


Abstract
Tacrolimus is a macrolide lactone with potent immunosuppressive properties. It has been shown in clinical studies to prevent allograft rejection. The pharmacokinetics of tacrolimus in healthy subjects and transplant patients has been described in earlier studies using immunoassay methods; however, detailed information on the absorption, distribution, metabolism, and excretion of tacrolimus using a radiolabeled drug is lacking. The objective of the present study was to characterize the disposition of tacrolimus after single i.v. (0.01 mg/kg) and oral (0.05 mg/kg) administration of 14C-labeled drug in six healthy subjects. Tacrolimus was absorbed rapidly after oral dosing with a mean Cmax andTmax of 42 ng/ml and 1 h, respectively. The oral bioavailability was about 20%. After i.v. and oral dosing, most of the administered dose was recovered in feces, suggesting that bile is the principal route of elimination. Urinary excretion accounted for less than 3% of total administered dose. In systemic circulation, unchanged parent compound accounted for nearly all the radioactivity; however, less than 0.5% of unchanged drug was detectable in feces and urine. The excretion of the metabolites was formation-rate-limited. The mean total body clearance at 37.5 ml/min was equivalent to about 3% of the liver blood flow. Renal clearance was less than 1% of the total body clearance. The mean elimination half-life was 44 h.
Tacrolimus, [3S-[3R*[E(1S*,3S*,4S*)]4S*,5R*,8S*,9E,12R*, 14R*,15S*,16R*,18S*,19S*,26aR*]]-5,6,8,11,12,13,14,15,16, 17,18,19,24,25,26,26a-hexadecahydro-5,19-dihydroxy-3-[2-(4-hydroxy-3-methoxycyclohexyl)-1-methylethylenyl]-14,16-dimethoxy-4,10,12,18-tetramethyl-8-(2-propenyl)-15,19-epoxy-3H-pyrido[2,1-c] [1,4]oxaazacyclotricosine-1,7,20,21(4H,23H)-tetrone, monohydrate (FK506; Prograf), is a potent immunosuppressive agent developed by Fujisawa, Japan. Tacrolimus is a macrolide obtained from Streptomyces tsukubaensis, which has been shown in preclinical and clinical studies to prevent allograft rejection (Yoshimura and Oka, 1990; European FK506 Multicentre Liver Study Group, 1994; European Tacrolimus Multicentre Renal Study Group, 1997). After binding to the intracellular immunophilin FK506-binding protein, tacrolimus blocks intermediate steps in the pathway that links early membrane-associated events with gene expression and inhibits T and B cell proliferation. Its selective effect on the immune system appears to be due to selectivity for a subset of calcium-associated signal transduction pathways that may predominate in the T cell receptor-mediated cascade leading to cytokine production.
The pharmacokinetics of tacrolimus in healthy volunteers and transplant patients has been described in studies using enzyme immunoassays to determine concentrations of tacrolimus in blood and plasma. These studies indicate that tacrolimus is bound strongly to plasma proteins and erythrocytes, that the ratio of blood/plasma concentrations is 20:1 (Venkataramanan et al., 1990; Beysens et al., 1991), and that pharmacokinetic parameters calculated from measurement of whole-blood concentrations are different from those derived from plasma concentrations. The terminal elimination half-life calculated from whole blood and from plasma was 23 to 40 h and 27 to 65 h, respectively. Estimates of total body clearance also differ greatly (1.2–2.2 liters/h in whole blood and 18–49 liters/h in plasma). Detailed information on the absorption, distribution, metabolism, and excretion of tacrolimus using a radiolabel assay is lacking.
The objectives of the present study were to characterize the pharmacokinetics of tacrolimus in whole blood after i.v. and oral administration of 14C-labeled drug in healthy volunteers.
Materials and Methods
Subjects.
After being informed of the purpose, design, and potential risks of the study, six subjects gave written informed consent to participate. The study was approved by the local Ethics Committee. The subjects were healthy, male, and white nonsmokers, aged 49 to 67 years, with no evidence of metabolic or other disease or drug abuse, who had alcohol consumption of <1 liter of beer/day or the equivalent and had not received a radioisotope within the previous 12 months. Medication other than paracetamol was not permitted; however, one subject received treatment for facial paresis (not drug-related) from day 8 of the study. All subjects were negative for HIV and hepatitis B surface antigen.
Subjects were randomized in a nonblinded crossover study design to receive single doses of 14C-labeled tacrolimus i.v. and orally separated by a washout period of 14 days. Each was admitted on the evening before administration and remained in the unit until the morning of the 12th day thereafter. Samples of blood and feces were taken until discharge from the unit.
The subjects’ mean age was 59.0 years (S.D. = 6.2; range, 49–62) and their mean weight was 77.5 kg (S.D. = 3.6; range, 72–82). Laboratory and ECG parameters were appropriate for this age group in all subjects and did not differ significantly from normal. There was no difference between pre- and poststudy parameters. No subject withdrew from the study.
Dosage and Administration.
The i.v. dose was administered as a 4-h infusion (5% dextrose solution) at a nominal dose of 20 μg/kg14C-labeled tacrolimus adjusted to a maximum dose of radioactivity of 30 μCi. Blood samples were taken from an appropriate vein in the opposite arm.
The oral dose was formulated in polyethylene glycol solution and administered in a single hard-gelatin capsule with 100 ml of water while the subject was in an upright sitting position that was maintained for 30 min after administration. The oral dose of14C-labeled tacrolimus was approximately 50 μg/kg (range, 44–51 μg/kg), adjusted to a maximum dose of radioactivity of 70 μCi.
The volunteers had fasted for at least 10 h before and for 4 h after the dose, at which time they received a light lunch; they received decaffeinated coffee or rose hip tea after 8 h and dinner after 10 h. On the remaining days, they received standardized hospital meals.
Assays.
Total radioactivity (tacrolimus and metabolites) was measured by liquid scintillation counting (LSC)1 (Iwasaki et al., 1995); the concentration of tacrolimus was measured by enzyme-linked immunosorbent assay (ELISA) in blood, plasma, feces, and urine (Iwasaki et al., 1991).
Pharmacokinetics.
Pharmacokinetic parameters were calculated by a compartment-independent model. The area under the concentration–time profile (AUC) was estimated using the log/linear trapezoidal rule. The AUC to infinite time (AUC0-∞) was defined as the area under the curve to the time of the last measured concentration above the limit of quantification (AUC0-t) plus the remaining area extrapolated to infinity (AUCt-∞) estimated using the following equation:
The total clearance (CL) was determined after i.v. administration by the ratio; CL = D/AUC(0-∞) and the volume of distribution (Vd) was determined by CL*MRT, with MRT being the mean residence time after i.v. infusion.
The terminal half-life was calculated using an unweighted log/linear regression of the last concentration–time points of the monoexponential terminal phase. Cumulative excretion in urine and feces was calculated from the time of administration up to 264 h after dosing or until the last measurement at which the concentration was above the lower limit of quantification.
Means of total radioactivity and tacrolimus concentrations in blood and plasma were calculated only if at least four individual data points were above the lower limit of quantification. Statistical analysis was carried out with the Statistical Analysis System, version 6.0 (SAS Institute, Inc., Cary, NC).
Results
The mean total radioactivity (expressed as tacrolimus equivalents), measured by LSC, and the mean concentrations of tacrolimus, measured by ELISA, in whole blood after i.v. and oral dosing are summarized in Table 1, and the corresponding plasma values are shown in Table2. No radioactivity was detectable in plasma after i.v. infusion. These data are illustrated graphically in Fig. 1.
Mean (±S.D.) total radioactivity (LSC), expressed as tacrolimus equivalents, and concentrations of tacrolimus (ELISA) in whole blood after a single i.v. infusion and a single oral dose of14C-labeled tacrolimus
Mean (±S.D.) total radioactivity (LSC), expressed as tacrolimus equivalents, and concentrations of tacrolimus (ELISA) in plasma after a single i.v. infusion and a single oral dose of 14C-labeled tacrolimus
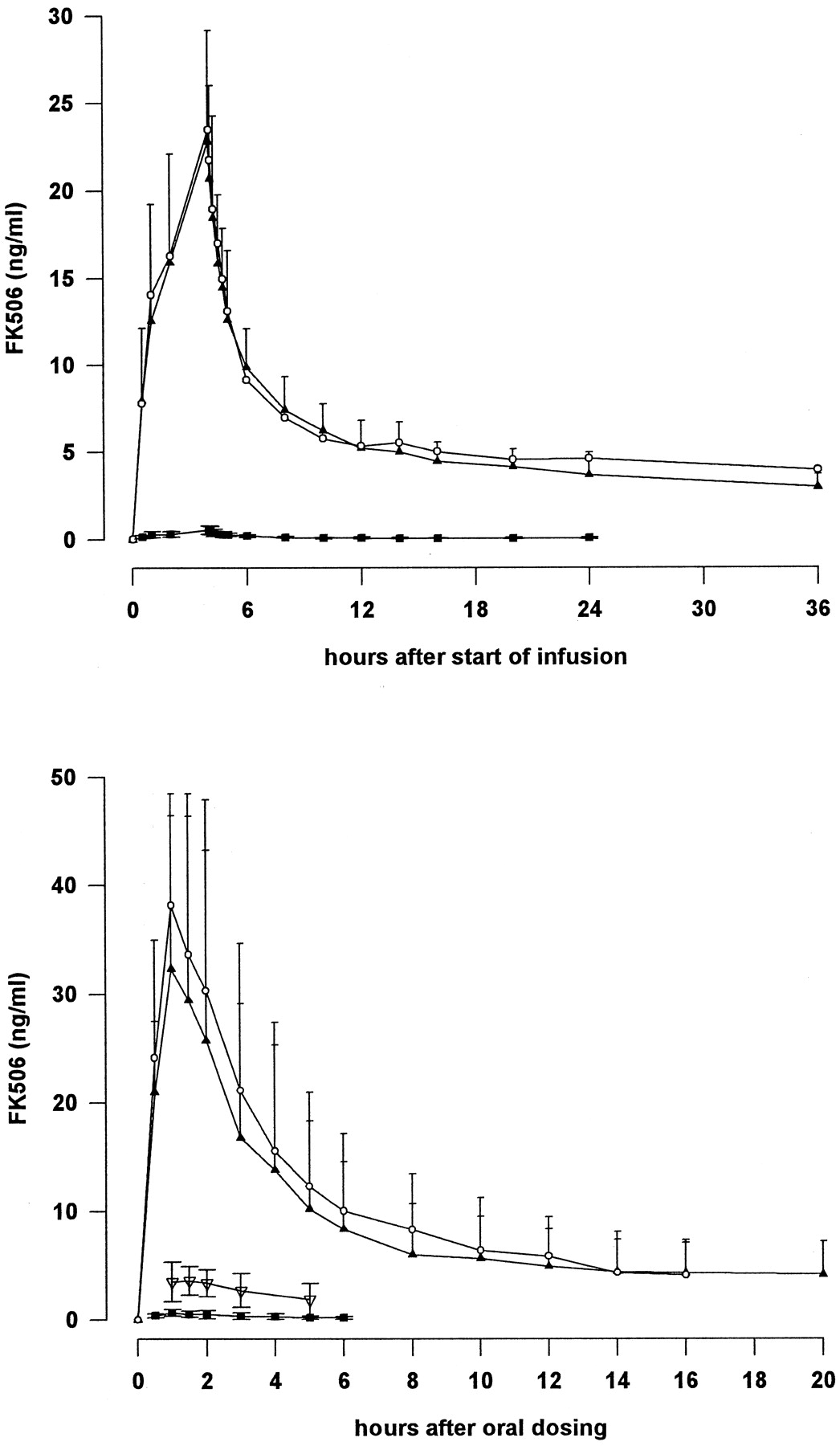
Mean total radioactivity (LSC) and tacrolimus concentration (ELISA) in whole blood and plasma after single i.v. infusion and single oral administration of 14C-labeled tacrolimus. ▴, ELISA (blood); ○, LSC (blood); ▪, ELISA (plasma); ▿, LSC (plasma).
After i.v. administration, peak concentrations of tacrolimus in blood (22.8 ng/ml) were observed at the end of the 4-h infusion. After cessation of infusion, the concentration of tacrolimus decreased over the next hour to a mean of 12.6 ng/ml and continued to decline more slowly thereafter.
After oral administration, the concentration of tacrolimus reached a maximum at a mean of 1 h. The mean concentration then declined rapidly up to 8 h postadministration. Tacrolimus was measurable in all subjects at 108 h after oral administration but in only 3 after 264 h. The concentration–time curves after oral and i.v. administration were similar between 10 and 108 h.
The concentration of tacrolimus in plasma was low after both routes of administration, and mean concentrations after oral administration could be calculated only up to 6 h postdose because plasma concentrations were above the limit of quantification in fewer than four subjects.
Pharmacokinetic Parameters.
Plasma concentrations measured by LSC were very low with marked intersubject variation, and it was not appropriate to calculate pharmacokinetic parameters from these data. Furthermore, LSC was not sufficiently sensitive to monitor tacrolimus concentrations in plasma for more than 6 h or in whole blood for more than 36 h. By contrast, ELISA enabled measurements to continue for up to 24 h in plasma and up to 264 h in whole blood.
The parameters calculated from tacrolimus concentrations in whole blood after oral and i.v. administration are listed in Table3. There is close agreement between parameters derived from measurements by LSC and ELISA. The similarity in blood concentrations indicates that most of the radioactivity in the systemic circulation is the parent compound.
Mean pharmacokinetic parameters after a single 4-h infusion and a single oral dose of 14C-labeled tacrolimus measured in whole blood by LSC and ELISA (±S.D.)
The AUC values were also similar although concentrations by LSC were determined up to 36 h after administration compared with 264 h with ELISA. Total radioactivity could not be detected 20 h after oral administration; therefore, it was not possible to estimate the terminal half-life of total radioactivity after this route of administration.
Oral bioavailability, Vd, and total clearance calculated from each method were similar, but the renal clearance derived from LSC data was greater than that obtained from ELISA measurements.
Tacrolimus Excretion in Feces and Urine.
After i.v. and oral administration, most of the radioactivity was excreted in the feces, with less than 3% of the dose excreted in the urine. There was marked intersubject variation in fecal excretion of tacrolimus (Table 4). Within approximately 11 days, 75% of the i.v. dose and 93% of the oral dose had been excreted in the feces. This indicates that biliary excretion is the principal route of elimination. ELISA measurement indicates that only trace concentrations of unchanged tacrolimus occur in the feces, suggesting that tacrolimus undergoes extensive metabolism before excretion.
Mean cumulative excretion of total radioactivity (LSC) and tacrolimus (ELISA) in feces after a single infusion and a single oral dose of tacrolimus, expressed as % of administered dose ±S.D.
Total urinary excretion measured by LSC was 2.38 ± 1.02% after infusion and 2.28 ± 1.08% after oral administration; the corresponding figures for ELISA measurement were 0.04 ± 0.02% and 0.02 ± 0.01%, respectively (Table 4). Approximately 50% of total urinary recovery had been excreted by 48 h after administration, but low levels of radioactivity were detectable by LSC on the last collection day (264 h postdose). Measurement of tacrolimus by ELISA indicated that urinary excretion was complete 96 h after i.v. infusion and 120 h after oral administration.
The total recovery of radioactivity in urine and feces within 264 h therefore was 77.76 ± 12.67% after infusion and 94.93 ± 30.72% after oral administration. The proportion of unchanged tacrolimus, indicated by ELISA, was 0.33 ± 0.27% after infusion and 0.14 ± 0.13% after oral administration.
Safety.
Four subjects reported adverse events; only headache (in two subjects after oral and i.v. administration) was considered drug-related. Other adverse events not believed to be causally related to drug administration included headache, nausea, and facial nerve paresis. Mean systolic blood pressure increased slightly after i.v. infusion but the increase was not clinically relevant; there was no change in diastolic blood pressure or pulse rate during the first 12 h after administration and no further changes in blood pressure or pulse rate on any other day of the study. ECG, clinical chemistry, and hematology parameters remained normal throughout the study.
Discussion
This study describes the pharmacokinetic characteristics of tacrolimus in whole blood after single oral and i.v. doses using measurements obtained over 11 days after administration. Both ELISA, which is specific for tacrolimus, and LSC, which measures total radioactivity from 14C-labeled tacrolimus and its metabolites, were used for the measurements. During this 11-day period, 78 to 95% of the dose was excreted predominantly in the feces (urinary excretion accounted for less than 3% of total radioactivity). In both urine and feces, only trace amounts of unchanged drug were detected (0.3 and 0.5%, respectively) indicating extensive metabolism. These data indicate that bile is the principal route of elimination. Renal clearance accounts for less than 1% of the total body clearance, suggesting that renal impairment is unlikely to affect the disposition of tacrolimus.
The oral bioavailability of 20 to 23% is in good agreement with data obtained by ELISA in patients after kidney or liver transplantation (Mekki et al., 1993; Jusko et al., 1995). The AUC and half-life were comparable after oral and i.v. administration. The total body clearance was estimated at 37.5 ml/min; this is low, being equivalent to 3% of liver blood flow. The low clearance and long half-life of tacrolimus suggest that, in clinical practice, steady-state concentrations will not be achieved until after several days’ administration.
The finding that the concentration of tacrolimus is similar when measured by both ELISA LC/mass spectrometry and LSC suggests that there is a low concentration of metabolites in whole blood after administration of a single dose. Only trace levels of unchanged tacrolimus were detected in feces, indicating that the rate of elimination of metabolites from the systemic circulation is formation-rate-limited. This suggests that liver function plays an important role in the elimination of tacrolimus from the body in organ transplant patients.
Footnotes
-
Send reprint requests to: Dr. N. A. Undre, Fujisawa GmbH, Levelingstrasse 12, D-81606, Munich, Germany. E-mail:Nas.Undre{at}Fujisawa.DE
-
This study was supported by Fujisawa GmbH, Munich.
- Abbreviations used are::
- LSC
- liquid scintillation counting
- ELISA
- enzyme-linked immunosorbent assay
- AUC
- area under the concentration–time profile
- Received May 8, 1998.
- Accepted September 28, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}