


Abstract
Strategies for the prediction of in vivo drug clearance from in vitro drug metabolite kinetic data are well established for the rat. In this animal species, metabolism rate-substrate concentration relationships can commonly be described by the classic hyperbola consistent with the Michaelis-Menten model and simple scaling of the parameter intrinsic clearance (CLint − the ratio of Vmax to Km) is particularly valuable. The in vitro scaling of kinetic data from human tissue is more complex, particularly as many substrates for cytochrome P450 (CYP) 3A4, the dominant human CYP, show nonhyperbolic metabolism rate-substrate concentration curves. This review critically examines these types of data, which require the adoption of an enzyme model with multiple sites showing cooperative binding for the drug substrate, and considers the constraints this kinetic behavior places on the prediction of in vivo pharmacokinetic characteristics, such as metabolic stability and inhibitory drug interaction potential. The cases of autoactivation and autoinhibition are discussed; the former results in an initial lag in the rate-substrate concentration profile to generate a sigmoidal curve whereas the latter is characterized by a convex curve as Vmax is not maintained at high substrate concentrations. When positive cooperativity occurs, we suggest the use of CLmax, the maximal clearance resulting from autoactivation, as a substitute forCLint. The impact of heteroactivation on this approach is also of importance. In the case of negative cooperativity, care in using theVmax/Km approach toCLint determination must be taken. Examples of substrates displaying each type of kinetic behavior are discussed for various recombinant CYP enzymes, and possible artifactual sources of atypical rate-concentration curves are outlined. Finally, the consequences of ignoring atypical Michaelis-Menten kinetic relationships are examined, and the inconsistencies reported for both different substrates and sources of recombinant CYP3A noted.
There have been several recent reports highlighting unusual in vitro kinetic behavior for the metabolism of various drugs by certain members of the cytochrome P450 (CYP)2 enzyme system, in particular CYP3A4 (Ueng et al., 1997; Korzekwa et al., 1998; Shou et al., 1999). This review provides a critical examination of metabolism rate-substrate concentration relationships that cannot be described by the classic hyperbola consistent with the Michaelis-Menten model and considers some of the consequences that arise from this kinetic behavior. Specifically, the cases of autoactivation and autoinhibition are discussed. The former results in an initial lag in the rate-substrate concentration profile generating a sigmoidal curve (Fig. 1A) and the latter is characterized by a convex curve due toVmax not being maintained at high substrate concentrations (Fig. 1B). Emphasis is placed on the possible constraints these findings place on the ability to extrapolate in vitro data on drug metabolism to predict in vivo pharmacokinetic characteristics, such as metabolic stability and inhibitory drug interaction potential.
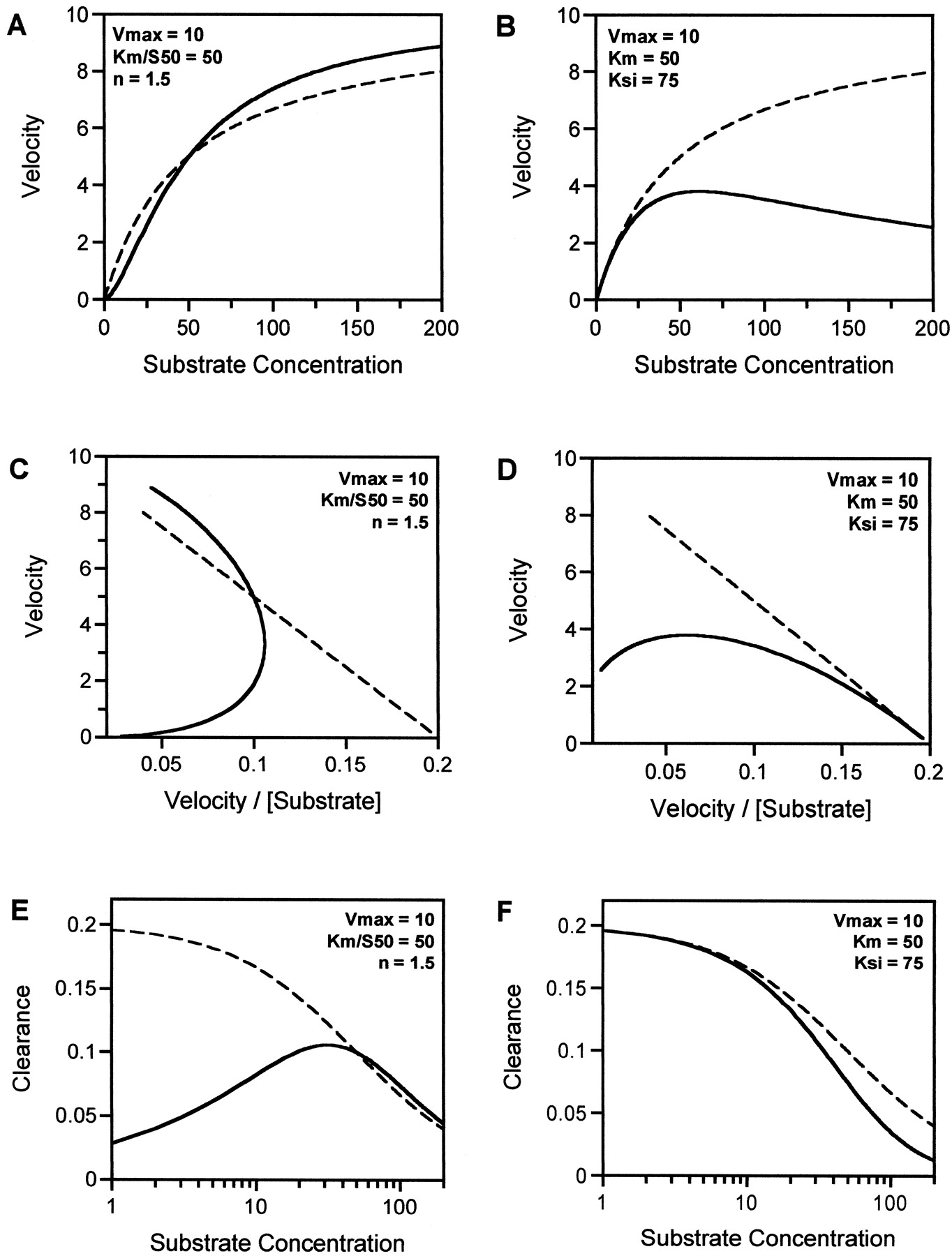
Hyperbolic and nonhyperbolic relationships between rate of metabolic formation and substrate concentration.
Substrate dependence for rates of metabolism (A and B), Eadie-Hofstee plots (C and D), and clearance plots (E and F) for enzymes showing either standard Michaelis-Menten (dashed curves), sigmoidal (solid curves in A, C, and E), or substrate inhibition (solid curves in B, D, and F) kinetics. For each curve both Vmaxand Km (S50) are constant (10 and 50, respectively), and n andKsi values are 1.5 and 75, respectively.
Use of the Michaelis-Menten Model for Describing Drug Metabolite Kinetics In Vitro
CYPs are responsible for the metabolism of a wide variety of drugs and other foreign compounds and their unusual lack of substrate specificity may explain many of the complexities in the operation of this family of isoforms (Guengerich, 1995, 1997; Lewis, 1996). However, the kinetic properties of these enzymes are often described satisfactorily by the classical Michaelis-Menten model (eq. 1):
Most metabolite kinetic studies involve the use of hepatic microsomes, which contain a mixture of several CYP isoforms with overlapping specificity, and the observed rates of metabolism reflect the net effect of several protein-drug interactions. In some cases there may be a smoothing over of any irregularities and the kinetics may look hyperbolic due to the ‘canceling out’ of different kinetic features. In other cases, complications can arise due to the differing impact of several isoforms at different substrate concentrations. Such complications are absent when purified and recombinant enzymes are used, and for many drugs metabolic kinetics can be analyzed appropriately by the Michaelis-Menten equation (for example,Tassaneeyakul et al., 1993; Veronese et al., 1993; Zhang and Kaminsky, 1995; Ellis et al., 1996; Rodrigues et al., 1996; Yamazaki et al., 1996a; Olesen and Linnet, 1997; Lasker et al., 1998).
One valuable application of the Michaelis-Menten model has been in the area of scaling in vitro kinetic data to predict the in vivo clearance of drugs (Houston, 1994). As therapeutic drug concentrations rarely approach the Km, the first order limit of eq. 1 is applicable to describe the rate of metabolism in vivo. The ratio of Vmax toKm provides the parameter, intrinsic clearance (CLint), which defines the rate of metabolism for a given drug concentration (eq. 2):
As an in vitro parameter, CLint, expresses inherent metabolic activity in terms of unit of enzyme (often per picomole for the CYP recombinant enzyme, but more frequently per milligram of microsomal protein or per million cells). This descriptor of the subsystem (whether enzyme, microsomes, or isolated cells) can be scaled to the corresponding in vivo parameter when the total content of enzyme (microsomal protein or hepatocellularity) for the liver is known (Houston, 1994). However, the full capacity of the organ will only be estimated when appropriate allowance is made for the consequences of both parallel and sequential pathways of metabolism. The integration of the total hepatocellular activity with the other physiological determinants of liver clearance, namely, blood flow and drug binding in the blood matrix, requires the use of a pharmacokinetic model (e.g., the venous equilibration liver model; Wilkinson, 1987) and the assumptions of these models should be fully appreciated. To complete the sequence of data manipulations and provide the in vivo clearance prediction, consideration must be given to parallel routes of elimination (e.g., renal excretion) as well as to extrahepatic sites of metabolism. Despite its simplistic view of a complex process, this scaling strategy has been found to be valuable for predicting in vivo clearance from both microsomes and freshly isolated hepatocytes from rat liver (Houston and Carlile, 1997; Iwatsubo et al., 1997; Lin and Lu, 1997). However, there has yet be general agreement on the level of precision that can be realistically accepted from such a crude procedure (Houston and Carlile, 1997), and the issue of variability must be addressed before routine application can be extended to human tissue (Carlile et al., 1999).
Deviations from the Michaelis-Menten Relationship for CYP
One of the assumptions of the Michaelis-Menten model implicit in applying the above scaling strategy is the premise that substrate-enzyme interactions occur at only one site per enzyme and that each site operates independently from the others. There is abundant evidence that for at least one major human cytochrome, CYP3A4, this is not the case (for example: Schwab et al., 1988; Andersson et al., 1994; Shou et al., 1994, 1999; Lee et al., 1995; Ueng et al., 1997; Wang et al., 1997; Korzekwa et al., 1998). There are particular kinetic features for several CYP3A4 substrates that cannot be explained within the context of the Michaelis-Menten model and require the adoption of an enzyme model with multiple sites showing cooperative binding for the drug substrate. Recent evidence suggests that CYP3A may not be the only CYP isoform prone to these features (Ekins et al., 1998; Korzekwa et al., 1998; Venkatakrishnan et al., 1998).
The number of drugs whose in vitro kinetics show deviations from the standard hyperbolic rate-substrate concentration relationship has grown considerably since the first demonstration of autoactivation for 6β-hydroxylation of progesterone in 1988 (Schwab et al., 1988). Examples of drugs showing both positive or negative cooperative effects are given in Table 1. As stated earlier, two characteristic types of curves have been reported: 1) sigmoidal, believed to result from autoactivation, and 2) convex, resulting from substrate inhibition. Both give characteristic curved Eadie-Hofstee plots (see Fig. 1, C and D) that deviate from the linear relationship expected from the Michaelis-Menten model and are useful diagnostic plots for identifying such behavior. This can be particularly valuable when a wide substrate concentration range has been studied and sigmoidicity is occurring at relatively low concentrations and can be easily missed on the conventional rate plot.
Examples of nonhyperbolic rate-substrate concentration profiles for CYP substrates using microsomal preparations of human liver and heterologously expressed systems
Negative cooperativity could alternatively lead to an apparent biphasic Michaelis-Menten curve, identical in form to that frequently observed when two enzymes (a high-affinity, low-capacity enzyme and a low-affinity, high-capacity enzyme) contribute to a particular metabolic reaction. However, to date there appear to be only two examples of this behavior in recombinant CYP3A4 systems (Clarke, 1998;Korzekwa et al., 1998). However, this situation may be a reflection of the more extensive use of human liver microsomes rather than pure enzyme systems, as well as the lack of sufficient data points. Our considerations center on the two more commonly reported effects illustrated in Fig. 1. Although there is much to be elucidated at the molecular level concerning the actual binding processes responsible for these multisite interactions, the net consequences in the kinetic profile can be addressed now.
Two Cases of Nonhyperbolic In Vitro Kinetics.
These two cases of homotropic effects are of importance as neither allow estimation of CLint in vitro by the standard method previously described. Figure 1 contrasts the kinetic features of autoactivation and substrate inhibition with the hyperbolic case and shows the relationships between rate of metabolism and substrate concentration for the three cases. The dashed and solid curves (Fig. 1A) refer to the Michaelis-Menten model and the sigmoidal rate plot, respectively. The latter can be described by the Hill equation (eq. 3):
For the solid curve in Fig. 1B (substrate inhibition case), although a hyperbolic-type curve is apparent at low concentrations, there is no clearly defined plateau at high substrate concentrations and rates decrease as substrate concentrations are further increased. Hence with substrate inhibition, the rate plot is convex, and it is notable that the maximum rate falls short of the trueVmax for the reaction. Substrate inhibition can be considered to be analogous to an uncompetitive type of inhibition mechanism and can be described by eq. 4:
The corresponding Eadie-Hofstee plots, which are valuable diagnostic plots, are shown in Fig. 1, C and D. Whereas hyperbolic curves transform to a linear function, characteristic curves are evident for both sigmoidicity and substrate inhibition. Again dashed lines and solid lines denote the Michaelis-Menten and nonMichaelis-Menten cases, respectively. Figure 1, E and F, shows these cases in the form of clearance plots (v/S versus S), which is helpful within the present context for equating in vitro with potential in vivo characteristics. The clearance relationship is plotted against the log of the substrate concentration to accentuate the independence of clearance on concentration in the initial section of the curve (substrate concentrations < 10% of theKm value) which relates to theVmax/Km orCLint term (dashed curve for the Michaelis-Menten case).
With autoactivation, the sigmoidal rate plot translates to a gradual increase in clearance as substrate concentration is increased to reach a maximum (solid curve in Fig. 1E) followed by a decrease in clearance due to saturation, as seen for the Michaelis-Menten case. This relationship can be described by eq. 5:
Mechanistic View.
Equations 3 and 4 are empirical in nature, and there are advantages in not assigning a more detailed enzyme model where one is not required. However, the unusual kinetics associated with CYP3A have been attributed to the binding of multiple substrate molecules to the enzyme (Shou et al., 1994; Ueng et al., 1997; Harlow and Halpert, 1998;Korzekwa et al., 1998), and it is important to consider the interactions occurring between these multiple sites rather than purely assigning a curve to the data. A more precise description of molecular events incorporating the binding of multiple substrate molecules can be achieved with a steady-state, rapid equilibrium approach to the analysis of the interactions (Segel, 1975). However, it is important to be aware that such models do not distinguish between the simultaneous binding of multiple molecules within a single active site and the binding of two molecules to two distinct sites, both situations may result in sigmoidal kinetics or substrate inhibition. The following kinetic scheme and equation (eq. 7) can be derived for substrate interactions at two separate binding sites.
 Equation 7In this scheme Ks represents the substrate dissociation constant and Kp is the effective catalytic rate constant. For enzymes with two binding sites, Vmax is equivalent to 2Kp/[E]t, where [E]t is the total enzyme concentration. TheKs changes by the factor α when a second substrate molecule binds to the enzyme. When α < 1, the binding affinity for the second substrate molecule is increased, enhancing the overall product formation rate resulting in autoactivation. An alternative mechanism for autoactivation involves a change to theKp by the factor β when both substrate sites are occupied. When β > 1, the overall rate of the reaction is increased and if β < 1 the overall rate is decreased. Thus this model can be used to describe data from substrates showing both sigmoidicity and substrate inhibition. It is possible that some cases of normal hyperbolic kinetics may result from situations where α and β are equivalent to 1, or when the net effects of interactions with Ks andKp are canceled out.
Equation 7In this scheme Ks represents the substrate dissociation constant and Kp is the effective catalytic rate constant. For enzymes with two binding sites, Vmax is equivalent to 2Kp/[E]t, where [E]t is the total enzyme concentration. TheKs changes by the factor α when a second substrate molecule binds to the enzyme. When α < 1, the binding affinity for the second substrate molecule is increased, enhancing the overall product formation rate resulting in autoactivation. An alternative mechanism for autoactivation involves a change to theKp by the factor β when both substrate sites are occupied. When β > 1, the overall rate of the reaction is increased and if β < 1 the overall rate is decreased. Thus this model can be used to describe data from substrates showing both sigmoidicity and substrate inhibition. It is possible that some cases of normal hyperbolic kinetics may result from situations where α and β are equivalent to 1, or when the net effects of interactions with Ks andKp are canceled out.
There are no direct relationships between parameters from this model and the Hill coefficient, n, or the substrate inhibition constant, Ksi. A positive cooperative effect or sigmoidicity can be observed when the value of α is <1 or the value of β is >1. Generally using a value of α < 1 to describe sigmoidal data gives a more realistic approximation ofVmax that is equivalent to the maximal velocity calculated from the Hill equation. A negative cooperative effect is observed when the value of α is >1, resulting in a biphasic kinetic profile, or when the value of β is <1, resulting in substrate inhibition. In theory a combination of both positive and negative effects may be observed resulting from a change to both α and β simultaneously.
Other substrates of CYP3A4 or activators/inhibitors may also interact with α and β and can result in activation or atypical inhibition profiles. Heterotropic effectors of CYP3A substrates displaying nonhyperbolic curves commonly may alter the kinetic profile to generate a hyperbolic curve; for example, activation by α-naphthoflavone (ANF) of estradiol (Kerlan et al., 1992; Ueng et al., 1997), progesterone (Domanski et al., 1998), diazepam (Andersson et al., 1994), and carbamazepine (Kerr et al., 1994) metabolism. Some inhibitors may also produce a similar effect, for example, diazepam and testosterone inhibition of terfenadine metabolism (Kenworthy, 1999); and ANF inhibition of testosterone and amitriptyline metabolism (Ueng et al., 1997).
The difficulty in assigning unique models to explain cooperative effects has long been appreciated, and there may be several solutions that satisfactorily model the same data set (Schmider et al., 1996). The correct solution cannot be fully identified without additional knowledge of the substrate-binding characteristics of the enzyme. Some workers have favored a model-independent approach through the use of polynomials (Childs and Bardsley, 1975; Mayhew et al., 1995).
Are There In Vivo Consequences?
It is important to address the practical problem of dealing with data that cannot be described by the Michaelis-Menten model with a view to making in vivo predictions, particularly as it is the major human cytochrome, CYP3A4, for which these complications have been first identified. Two questions need to be considered. First, are these kinetic characteristics solely an in vitro phenomenon, and second, how can we accommodate these characteristics into strategies for in vitro-in vivo scaling? Whether autoactivation and/or substrate inhibition occur in vivo is not the moot point, as the initial steps of any in vitro-in vivo scaling strategy are to describe fully the in vitro data and then abstract a useful parameter(s) for extrapolation. Thus the in vitro subsystem will always need to be characterized in a fully comprehensive manner with limits that may extend beyond those observed in vivo.
The phenomenon of substrate inhibition is unlikely to be of consequence in vivo due to the high concentrations required. The in vivo importance of autoactivation is difficult to gauge, however, as there is no strong evidence for the manifestation of this cooperative effect in vivo. The in vivo detection of autoactivation would require detailed and judiciously planned studies to provide unequivocal evidence for its reality. This has yet to be carried out; however, an early report of CYP heteroactivation in vivo came from the group of Conney (Lasker et al., 1984). These investigators provided evidence for in vivo activation of radiolabeled zoxazolamine metabolism by flavone in neonatal rats based on metabolite formation (measured by recovery of tritiated water in the total body homogenate). Coadministration of flavone resulted in a dose-dependent increase in metabolite recovery at a fixed time point and a time course consistent with activation. However, in view of the crude nature of the study, additional experimentation is required to eliminate alternative or additional explanations that may contribute to these observations.
Autoactivation is not a phenomenon limited to microsomal incubations. A recent study has shown sigmoidicity for N-demethylation of dextromethorphan in freshly isolated rat hepatocytes as well as in rat hepatic microsomes (Witherow and Houston, 1999). It was proposed that any endogenous activator(s) would be washed out of both in vitro preparations during either the isolation procedures, in the case of hepatocytes, or the homogenization/centrifugation steps, in the microsomal case. It is of interest that the extent of sigmoidicity (as judged by the Hill coefficient) for this reaction is more pronounced in isolated hepatocytes than in microsomes. A similar situation has been observed for both the N-demethylation and the 3-hydroxylation of diazepam in rat in vitro systems (L. E. Witherow, unpublished observations). Also, heteroactivation of midazolam metabolism by ANF has been demonstrated in human hepatocytes (Maenpaa et al., 1998).
It would appear prudent to assume that in vivo the CYP3A system is activated as endogenous steroid hormones (e.g., testosterone and progesterone) and dietary flavones are established as the prototypic activators. Thus the phenomenon of activation must be incorporated into the treatment of in vitro data when prediction of in vivo events is the aim of the study. This will not be a trivial issue and heteroactivation is likely to be an important source of variability between individuals due to different dietary intakes and hormonal changes, which will compound further the issue of variability in expression of these enzymes.
Calculation of Maximal Clearance due to Autoactivation (CLmax)
Recent reviews assessing the utility of in vitro predictions of drug clearance (Houston and Carlile, 1997; Iwatsubo et al., 1997) have shown that unsuccessful examples are usually cases of underprediction. This is particularly evident for human hepatic microsomes. One explanation may lie in unidentified sigmoidal kinetic characteristics and the lack of any allowance for autoactivation.
Consideration of Fig. 1E shows there to be a well defined maximum for the clearance of a drug which is subject to autoactivation. ThusCLmax provides an estimate of the highest clearance attained as substrate concentration increases before any saturation of the enzyme sites. Thus if the assumption is made that in vivo activation occurs via endogenous activators (e.g., steroids), thenCLmax may be an appropriate parameter for describing the salient feature of the subsystem that can be used for predictive purposes. Equation 8 describes the relationship between the various parameters in the Hill equation andCLmax (derivation shown in ). Equation 8For simplicity, the second term containing the n values can be defined as H, the Hill factor, thus eq. 8 can be rewritten as eq. 9.
Equation 8For simplicity, the second term containing the n values can be defined as H, the Hill factor, thus eq. 8 can be rewritten as eq. 9. Equation 9There is a minimum value for H of 0.5, which corresponds ton = 2. When 1 < n < 2, H ranges from 1 to 0.5 and as n increases from 2 to 5 the value of H gradually increases from 0.5 to 0.6.
Equation 9There is a minimum value for H of 0.5, which corresponds ton = 2. When 1 < n < 2, H ranges from 1 to 0.5 and as n increases from 2 to 5 the value of H gradually increases from 0.5 to 0.6.
It must be recognized that there are several artifactual sources of sigmoidicity. Therefore it is essential to eliminate the effect of any nonenzymatic processes that may impinge on the shape of the rate-substrate concentration profile (Witherow and Houston, 1999). Table 2 lists some of the processes that would lower the concentration of substrate available to the enzyme relative to the concentration calculated, after the addition of a particular quantity of substrate to the incubation. Three of these processes involve saturable events, and the impact would be concentration-dependent—maximal at the low concentrations and tapering off to no effect at high concentrations.
Examples of artifactual sources for sigmoidicity and convexity in rate-substrate concentration profiles
Ideally turnover of substrate should be <10% to comply with initial rate conditions, yet analytical limitations may prevent this, and correction for loss of substrate during the incubation is required to avoid artifactual conclusions. Similar care is required to avoid sequential metabolism complications.
Futile binding (Obach, 1996, 1997) has recently received attention as a source of complications for in vitro work and it is frequently related to the protein content of the incubation. However, this may be of less concern with the use of recombinant enzymes as the high level of expression removes the need to incubate with high protein concentrations and reduces the opportunity for futile binding to protein/lipid sites. Nevertheless, cases of sigmoidicity for all microsomal preparations should be confirmed at more than one protein concentration to eliminate this artifact. An additional consideration that arises with the use of cellular systems is the need to evaluate the role of active transporter systems to detect any inconsistency between cellular and media concentrations of drug substrates. If the dissociation constant associated with either futile binding or cellular efflux is significantly less than the Kmfor metabolism, sigmoidicity could arise in the rate-substrate concentration profile in the absence of any enzyme autoactivation.
Other reasons for sigmoidicity or convex rate-substrate concentration profiles include analytical and solubility issues, which result in a lack confidence in the data for the extremes of the concentration range. Finally it must be stated that a minimum of ten data points, suitably dispersed over the curve, are required before sigmoidicity can be considered as a suitable description of a data set.
It is of interest to note that the substrate concentration at whichCLmax occurs (Smax) is a function of bothS50 and n (see ). This will vary considerably among drugs, as illustrated in Table 3; the lowest being 4 μM for progesterone and the highest over 100 μM for amitriptyline. Smax is in all cases below the S50 except for the aflatoxin B1 metabolites for which the n values are greater than for the other substrates shown. For four drugs there are data in more than one expression system. In all cases there is good agreement between the parameters, Smax andS50, although bothVmax and CLmaxdiffer for each expression system. The same trend for these four parameters is apparent when the two pathways of metabolism for diazepam and aflatoxin B1 are compared. The full impact of the activation of CYP3A4 in vivo may be difficult to assess as the concentrations of heteroactivator(s) as well as drug concentrations will need to be taken into account.
Kinetic parameters for CYP3A4 substrates showing sigmoidicity
Heteroactivation of CYP3A Reactions.
It is of interest to speculate that the effects of a heteroactivator on a CYP3A reaction can be additive to autoactivation. As the binding site(s) and/or the affinity for heteroactivators is likely to be different from those of the substrate, a much greater effect may result. The additive effect may be restricted to low substrate concentrations and may plateau as the enzyme becomes saturated and at high concentrations of activator inhibition may be seen due to competition at the binding site(s). However, this will not necessarily be the case as heteroactivation may be additive at all concentrations if the activator binds at a separate site, or if the allosteric change in the binding of the activator is much greater that with the substrate itself.
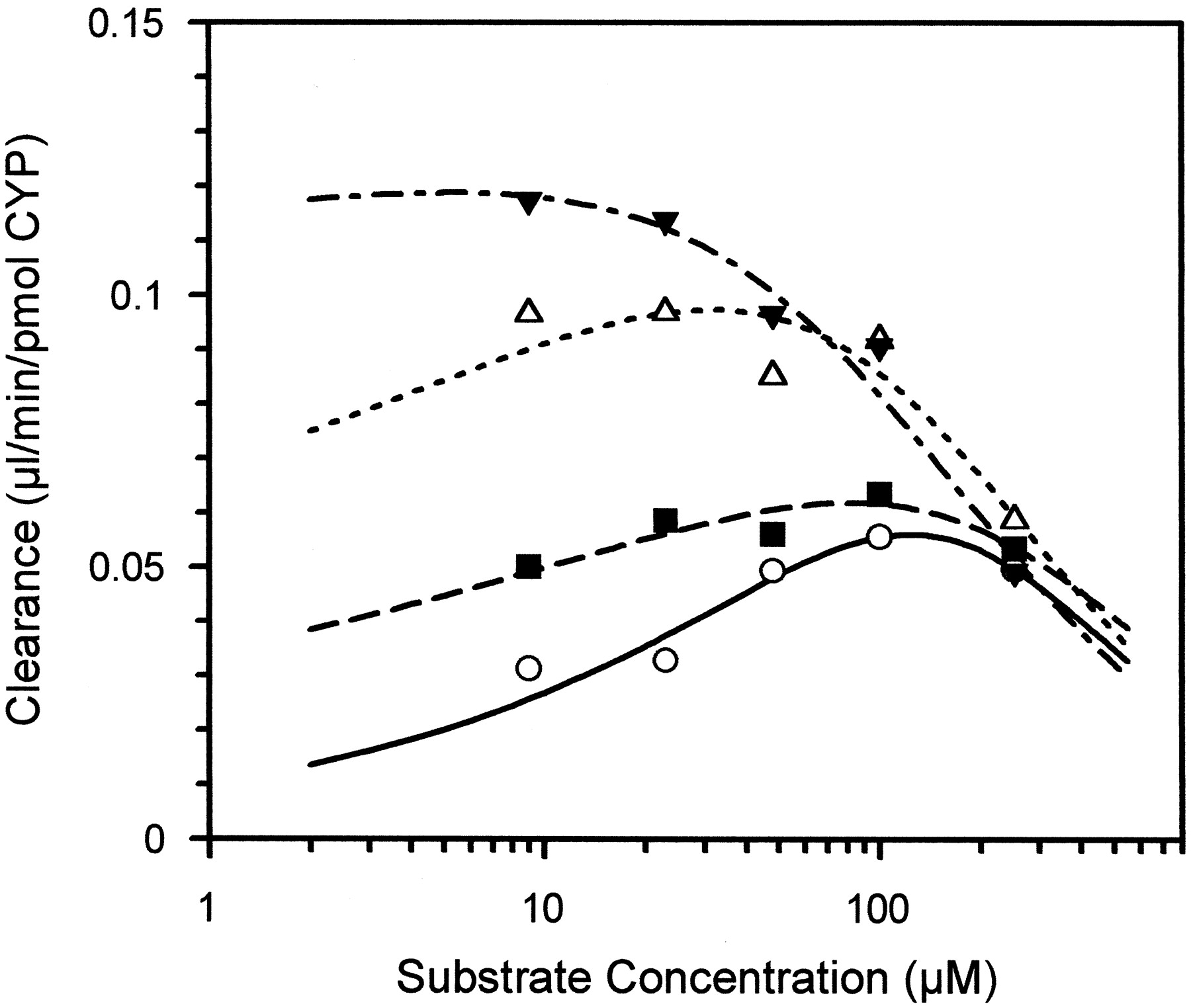
Figure 2 shows an example of one type of situation with the 3-hydroxylation of diazepam by microsomes (obtained from the Gentest Corporation) from β-lymphoblastoid (β-LM) cells expressing human CYP3A4 and CYP reductase (Kenworthy, 1999). At 120 μM, the enzyme is fully autoactivated and the maximum clearance is attained (0.06 μl/min/pmol CYP). More activation may be observed at this concentration by the addition of testosterone as a heteroactivator. However, more substantial activation is seen at lower substrate concentrations and there is a clear trend to lower values ofSmax as the testosterone concentration andCLmax increase 2-fold.

Clearance plot for 3-hydroxylation of diazepam as a function of substrate concentration in microsomes from β-LM cells expressing human CYP3A4 and CYP reductase.
Data (Kenworthy, 1999) in the absence (○) and presence of testosterone: 5 (▪), 25 (▵), and 150 μM (▴).
This raises the philosophical issue of how to define activation. Is it full activation by whatever means, or maximal attainable for the substrate without more supplementation of activators? This will always be an imponderable issue for in vitro studies. Until the precise nature of in vivo activators are known, their appropriate concentrations and the likely extent of their effects cannot be addressed.
Consequences of Ignoring Nonhyperbolic Kinetic Behavior
Although the above phenomena are commonly seen in kinetic profiles, they are not always appreciated by the investigators, and several examples exist of standard Michaelis-Menten hyperbolic curves forced through the data rather than the adoption of more suitable models. In other cases the paucity of data points precludes any meaningful selection of an alternative model. What will the consequences be of ignoring the nonhyperbolic nature of a kinetic profile and fitting the Michaelis-Menten equation? The extent of error and the consequences when scaled for in vivo prediction can be considered for three situations:
1. For substrate inhibition, the consequences are clear in Fig. 1B. Substantial underestimation of Vmax will occur by merely ignoring the high concentration data points and forcing a standard Michaelis-Menten model through the remaining lower substrate concentration data. Also, Km would be poorly estimated. Thus there is a need for full description of the profile to allow for the impact of this phenomenon ifCLint is to be calculated from theVmax/Km. Alternatively v/S for low substrate concentrations could be used, providing the substrate concentration selected is below theKm. (see Fig. 1F).
2. For a sigmoidal curve, there may be either an underestimation or overestimation of CLint if a hyperbolic curve is forced through the data and the parametersVmax and Km are used to calculate CLint. How precisely the clearance estimate will be altered by the model misspecification will vary from case to case and will be dependent on the number and quality of the data points. The Vmax value may be overestimated but it is likely that the Km(S50) term will be most affected. Thus it is probable that CLint calculated from a misspecifiedVmax/Km ratio will underestimate CLmax.
Another important consideration is the estimation of metabolic stability of new chemical entities in terms of in vitro clearance, a common practice within the pharmaceutical industry. This is usually achieved through the use of substrate concentration depletion time profiles, and clearance values are obtained either directly from the area under the curve or from the microsomal half-life. This is usually carried out at one substrate concentration, often in the 1-μM region, based on the rationale that this concentration should be well below the (unknown) Km value. If consideration is not given to the phenomenon of activation, underestimates of clearance will occur. Taking the example of diazepam illustrated in Fig. 2 and discussed above, the microsomal half-life at 1 μM is five times longer than at 100 μM (close to theSmax), reflecting the nonactivated and fully activated clearance.
3. For inhibition studies, models are fitted to data that account for the effect of various concentrations of inhibitor, including the absence of inhibitor. It is clear from the literature that in several inhibition studies there are insufficient data points to allow any detailed examination of the effects of atypical kinetic profiles. If atypical kinetic data are misspecified because in the presence of inhibitor, hyperbolic curves are seen and falseKi values may be obtained. Once again the degree of inaccuracy is hard to predict, however, if the objective is to place compounds in rank order for comparative purposes, sensible conclusions may not always be obtained.
These concerns are of particular relevance within the current trend for adopting high-throughput screening approaches. Many investigators work with only one substrate concentration and assess inhibitory potential by the concentration responsible for a 50% inhibition value (I50). Inhibitory screens may be subject to error when due consideration is not given to the substrate concentration used, particularly if CYP3A has a major involvement in the metabolism of the drugs of interest and multisite effects are apparent. The shape of the velocity curve for a CYP3A4 substrate should be taken into account in the design of inhibition screens. The consequences of ignoring atypical concentration-effect profiles have been highlighted by Leff and Dougall (1993). When characterizing inhibition curves for CYP3A4, the importance of the substrates should be placed on obtaining a sufficiently large number of data points at several substrate concentrations to avoid misinterpretation of the data. Detailed consideration should be given to the fractional inhibition-inhibitor concentration plot, as this may display characteristics indicative of multisite effects for both substrate and inhibitor. The logistic function (eq. 10) routinely used may require a slope factor different from 1 (for the same reason as the nvalue for a sigmoidal rate-concentration plot): Equation 10where fI is the fractional inhibition for a given inhibitor concentration, I. Figure3 shows three examples of inhibitor-substrate interactions in microsomes (obtained from the Gentest Corporation) from β-LM cells expressing human CYP3A4 and CYP reductase (Kenworthy, 1999). They illustrate the range of different inhibitory responses, which may occur when inhibitors and/or substrates bind to multiple sites. For example, the inhibition of testosterone by terfenadine shows no cooperativity (n = 1), the inhibition of diazepam by terfenadine shows negative cooperativity (n = 0.58), and the inhibition of erythromycin by testosterone shows positive cooperativity (n = 1.55).
Equation 10where fI is the fractional inhibition for a given inhibitor concentration, I. Figure3 shows three examples of inhibitor-substrate interactions in microsomes (obtained from the Gentest Corporation) from β-LM cells expressing human CYP3A4 and CYP reductase (Kenworthy, 1999). They illustrate the range of different inhibitory responses, which may occur when inhibitors and/or substrates bind to multiple sites. For example, the inhibition of testosterone by terfenadine shows no cooperativity (n = 1), the inhibition of diazepam by terfenadine shows negative cooperativity (n = 0.58), and the inhibition of erythromycin by testosterone shows positive cooperativity (n = 1.55).
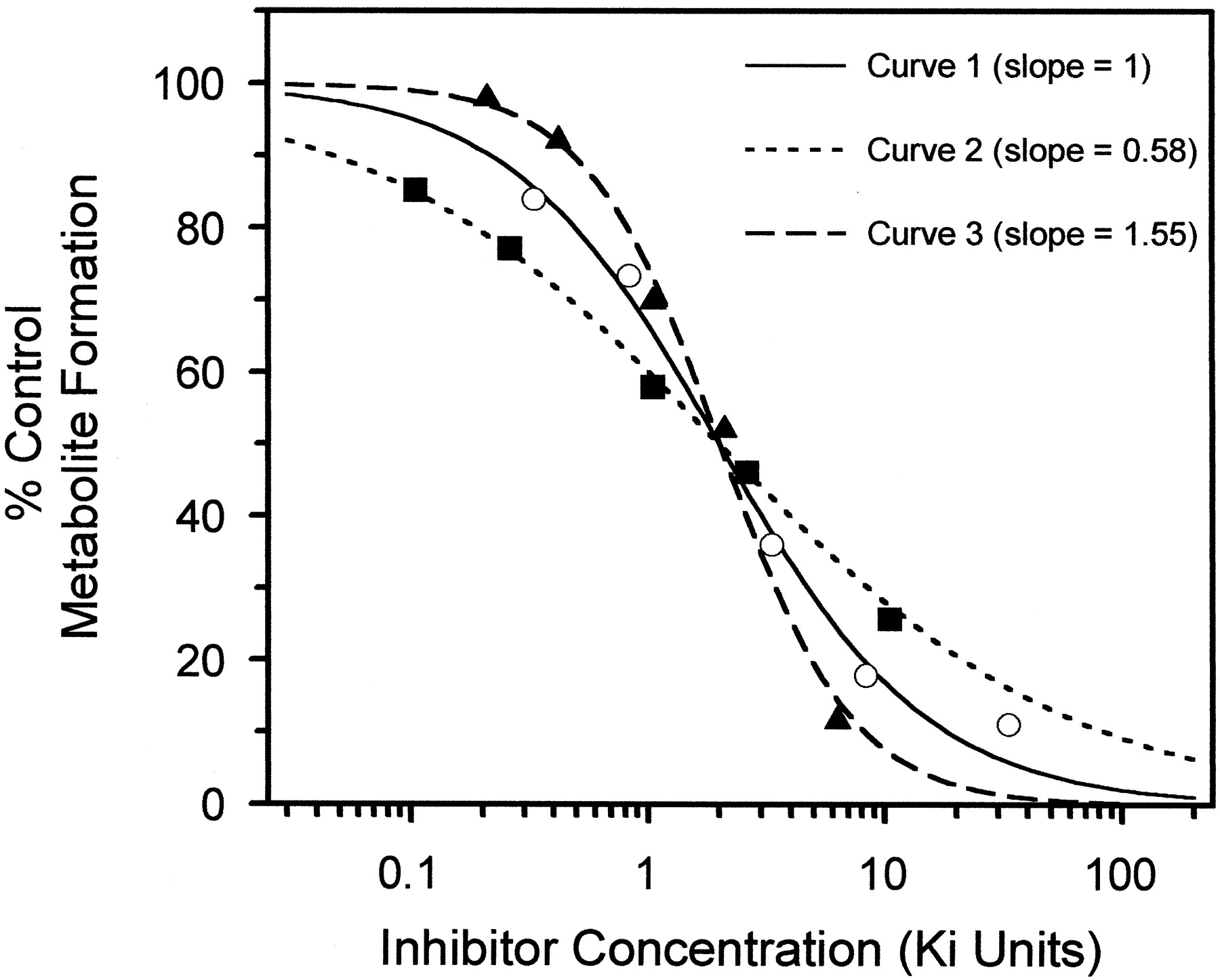
Examples of fractional inhibition of metabolism in microsomes from β-LM cells expressing human CYP3A4 and CYP reductase.
Data (Kenworthy, 1999) show three substrate-inhibitor interactions: curve 1, where the inhibitor shows no cooperativity [inhibition of testosterone metabolism (50 μM) by terfenadine], curve 2, where the inhibitor shows negative cooperativity [inhibition of diazepam metabolism (10 μM) by terfenadine], and curve 3, where the inhibitor shows positive cooperativity [inhibition of erythromycin metabolism (50 μM) by testosterone]. Inhibitor concentrations are normalized for their Ki values (3, 10, and 24 μM for curves 1, 2, and 3, respectively).
Inhibition or activation at multiple sites may also be apparent, resulting in partial or cooperative inhibition or sigmoidal curves that revert to hyperbolic curves in the presence of an activator competing at two binding sites. Additionally, the effect of some CYP3A4 modifiers may differ according to the particular CYP3A4 substrate selected (Kenworthy et al., 1999), stressing the importance of investigating more than one CYP3A4 substrate in inhibition screens. At present there is not a simple solution for accurately predicting CYP3A4 interactions. If a meaningful understanding of the interactions is required, there appears to be little alternative to investigating a wide concentration range of inhibitor and substrates and analyzing the data using a model incorporating the binding of multiple substrate molecules.
Inconsistencies between Substrates and Sources of Recombinant CYP3A4
Interpretation of the nonhyperbolic type behavior discussed above is complicated by the fact that such effects are not consistently seen with all CYP3A substrates. The variety of observed effects may result from a number of scenarios, for example, some substrates may only be able to interact with one binding site, due to steric restrictions, for example, the macrolide antibiotics. In the case of small molecular weight substrates, two or more molecules may bind with only one site, resulting in metabolism, and the second substrate molecule having no effect on the first. As highlighted earlier, some substrates may interact with two sites without causing any significant change to α and β, generating a hyperbolic curve.
Another complicating factor in the study of atypical kinetics is the apparent variability in kinetic behavior between different enzyme sources. It is hard to pinpoint if this is due to the variability in the experimental conditions used, including the use of different buffers as noted by Maenpaa et al. (1998) and the role of various cofactors including CYP reductase and cytochromeb5 (Peyronneau et al., 1992; Yamazaki et al., 1996b), or if it is due to discreet differences in the enzyme active site between different recombinant sources. The activation and substrate inhibition phenomena may be highly dependent on a particular folding pattern of the protein or the location of the protein in different membrane sources. The effects of substrates on the binding kinetics of carbon monoxide (Koley et al., 1996) suggests that CYP3A4 may exist as multiple conformers with different kinetic properties. Slight changes in the flexible structure of CYP3A4 may alter the active site and influence the complex interactions between multiple molecules. It is of interest to note that for certain drugs (amitriptyline, carbamazepine, nifedipine, and testosterone) sigmoidicity is seen in some expression systems, but not others (see Table 1).
In the past a lack of analytical sensitivity has restricted the substrate concentration range used, but this is not a current limitation. Comprehensive data sets are particularly important for substrates showing atypical kinetics to ensure a reasonable degree of confidence in the model parameters. Recent kinetic observations with CYP3A substrates highlight the complexity of this enzyme and our comparatively superficial approach. Absence of nonhyperbolic characteristics for a particular substrate may arise as a result of a lack of cooperative effects or, as discussed earlier, the canceling out of different interaction factors, thus the phenomenon becomes nonidentifiable rather than being absent.
Conclusion
A strategy for in vitro-in vivo extrapolation is well established for use with rat data. In this animal species hyperbolic profiles are very common. Extension to the human situation where CYP3A dominates brings more complexities, one of which is nonhyperbolic curves. The question of whether it occurs in vivo is unanswered yet the problem of equating in vitro with in vivo for CYP3A substrates remains a topical issue that should be addressed now and not delayed until the intricacies of CYP3A are resolved. Recent articles (Ekins et al., 1998;Korzekwa et al., 1998) have indicated that there may be other CYPs, in addition to 3A4, that show nonhyperbolic kinetic behavior. Furthermore, with the use of human liver microsomes, there will always be a high likelihood that CYP3A4, due to its particularly broad substrate specificity, will play some role in the metabolism of the vast majority of drugs. Thus even a relatively selective substrate for CYP2C9 or CYP2D6 may have its microsomal kinetics 'contaminated' with CYP3A4 complexities, particularly when activation comes into play.
The use of recombinant and/or purified enzymes, as opposed to native hepatic mixes, has allowed the identification of unequivocal kinetics under well controlled conditions. When negative cooperativity is manifested as a convex rate-substrate concentration profile, theVmax/Kmapproach to CLint determination is only valid if the correct equation (eq. 6) is used to obtain the parameters. In the case of positive cooperativity, this review suggestsCLmax as a pragmatic solution. Time will tell whether this particular simplification offers a solution that is both robust and comprehensive. The high level of activity surrounding the CYP3A subfamily of enzymes is providing much molecular information that will necessitate continued refinement of our approaches to analyzing and making optimal use of in vitro kinetic data.
Acknowledgments
We thank Drs. S. Clarke, D. Carlile, and L. Witherow for valuable discussions.
Derivation of Clmax
CLmax (the maximum point of thev/S plot) can be derived from the Hill equation and used for comparison with CLint values. The Hill equation can be used to describe sigmoidal data (eq. 2) and the equation of the corresponding clearance plot is shown in eq. 5. The first derivative of this equation, d v/S/d S gives the slope of the clearance plot (eq. 11). Equation 11When the slope, d v/S/d S is set to zero, thex and y coordinates for the inflection point of the curve (CLmax) can be calculated. Solving for Sn and S:
Equation 11When the slope, d v/S/d S is set to zero, thex and y coordinates for the inflection point of the curve (CLmax) can be calculated. Solving for Sn and S: Equation 12Substituting the value of S into the equation for the clearance plot (eq. 5), the y value (v/S) at the inflection point can be obtained (eq. 13).
Equation 12Substituting the value of S into the equation for the clearance plot (eq. 5), the y value (v/S) at the inflection point can be obtained (eq. 13). Equation 13Equation 13 can be rearranged into the form shown in eq. 8.
Equation 13Equation 13 can be rearranged into the form shown in eq. 8.
Footnotes
-
Send reprint requests to: Dr. J.B. Houston, School of Pharmacy and Pharmaceutical Sciences, University of Manchester, Manchester M13 9PL UK. E-mail: Bhouston{at}fs1.pa.man.ac.uk
-
↵1 Present address: Department of Drug Metabolism and Pharmacokinetics, SmithKline Beecham Pharmaceuticals, The Frythe, Welwyn, Herts AL6 9AR, UK.
-
K.E.K. was supported by a SmithKline Beecham studentship.
- Abbreviations used are::
- CYP
- cytochrome P450
- ANF
- α-naphthoflavone
- β-LM
- β-lymphoblastoid
- CLint
- intrinsic clearance
- CLmax
- maximal clearance due to autoactivation
- Ksi
- inhibition constant for substrate inhibition
- n
- the Hill coefficient
- S50
- substrate concentration resulting in 50% of Vmax
- Smax
- substrate concentration resulting in maximal clearance due to autoactivation
- Received August 11, 1999.
- Accepted November 2, 1999.
- The American Society for Pharmacology and Experimental Therapeutics
References




{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Use of the Michaelis-Menten Model for Describing Drug Metabolite Kinetics In Vitro
- Deviations from the Michaelis-Menten Relationship for CYP
- Are There In Vivo Consequences?
- Calculation of Maximal Clearance due to Autoactivation (CLmax)
- Consequences of Ignoring Nonhyperbolic Kinetic Behavior
- Inconsistencies between Substrates and Sources of Recombinant CYP3A4
- Conclusion
- Acknowledgments
- Derivation of Clmax
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters