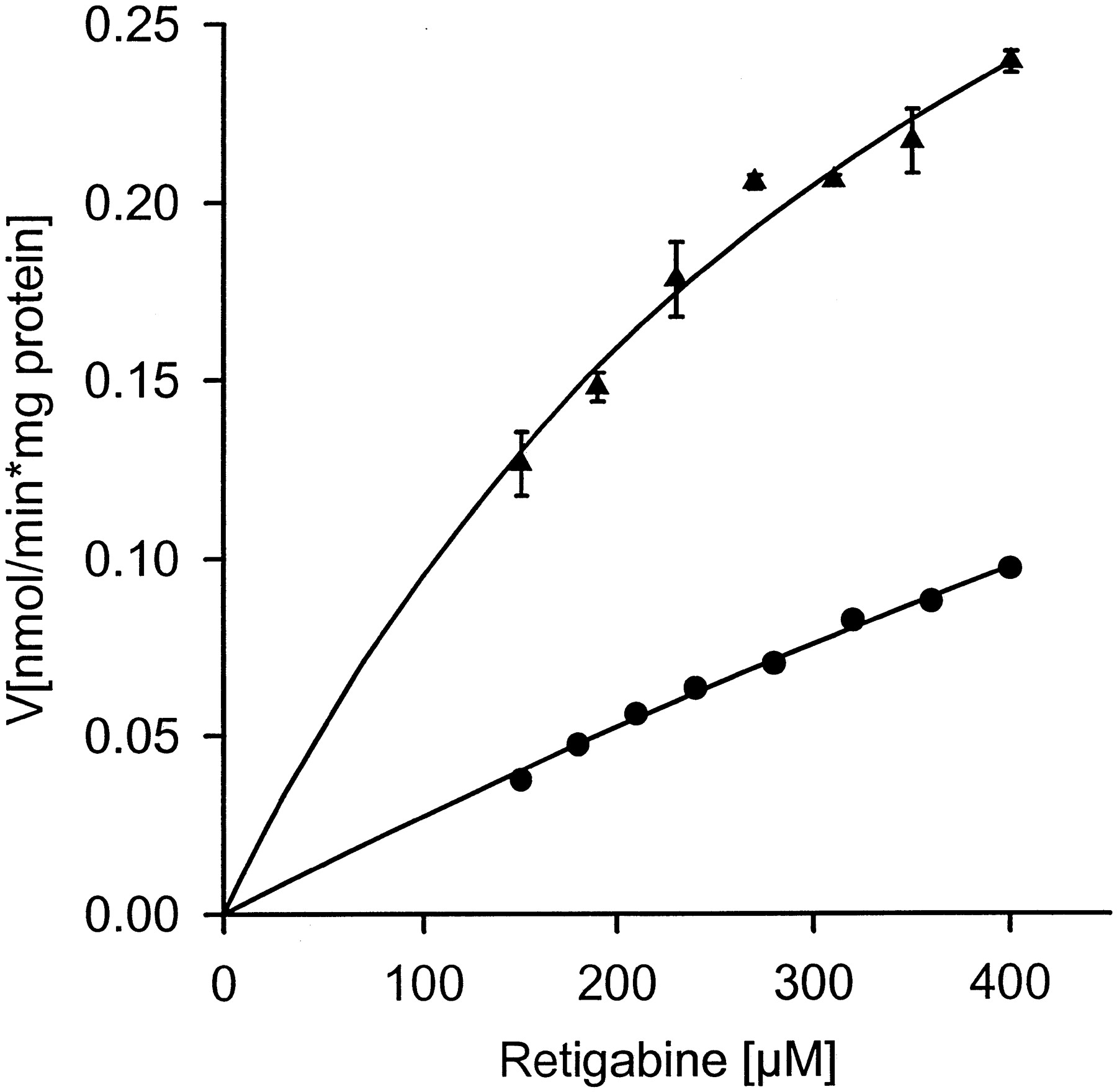
Retigabine was glucuronidated by liver microsomes from Gunn rats, which are deficient in the functional expression of the family of UGT1 enzymes. The plot of N-glucuronidation activity versus substrate concentration (Fig. 9) was nearly linear up to the maximal retigabine concentration of 400 μM. Hence the Km is much higher than 400 μM. A nonlinear regression yielded an apparentKm of 1800 ± 600 μM. Thus an exact determination of the Michaelis-Menten constant, which should be in the millimolar range, was not possible. The glucuronidation activity in Gunn rat liver microsomes at a retigabine concentration of 400 μM was 0.10 nmol · min−1 · mg−1protein, i.e., 42% of the glucuronidation activity in liver microsomes of wild-type Wistar rats.

Kinetics of retigabine glucuronidation in Wistar (▴) and Gunn rat liver microsomes (●).
100 μg total protein were incubated 40 min in the presence of 2.5 mM UDPGA and increasing amounts of retigabine. Each curve represents the results of a single experiment, whereas each data point is shown as the mean ± S.D. of two (Wistar rat) and three (Gunn rat) determinations, respectively. The error bars of the data from Gunn rat liver microsomes are almost completely invisible.
We tested a range of human recombinant UGTs expressed in insect cells for their ability to glucuronidate retigabine (Table4). Among ten different isoenzymes UGT1A1, UGT1A3, UGT1A4, and UGT1A9 produced retigabineN-glucuronide. UGT1A6, UGT1A7, UGT1A10, UGT2B4, UGT2B7, and UGT2B15 did not catalyze retigabine N-glucuronidation. It is known that UGT1A4 is involved in the N-glucuronidation of a number of substrates (Green and Tephly, 1998). Therefore, we tested the inhibition of retigabine N-glucuronidation by lamotrigine, which is mainly glucuronidated by UGT1A4 (Green and Tephly, 1998). In the presence of 4 mM lamotrigine the formation of retigabineN-glucuronide is decreased to 21 ± 3% of control values, as tested with liver microsomes of three subjects. UGT1A1 and UGT1A4 are the two bilirubin-glucuronidating enzymes in humans. Therefore, we tested the inhibition of retigabine glucuronidation in human liver microsomes of four subjects. In the presence of 330 μM bilirubin the formation of retigabine N-glucuronide is decreased to about 75 ± 7% of control values (Fig.10). However, in rat liver microsomes the presence of 330 μM bilirubin decreases the amount of formed retigabine N-glucuronide to about 10% in two independent determinations of different animals (Fig. 10). Control experiments were performed with recombinant human UGT1A1, whereas retigabine glucuronidation was completely inhibited in the presence of 330 μM bilirubin. The retigabine N-glucuronidation activity by human UGT1A4 was decreased under these conditions to about 30% (data not shown).
Retigabine N-glucuronidation by human recombinant UGTs. Incubations contained 100 μg of total protein and 400 μM of retigabine and were incubated for 90 min at 37°C. UGT1A6, UGT1A7, UGT1A10, UGT2B4, UGT2B7, and UGT2B15 did not catalyze retigabine-N-glucuronidation.

Inhibition of retigabine N-glucuronidation by bilirubin in human and rat liver microsomes.
Values represent the percentage of retigabineN-glucuronide formed in the presence of 330 μM bilirubin/111 μM BSA normalized to control values without bilirubin. Data are the mean ± S.D. from four (human) and two (rat) experiments performed in duplicate, respectively.
The apparent Km of retigabineN-glucuronidation in human UGT1A1 and UGT1A4 was determined to be 460 μM and 322 μM, respectively. These values represent the mean of two independent determinations. Because of the limited solubility of retigabine these values are only rough estimates.
Discussion
In this study we present evidence that the new anticonvulsant retigabine is extensively N-glucuronidated in humans and that the ratio of retigabine to retigabine N-glucuronide is constant in all volunteers tested. This constant ratio between retigabine and its N-glucuronide suggests that the concentrations of these two compounds are coupled via enterohepatic circulation (EHC) and glucuronidation/deglucuronidation reactions. Here we have identified components of this potential mechanism. On the one hand, efficient glucuronidation of retigabine can be attributed to microsomal UDPGA-dependent glucuronosyl transferases. On the other hand, we show that bacterial glucuronidases cleave retigabineN-glucuronide to form retigabine. Such a mechanism could be the enzymatic basis for an efficient EHC in humans where retigabineN-glucuronide is formed in the liver (or other tissues) and is cleaved subsequently in the gut. Reabsorption of the cleaved retigabine would complete the enterohepatic cycle. Because of the quantitative importance and to explain distinctive species differences of N-glucuronide formation in vivo, we have analyzed the mechanism of retigabine N-glucuronidation in more detail using in vitro human and animal experiments.
One aim of the present study was to investigate the pharmacokinetics of retigabine N-glucuronide in human blood plasma. The concentrations of retigabine N-glucuronide and the unchanged drug after oral dosing of 200 or 600 mg retigabine are shown in Fig. 4. As can be seen from the AUC ratio of retigabineN-glucuronide and retigabine, the metabolite exceeds retigabine by about 25-fold at the 200-mg dose (Table 2). The terminal half-lives for both retigabine and retigabine N-glucuronide are very similar with a T1/2 of about 12 h. Moreover, retigabine N-glucuronide concentration normalized to the unchanged drug remained nearly constant in human blood plasma of ten volunteers after multiple oral dosage of retigabine. We speculate that the ratio between the two compounds is regulated through an EHC of retigabine in humans. The pharmacokinetic parameters from dogs for retigabine and its N-glucuronide were similar to our findings in humans (see Results, Fig.6).
A parallel decay of the unchanged drug and its N-glucuronide in a semilogarithmic plot was also reported for lorazepam in ponies (Greenblatt and Engelking, 1988) and for lamotrigine in guinea pigs (Remmel and Sinz, 1991). This pharmacokinetic behavior is probably due to an EHC. An EHC was also described for lorazepam glucuronide in humans (Herman et al., 1989) and acetaminophen glucuronide in rats (Watari et al., 1984). Siegers et al. (1983) reported the EHC of paracetamol and its glucuronide and sulfate conjugates.
The similarity of pharmacokinetics of retigabine and itsN-glucuronide in humans and dogs and the dissimilarity to the findings in rats led us to study the kinetic data for in vitro glucuronidation of retigabine in dog, rat, and human liver microsomes (Table 3). The apparent Km for retigabine glucuronidation in human liver microsomes was similar to the value obtained by McNeilly et al. (1997). We attribute the lowerVmax determined by McNeilly to a difference in the experimental protocol. The apparentVmax value of 1.2 ± 0.3 nmol · min−1 · mg−1was determined as the mean ± S.D. of the four human liver samples HGW10, HGW11, HGW11, and HGW12. Relative to the population of Fig. 8, these samples have relatively high Vmaxvalues. To determine the extent of interindividual variation in the human glucuronidation in vitro, microsomes from 16 human livers were analyzed for retigabine N-glucuronide formation. We found a 5-fold difference in the glucuronidation activity between the different livers. This corresponds to the in vivo observed variation of retigabine N-glucuronide concentrations in human blood plasma of ten different subjects (Fig. 5).
Because of the importance of the glucuronidation reaction for the metabolic fate of retigabine, we asked whether isoforms of UGTs that contribute to retigabine glucuronidation could be identified. The determination of the UGT isoforms that glucuronidate retigabine in rat and humans was approached in several ways: 1) we used expressed human UGT isoenzymes, 2) we inhibited retigabine glucuronidation by bilirubin and lamotrigine, and 3) we used genetically deficient Gunn rats (Owens and Ritter, 1992). We show that among ten different human recombinant UGT isoenzymes, UGT1A1, UGT1A3, UGT1A4, and UGT1A9 glucuronidated retigabine. Lamotrigine, which is mainly glucuronidated by UGT1A4 (Magdalou et al., 1992; Green and Tephly, 1998), inhibited the in vitro retigabine N-glucuronidation in human liver microsomes by about 80%. It is therefore likely that a major part of retigabineN-glucuronide is formed by this UGT isoenzyme. In humans, two isoenzymes, UGT1A1 and UGT1A4, are forming bilirubin glucuronides. Among these, UGT1A4 plays only a minor role as shown by a mRNA ratio of 5:2 (Ritter et al., 1992). Formation of retigabineN-glucuronidation was inhibited by only 25 ± 7% of control activity in the presence of 330 μM bilirubin. Because we identified UGT1A3 and UGT1A9 as retigabine N-glucuronidating enzymes, it is likely that these enzymes contribute to the residual activity.
In rat liver microsomes retigabine N-glucuronidation can be inhibited by 90% in the presence of 330 μM bilirubin. Bilirubin glucuronidation in rats is catalyzed mainly by UGT1A1 and to a lower extent by UGT1A2. The Km value for bilirubin-glucuronidation in rat liver microsomes is 0.9 μM (Vanstapel and Blanckaert, 1987). Hence, in rat liver microsomes the bilirubin glucuronidating enzymes UGT1A1 and UGT1A2 possess a greater importance for retigabine glucuronidation when compared with humans.
These findings were further confirmed with results from glucuronidation experiments with Gunn rat liver microsomes. The Gunn rat does not express functional isoenzymes of UGT family 1 and therefore completely lacks glucuronidation activity toward bilirubin and digitoxigenin monodigitoxoside (Owens and Ritter, 1992). Our finding that retigabineN-glucuronide is formed in Gunn rat liver microsomes with approximately 40% of the activity of Wistar rat liver microsomes indicates that family 2 isoenzymes contribute to retigabineN-glucuronidation in rats.
From a clinical point of view, the biotransformation of retigabine predominantly by glucuronidation is considered to be favorable to the future treatment of patients.
First, glucuronidation is a robust metabolic step preserved even in advanced stages of liver diseases (Hoyumpa and Schenker, 1991). Second, the isoenzymes of glucuronyltransferases are known to have a wide substrate specificity and the biotransformation of exogenous compounds is usually not related to one specific enzyme. This is also true for retigabine. Third, we have found a constant ratio between retigabine and its N-glucuronide in vivo in different volunteers. The observed glucuronidation variability in vitro is consequently not relevant for the plasma clearance of retigabine.
Therefore, it is unlikely that the presence of hepatic impairment or genetic lesions (Ritter et al., 1993) associated with a reduced hepatic glucuronidating activity of one isoenzyme (e.g., Gilbert’s syndrome, Crigler-Najar syndrome type II) may have substantial impact on retigabine clearance.
In summary we have shown that the constant ratio between retigabine and retigabine N-glucuronide in vivo is likely to be the result of an EHC that is mechanistically based on reversible glucuronidation-deglucuronidation reactions in humans and dog. This mechanism probably prolongs the terminal half-life of retigabine in the plasma of dogs and humans. Retigabine N-glucuronidation is mainly catalyzed by UGT1A4 in humans. Additionally, human UGT1A1, UGT1A3, and UGT1A9 also contribute to retigabineN-glucuronidation. In the rat most of retigabineN-glucuronidation is catalyzed by UGT1A1 and UGT1A2, whereas family 2 UGT enzymes also contribute to the metabolism of retigabine.
Acknowledgments
We thank U.A. Meyer, Biocenter of the University of Basel, Switzerland and W. Siegmund, Institute of Clinical Pharmacology, University of Greifswald, Germany, for the supply of human liver tissue.
Footnotes
-
Send reprint requests to: Dr. Thomas Kronbach, Corporate Research & Development ASTA Medica Group, Biochemistry Dresden, Arzneimittelwerk Dresden GmbH, Meiβner Str. 191, D-01445 Radebeul, Germany. E-mail: Dr_Thomas.Kronbach{at}astamedica.de
-
This work was supported in part by grants from the Sächsisches Ministerium für Arbeit und Wirtschaft (project number 1791) and by United States Public Health Service Grant GM49135 (R.H.T).
- Abbreviations used are::
- UGT
- UDP-glucuronosyltransferase
- AUC
- area under the plasma concentration-time curve from t0 up to t∞
- HPLC
- high-performance liquid chromatography
- EHC
- enterohepatic circulation
- UDPGA
- UDP-glucuronic acid
- Received July 22, 1998.
- Accepted January 25, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}