


Abstract
Fexofenadine, a nonsedating antihistamine, does not undergo significant metabolic biotransformation. Accordingly, it was hypothesized that uptake and efflux transporters could be importantly involved in the drug’s disposition. Utilizing a recombinant vaccinia expression system, members of the organic anion transporting polypeptide family, such as the human organic anion transporting polypeptide (OATP) and rat organic anion transporting polypeptides 1 and 2 (Oatp1 and Oatp2), were found to mediate [14C]fexofenadine cellular uptake. On the other hand, the bile acid transporter human sodium taurocholate cotransporting polypeptide (NTCP) and the rat organic cation transporter rOCT1 did not exhibit such activity. P-glycoprotein (P-gp) was identified as a fexofenadine efflux transporter, using the LLC-PK1 cell, a polarized epithelial cell line lacking P-gp, and the derivative cell line (L-MDR1), which overexpresses P-gp. In addition, oral and i.v. administration of [14C]fexofenadine to mice lackingmdr1a-encoded P-gp resulted in 5- and 9-fold increases in the drug’s plasma and brain levels, respectively, compared with wild-type mice. Also, a number of drug inhibitors of P-gp were found to be effective inhibitors of OATP. Because OATP transporters and P-gp colocalize in organs of importance to drug disposition such as the liver, their activity provides an explanation for the heretofore unknown mechanism(s) responsible for fexofenadine’s disposition and suggests potentially similar roles in the disposition of other xenobiotics.
Processes involved in metabolic biotransformation, especially those mediated by the cytochrome P-450 monooxygenases, are frequently critical determinants in the disposition of many drugs. However, it is increasingly recognized that additional processes such as membrane-bound transport systems may also be similarly important.
Fexofenadine (Allegra, Hoechst Marion Roussel, Kansas City, MO), a nonsedating antihistamine, has recently been approved for the symptomatic relief of seasonal allergy (Markham and Wagstaff, 1998). This compound is, in fact, the active metabolite of the, until recently, widely prescribed and related drug terfenadine (Seldane, Hoechst Marion Roussel), but has the advantage that it is not cardiotoxic and, therefore, does not cause the rare but potentially fatal adverse reaction associated with certain drug interactions involving terfenadine use (Woosley et al., 1993). Significantly, fexofenadine does not undergo significant biotransformation in humans because 95% of the dose is excreted unchanged either in the urine or feces after biliary excretion (Lippert et al., 1995). Although fexofenadine’s absorption, distribution, and elimination could occur by passive diffusion, this is unlikely because, for example, coadministration of other drugs with fexofenadine results in drug interactions. In the case of the cytochrome P-450 3A inhibitors erythromycin (500 mg every 8 h) and ketoconazole (400 mg once daily), the steady-state plasma levels of fexofenadine increase by 109 and 164%, respectively (product information, Hoechst Marion Roussel). However, the mechanism(s) by which such drugs cause the observed interactions is unknown.
Recent biochemical and molecular cloning methodologies have identified a number of transporter proteins expressed in organs of importance in drug disposition, such as the intestine, liver, and kidney. Modulation of the function of such membrane transporters may potentially be responsible for certain drug interactions. In this report, we provide evidence to support the role of human organic anion transporting polypeptide (OATP)1drug uptake transporters and the drug efflux transporter, P-glycoprotein (P-gp), in the cellular uptake and excretion of fexofenadine. Moreover, we show that drugs that alter P-gp transport activity also affect the function of OATP transporters, suggesting that combined inhibition of both OATP and P-gp may indeed be the mechanistic explanation to account for the observed drug interactions involving fexofenadine.
Materials and Methods
Materials.
[14C]Fexofenadine (96.3 μCi/mg, >98% purity), was a gift from Hoechst Marion Roussel. Indinavir, nelfinavir, ritonavir, saquinavir, and PSC-833 were provided by Merck Inc. (Rahway, NJ), Agouron Pharmaceuticals Inc. (La Jolla, CA), Abbott Laboratories (Abbott Park, IL), Roche Products Ltd. (Welwyn Garden City, UK), and Novartis (Basel, Switzerland), respectively. [3H]Digoxin (15 Ci/mmol, >98% purity), [14C]inulin (1.92 mCi/g, >97% purity), [3H]taurocholate (2.0 Ci/mmol >98% purity), and [14C]tetraethylammonium bromide (5.0 mCi/mmol, >99% purity) were purchased from DuPont-New England Nuclear (Boston, MA). All other chemical and reagents, unless stated otherwise, were obtained from Sigma-Aldrich Research (St. Louis, MO) and were of the highest grade available.
Transporter cDNA Constructs.
Rat organic anion transporting polypeptide 1 (Oatp1) was obtained by reverse transcription-polymerase chain reaction (Titan RT-PCR; Boehringer Mannheim, Indianapolis, IN), using primers specific to Oatp1 (5′-AAGACAGCAAAGCAAAGACTTTTAAAG-3′ and 5′-TAACTTTTCAATGTGGCTTAATGAG-3′) (Jacquemin et al., 1994) and rat liver-derived mRNA. The polymerase chain reaction (PCR) product was then subcloned into a pCR2.1-Topo vector (Invitrogen, Carlsbad, CA), and the sequence was verified. This insert was released by digestion with the restriction endonucleasesNcoI and SacI, and was ligated into theNcoI and SacI linearized expression vector pTM1, thus maintaining the open reading frame in a sense orientation to the T7-promoter region of pTM1, with the ATG sequence present in theNcoI site serving as the translation initiation codon. For Oatp2, the primers 5′-AACAGCAGTAAGATTATTTAAAGAATAG-3′ and 5′-GTTAACAACCTGATTAAAGTTTTCAGTG-3′ were used in the PCR of a rat liver cDNA library (Noe et al., 1997). Again, the PCR product was subcloned into pCR2.1-Topo, and its sequence was verified. The Oatp2 insert was released from the pCR2.1-Topo vector by digesting with the restriction endonucleases EcoRV and SpeI, followed by ligation of the insert into the target pTM1 vector, which had been prepared by digesting the plasmid with XmaI, whose cohesive ends were blunted using Klenow DNA polymerase (Promega, Madison, WI), then a second digest with SpeI to ensure directional cloning of the Oatp2 cDNA insert. OATP was a gift from Dr. Peter Meier (University Hospital, Zurich, Switzerland). This clone (Kullak-Ublick et al., 1995) was packaged in the pSPORT vector (Life Technologies, Gaithersburg, MD) with the OATP coding sequence downstream from the pSPORT T7-promoter region, thus making it suitable for expression studies without further subcloning. Similarly, the molecular cloning of human sodium taurocholate cotransporting polypeptide (NTCP) was carried out using the primers 5′-ATGGAGGCCCACAACGCGTCT-3′ and 5′-CTAGGCTGTGCAAGGGGAGCA-3′ and a sample of human liver-derived cDNA library (kindly provided by Dr. F. P. Guengerich, Vanderbilt University, Nashville, TN) and cloning into the pCR2.1 vector in a sense orientation to the T7 promoter region suitable for expression using the vaccinia system. Rat organic cation transporter 1 (rOCT1) was cloned in the same manner using mRNA derived from rat kidney by using the primers 5′-ATGCCCACCGTGGATGATGTCC-3′ and 5′-TCAGGTACTTGAGGACTTGCCTG-3′ ligated into the pCR2.1 vector, again in a form suitable for expression using the recombinant vaccinia system.
HeLa Cell Culture and Preparation of Vaccinia Virus.
HeLa (American Type Culture Collection, Manassas, VA; ATCC) cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum (HyClone, Logan, UT) and 1% penicillin/streptomycin (Life Technologies) at 37°C in 5% CO2 in 150-mm tissue culture plates. These cells were used in the vaccinia viral stock preparation as well as for vaccinia-mediated transport studies. High titer vaccinia stocks (VTF-7, kindly provided by Dr. Bernard Moss, National Institutes of Health, Bethesda, MD) were obtained by infecting HeLa cells grown to confluence (1 × 107) in 150-mm plates. Cells were incubated with virus (10 plaque-forming units (pfu) per HeLa cell) in serum-free media for 30 min. Virus was removed, then the cells were washed with Dulbecco’s modified Eagle’s medium and replaced with standard media. Infected cells were incubated at 37°C for 48 h. The cells were harvested in PBS with a rubber policeman. After pelleting using a bench-top centrifuge at 1600g for 5 min at 4°C, the pellet was resuspended in ice-cold 10 mM Tris (pH 9.0) and homogenized with 10 strokes in a Dounce homogenizer, followed by centrifugation at 1000g for 5 min at 4°C. The supernatant containing the viral particles underwent three rounds of freeze-thaw, first in liquid nitrogen, then in a 37°C bath, after which it was layered on top of 36% sucrose and centrifuged at 130,000g for 20 min at 4°C. The resulting pellet was then resuspended in 10 mM Tris (pH 9.0) and aliquoted before storage at −20°C. Before using these viral preparations in transport experiments, titering was carried out by infecting near confluent HeLa cells (6-well plates with 1 × 106 cell/well) with various dilutions of the viral preparation.
Transport Studies Using Recombinant Vaccinia Virus-Transfected Cells.
HeLa cells grown in 12-well plates (∼0.8 × 106 cells/well) were infected with vaccinia at a multiplicity of infection of 10 pfu/cell in serum-free Optimem I medium (Life Technologies) and allowed to adsorb for 30 min at 37°C. Cells in each well were then transfected with 1 μg of plasmid cDNA or the parental plasmid lacking any insert by combining with the cationic lipid agent Lipofectin (Life Technologies). A lipofectin-to-plasmid DNA ratio of ∼3:1 was found to yield the greatest efficiency of transfection. After the application of this mixture, the cells were incubated at 37°C for 16 h. Transport was then evaluated by using [14C]fexofenadine. Cells were washed with 2 ml of Optimem and preincubated at 37°C for 15 min with 0.3 ml/well of this medium. Transport study was initiated by adding a 0.1 ml/well of [14C]-fexofenadine (2 μM) to the wells. Uptake was allowed to occur at 37°C, then transport activity was stopped at predetermined intervals by washing each well with 1 ml of ice-cold Optimem, repeated three times. The cells were lysed by the addition of 0.5 ml of 1% SDS to each well, followed by measurement of lysate radioactivity by using a liquid scintillation counter (model 1219, Rackbeta, LKB Instruments Inc., Gaithersburg, MD) after the addition of 5 ml of scintillation fluid (ScintiVerse BD; Fisher Scientific, Fairlawn, NJ). To calculate the fexofenadine transport kinetics, [14C]fexofenadine uptake during the linear phase (first 5 min for OATP and 10 min for Oatp1 and Oatp2) was assessed in the presence of varying concentrations of unlabeled fexofenadine. Passive diffusion was determined by carrying out parallel experiments using the parental plasmid DNA lacking the transporter cDNA, and this value was then subtracted from the total uptake rate seen in the presence of the transporter cDNA. Michaelis-Menten type nonlinear curve-fitting was carried out to obtain estimates of the maximal uptake rate, Vmax, and the concentration at which half the maximal uptake occurs,Km (Prism, GraphPad, San Diego, CA). Also, fexofenadine uptake, in the absence and presence of added inhibitors (1, 10, and 100 μM) was assessed. Protein concentrations were determined by Coomassie Protein Assay Reagent (Pierce Chemical Co., Rockford, IL) using BSA as the standard. All experiments were carried out in duplicate on at least three separate experimental days. Data are shown as mean ± S.E., and an unpaired Student’st test or Mann-Whitney U test were used to assess statistical significance (p < .05).
Transport in Cultured LLC-PK1, L-MDR1, and Caco-2 Cells.
LLC-PK1, L-MDR1, and Caco-2 cells were grown under identical conditions to those described previously (Kim et al., 1998). Transepithelial resistance was measured in each well using a Millicell ERS ohmmeter (Millipore, Bedford, MA); wells registering a resistance of 200 ohms or greater, after correcting for the resistance obtained in control blank wells, were used in the transport experiments. About 1 to 2 h before the start of the transport experiments, the medium in each compartment was replaced with a serum-free medium (Optimem). Then, the transport of fexofenadine was measured after replacing the medium in each compartment with 700 μl serum-free medium with or without14C-radiolabeled drug (5 μM). Radioactivity appearing in the opposite compartment after 1, 2, 3, and 4 h was measured in 25-μl aliquots taken from each compartment, after the addition of 5 ml of scintillation fluid (ScintiVerse BD). In addition, tracer amounts of [14C]inulin were used in a separate set of these cells in a similar manner to further verify absence of significant paracellular leak. Data shown represent results obtained from studies carried out on at least three different experimental days.
Determination of Tissue Distribution in mdr1a (+/+) and (−/−) Mice.
Male mdr1a (−/−) mice (FVB/TacfBR-[KO]mdr1aN7; 6 to 12 weeks of age) and genetically matched male mdr1a (+/+) mice (FVB/NTtacfBR) were obtained from Taconic Farms (Germantown, NY). Radiolabeled fexofenadine (96.3 μCi/mg), dissolved in 2% ethanol/0.9% saline solution in a total volume of 2.1 μl/g of body weight, was injected i.v. (1 mg/kg) over 10 min into the tail vein of groups of three mice and orally by gastric intubation (2.6 mg/kg, total volume of 3.5 μl/g of body weight) in groups of five mice. After 4 h, the animals were anesthetized using isoflurane (IsoFlo; Abbott) and blood was removed by orbital bleeding. The mice were then sacrificed, and the harvested tissues were weighed and homogenized with 4% (w/v) BSA. Total radioactivity was determined after the addition of 100 μl of plasma or tissue homogenate (500 μl) to vials containing 4 ml of scintillation fluid (ScintiVerse, BD). The contents of the small intestine and colon were removed before homogenizing and tissues were blotted with filter paper to remove any blood. The protocols for the animal experiments were approved by Vanderbilt University Animal Care Committee and they were cared for in accordance with the U.S. Public Health Service policy for the Care and Use of Laboratory Animals.
Results
Heterologous expression of the selected hepatic and renal uptake transporters showed that members of the OATP transporter family, specifically OATP and both Oatp1 and Oatp2 (Fig.1), mediated fexofenadine uptake. Moreover, there were differences in the affinity and capacity of fexofenadine uptake among transporters. For example, Oatp2 (Km = 6 μM) had a 5-fold greater affinity for fexofenadine than Oatp1 (30 μM); however, theVmax/Kmratio for Oatp1 was 2.4-fold greater than that for Oatp2, assuming a similar extent of expressed transporters. Other uptake transporters such as rOCT1 and the human bile acid/anion transporter NTCP, although capable of transporting known substrates such as tetraethylammonium (rOCT1) and taurocholate (NTCP), did not transport fexofenadine (data not shown).

Fexofenadine uptake assessed using a recombinant vaccinia expression system.
OATP (A and B), Oatp1 (C and D), andOatp2 (E and F) cDNAs (▪) were transiently expressed in HeLa cells, and the uptake of [14C]fexofenadine (2 μM), relative to control cells transfected with only the parental plasmids (▴) over time (A, C, and E) and the transport kinetics (Vmax and Km; B, D, and F) were defined. Data are mean ± S.E. from three or more experiments.
The role of P-gp in fexofenadine efflux transport was assessed using the LLC-PK1 cells and the derivative L-MDR1 cell line stably transfected with human multidrug resistance (MDR)1 gene. When labeled fexofenadine was administered to the basal compartment of the L-MDR1 cells, its appearance measured on the apical side (B→A) was significantly greater than when the drug was added to opposite compartment and sampled on the basal side, i.e., A→B (Fig.2). In LLC-PK1 cells, such polarized transport was absent. Interestingly, the net movement of [14C]fexofenadine in these cell lines was very low (<1%/h; Fig. 2) and a similarly low transport rate was also seen in another polarized epithelial cell line, Caco-2 (<0.5%/h). Measures of cellular tight junction formation as determined by transepithelial resistance or [14C]inulin (<0.5%/h) did not differ significantly between LLC-PK1 cells and L-MDR1 cells in either the basal-to-apical or apical-to-basal direction.

Transepithelial transport of [14C]fexofenadine (5 μM) across L-MDR1 and LLC-PK1 cell culture monolayer.
Radiolabeled drug was added to either the basal or apical compartment and at the indicated time points, samples were obtained from both the apical and basal compartments. Appearance of radioactivity in the opposite compartment was measured and represented as the fraction (shown in percentage) of total radioactivity added at the beginning of the experiment. Translocation from basal to apical compartments, (▴) solid line; translocation from apical to basal compartments, (▪) dotted line. Data are mean ± S.E. from three or more experiments.
To determine the relative importance of P-gp in fexofenadine’s in vivo disposition, total radioactivity was determined 4 h after i.v. or oral administration of radiolabeled fexofenadine to wild-type [mdr1a (+/+)] and mice in which themdr1a gene had been disrupted [mdr1a (−/−)]. After both routes of administration, 4- to 5-fold higher plasma and tissue levels were seen in the mdr1a (−/−) mice compared with the mdr1a (+/+) wild-type mice (Table1). Brain uptake appeared to be particularly affected by mdr1a gene disruption because in this tissue the fexofenadine level was increased 2-fold above that in the plasma.
Tissue levels of radioactivity (ng/g tissue) in mdrla (+/+) and (−/−) mice at 4 h after i.v. injection of [14C]fexofenadine (1.0 mg/kg)
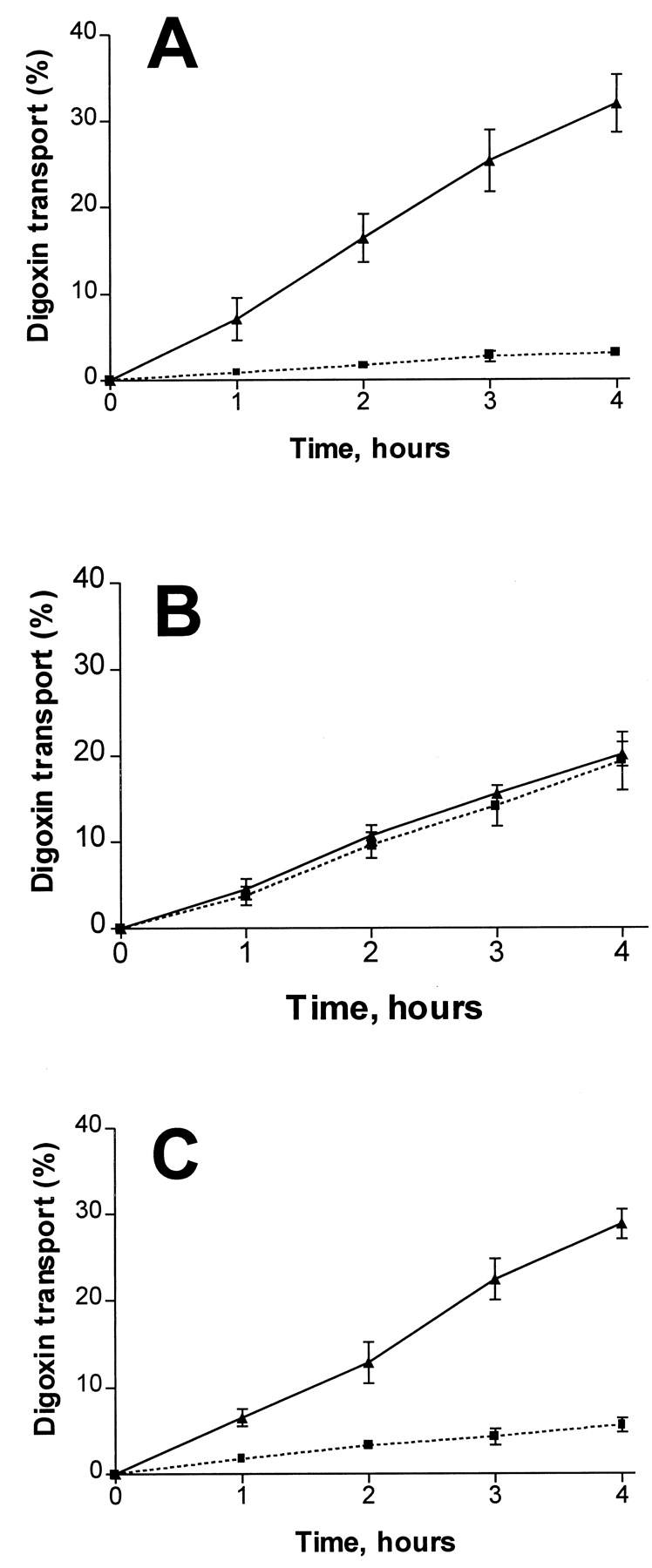
Although fexofenadine was a P-gp substrate, it did not appear to be a significant P-gp inhibitor because concentrations up to 100 μM lacked any inhibitory effect on P-gp-mediated digoxin transport in Caco-2 cells (Fig. 3). By contrast, all tested P-gp substrates/inhibitors were able to inhibit OATP-mediated fexofenadine uptake to a varying degree and in a concentration-dependent manner (Table 2).

Transepithelial transport of[3H]digoxin (5 μM) across a Caco-2 cell culture monolayer in the absence (A) or the presence (B) of 10 μM verapamil; or in the presence of 100 μM fexofenadine (C).
Translocation from basal to apical compartments, (▴) solid line; translocation from apical to basal compartments, (▪) dotted line. Data are mean ± S.E. from three or more experiments.
Inhibition of OATP-mediated fexofenadine uptake by drugs, an organic anion, and bile acids
Discussion
The role and importance of active carrier systems in the transport of drugs across biological membranes are well recognized, but it is only in the past several years that specific transporter proteins have been identified and begun to be characterized. Moreover, it is becoming increasingly clear that in certain situations, for example, translocation across the blood-brain barrier and other membranes important in drug disposition and for an increasing number of drugs such transport is critical (Schinkel et al., 1995; Kim et al., 1998). The described findings suggest that this is the case for fexofenadine. This drug is essentially eliminated from the body unchanged with biliary excretion followed by loss in the feces being the major route and urinary excretion also being involved but to a lesser extent (Lippert et al., 1995). Moreover, the uptake results in LLC-PK1 and Caco-2 cells indicate that fexofenadine is poorly permeable by passive diffusion. Therefore, it is not unreasonable to postulate that transporters known to be localized in the liver and kidney are involved. Accordingly, a number of cloned transporters present on the basolateral membrane of the hepatocyte and also in the kidney that could potentially be involved in fexofenadine’s uptake into the liver from the blood were investigated using a heterologous expression system.
Neither NTCP involved in bile acid uptake nor rOCT1 mediated the cellular transport of the antihistamine. This is not unexpected because NTCP appears to have a very restricted substrate specificity that is essentially limited to bile acids (Schroeder et al., 1998), and rOCT1, although capable of transporting small cationic molecules such as tetraethylammonium and choline, has not yet been shown to be capable of transporting larger entities such as drugs. By contrast, certain members of the OATP family of transporters were found to effectively mediate fexofenadine’s uptake, consistent with the anionic nature of this drug. In addition to fexofenadine, Oatp1 and Oatp2 also mediate the cellular uptake of a wide variety of compounds of different chemical structure and charge, such as bile acids, bromosulfophthalein, steroids, cardiac glycosides, peptidomimetics, and glucuronides (Bossuyt et al., 1996; Eckhardt et al., 1996; Noe et al., 1997). Thus, the substrate specificity of these transporters appears to extend beyond organic anions. In humans, only one OATP has currently been identified with 67% amino acid identity to Oatp1 (Kullak-Ublick et al., 1995), and like its rat homolog, it too is capable of mediating fexofenadine uptake. However, differences in the kinetics of fexofenadine uptake by the various OATP transporters were noted. In particular, the affinity of Oatp2 for fexofenadine (Km) was almost 5-fold greater than for Oatp1 but its maximal rate of transport was over 10-fold smaller. Thus, under first-order conditions of uptake (Vmax/Km) and assuming similar expressed levels of the transporters, Oatp1 would be about 2-fold more effective than Oatp2. TheKm values of OATP was nearly 5-fold lower than that of Oatp1, but its Vmaxwas nearly identical with that of the rat homolog.
In addition to uptake into the hepatocyte, subsequent excretion requires vectorial transport and secretion into the bile. Members of the ATP binding cassette superfamily of transporters localized in the canalicular membrane appear to be especially important in such cellular efflux processes. For example, the MDR1 gene product P-gp with its broad substrate specificity is considered to be importantly involved in the biliary excretion of many structurally divergent drugs (Pastan and Gottesman, 1991; Levêaque and Jehl, 1995). In fact, in L-MDR1 cells that overexpress this transporter compared with the parental LLC-PK1 cells, polarized efflux transport of fexofenadine was observed. However, the low net movement seen in both L-MDR1 and LLC-PK1 cells suggested that these cell lines lack functionally active fexofenadine uptake transport systems and passive diffusion of the drug into the cells is minimal. Nevertheless, the modest amount of fexofenadine that gains access into the cells appear to be efficiently removed from the cell by P-gp. This finding provides a mechanistic basis for the known excretion of the drug in the bile and feces. It is also possible that fexofenadine appears in the feces after direct secretion from blood into the lower intestine, where P-gp is also known to be located. In addition, the relatively poor bioavailability of fexofenadine, estimated to be about 30%, could also involve P-gp-mediated efflux in the small intestine as occurs with a number of other drugs (Mayer et al., 1996; Sparreboom et al., 1997). Another major site of P-gp expression is at the blood-brain barrier, where the transporter functions to limit access of drugs to the brain (Schinkel et al., 1996;Kim et al., 1998). Moreover, the absence of such a barrier, as occurs in mdr1a(−/−) mice (Schinkel et al., 1994), results in an increased brain-blood concentration ratio that appears to be specifically characteristics of P-gp substrates. Indeed, the brain-to-plasma ratio in the wild-type mice was 0.17, whereas in themdr1a(−/−) mice was 0.33. Thus, the observed 2-fold increase in this distribution ratio provides confirmation that fexofenadine is not only a P-gp substrate but that the involved transport is importantly involved in the drug’s disposition. However, the relatively modest increase in the brain-to-plasma ratio in these mice suggests that with fexofenadine translocation across the capillary endothelial cells that make up the blood-brain barrier may be less critical than that for other P-gp drug substrates like digoxin and HIV-1 protease inhibitors (Schinkel et al., 1996; Kim et al., 1998). Nevertheless, the 5-fold increase in fexofenadine’s plasma level 4 h after administration, indicative of impaired elimination in the mdr1a(−/−) mouse, indicates an important role of P-gp in the elimination of this compound. In fact, the extent of the increase in plasma level was considerably greater than the 1- to 2-fold increase observed with other P-gp drug substrates such as vincristine, dexamethasone, and cyclosporine (Schinkel et al., 1994, 1995). Most of these also undergo metabolism as well as P-gp mediated elimination, so this enhanced effect also emphasizes the critical involvement of the transporter in fexofenadine’s elimination from the body. In addition, the findings suggest that P-gp expression in organs such as the liver is not only critical in the drug’s elimination, but is more important than other efflux transporters including members of the multidrug resistance-associated protein transporter family (Cole et al., 1992;Kool et al., 1997) and sisP-gp (Childs et al., 1995; Gerloff et al., 1998).
Because of the broad substrate specificity of P-gp and apparently OATP, a potential exists for drug interactions to occur when multiple substrates are coadministered as drugs. The possibility of these types of interactions has not been widely appreciated despite the fact that the well-recognized interactions between digoxin and quinidine and various other drugs is entirely due to the fact that digoxin, which, like fexofenadine, is also eliminated without significant systemic metabolism, is also efficiently transported by P-gp (Fromm et al., 1999). Moreover, our data suggest that drug inhibitors/substrates of P-gp are also likely to inhibit OATP. In fact, all tested P-gp inhibitors and substrates were to a varying extent, inhibitors of OATP transport activity, although a potent P-gp inhibitor such as PSC-833 (Kim et al., 1999) was not the most potent OATP inhibitor. In fact, peptidomimetic HIV-1 protease inhibitors, such as ritonavir, nelfinavir, saquinavir, and the HMG-CoA reductase inhibitor, lovastatin, were better OATP inhibitors. Nevertheless, it appears that inhibitors of P-gp efflux also have the potential to inhibit OATP uptake; hence, inhibition of both systems may be the basis for some adverse drug interactions.
In conclusion, we show for the first time that certain members of OATP drug uptake transporter family and the drug efflux transporter P-gp appear to be involved in fexofenadine’s disposition and such transporters have a degree of substrate/inhibitor overlap. Considering the fact that OATP and P-gp colocalize in tissues of importance to drug disposition, their activity may be important in the disposition many drugs and their inhibition may be the basis of some drug-drug interactions.
Acknowledgments
We thank Hoescht Marrion Roussel Pharmaceutical (Kansas City, MO) for the generous supply of radiolabeled fexofenadine. In addition, we thank Dr. P. Meier (University Hospital, Zurich, Switzerland) for supplying the OATP cDNA, Dr. Bernard Moss (National Institute of Allergy and Immunology, Bethesda, MD) for the recombinant (VTF-7) vaccinia virus, and Holly Waldrop for expert technical assistance.
Footnotes
-
Send reprint requests to: Dr. Richard B. Kim, 572 MRB1, Division of Clinical Pharmacology, Vanderbilt University School of Medicine, Nashville, TN 37232-6602. E-mail:richard.kim{at}mcmail.vanderbilt.edu
-
This work was supported in part by U.S. Public Health Service Grants GM54724, GM31304, and GM07569, and the Deutsche Forschungsgemeinschaft (M.F.F.).
- Abbreviations used are::
- OATP
- human organic anion transporting polypeptide
- MDR
- multidrug resistance
- P-gp
- P-glycoprotein
- Oatp1
- rat organic anion transporting polypeptide 1
- Oatp2
- rat organic anion transporting polypeptide 2
- NTCP
- human sodium taurocholate cotransporting polypeptide
- rOCT1
- rat organic cation transporter 1
- PCR
- polymerase chain reaction
- Received February 4, 1999.
- Accepted May 3, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}