


Abstract
Tolbutamide is a sulfonylurea-type oral hypoglycemic agent whose action is terminated by hydroxylation of the tolylsulfonyl methyl moiety catalyzed by cytochrome P-450 (CYP) enzymes of the humanCYP2C subfamily. Although most studies have implicated CYP2C9 as the exclusive catalyst of hepatic tolbutamide hydroxylation in humans, there is evidence that other CYP2C enzymes (e.g., CYP2C19) may also participate. To that end, we used an immunochemical approach to assess the role of individual CYP2Cs in microsomal tolbutamide metabolism. Polyclonal antibodies were raised to CYP2C9 purified from human liver, and were then back-adsorbed against recombinant CYP2C19 coupled to a solid-phase support. Western blotting revealed that the absorbed anti-human CYP2C9 preparation reacted with only recombinant CYP2C9 and the corresponding native protein in hepatic microsomes, and no longer recognized CYP2C19 and CYP2C8. Monospecific anti-CYP2C9 not only retained the ability to inhibit CYP2C9-catalyzed reactions, as evidenced by its marked (90%) inhibition of diclofenac 4′-hydroxylation by purified CYP2C9 and by human liver microsomes, but also exhibited metabolic specificity, as indicated by its negligible (<15%) inhibitory effect on S-mephenytoin 4′-hydroxylation by purified CYP2C19 or hepatic microsomes containing CYP2C19. Monospecific anti-CYP2C9 was also found to inhibit rates of tolbutamide hydroxylation by 93 ± 4 and 78 ± 6% in CYP2C19-deficient and CYP2C19-containing human liver microsomes, respectively. Taken together, our results indicate that both CYP2C9 and CYP2C19 are involved in tolbutamide hydroxylation by human liver microsomes, and that CYP2C19 underlies at least 14 to 22% of tolbutamide metabolism. Although expression of CYP2C19 in human liver is less than that of CYP2C9, it may play an important role in tolbutamide disposition in subjects expressing either high levels of CYP2C19 or a catalytically deficient CYP2C9 enzyme.
Tolbutamide, an oral hypoglycemic agent, has been used extensively as a metabolic probe to study human cytochrome P-450 (CYP)3function. Previous studies led to the assignment of tolbutamide as a specific substrate for CYP2C9, a member of the CYP2Cgene subfamily (Srivastava et al., 1991; Veronese et al., 1991; Hall et al., 1994). Although CYP2C9 is indeed a strong catalyst of tolbutamide methylhydroxylation, we as well as other investigators have recently shown that a second CYP2C protein expressed in human liver, CYP2C19, catalyzes the same reaction (Richardson et al., 1995; Lasker et al., 1998; Venkatakrishnan et al., 1998). In fact, rates of hydroxytolbutamide (OHT) formation by purified CYP2C19 and the corresponding recombinant enzyme in reconstituted systems were similar to those exhibited by purified CYP2C9 and its corresponding heterologously expressed enzyme (Lasker et al., 1998). Despite such observations, it is still not known whether CYP2C19, like CYP2C9, plays a significant role in tolbutamide hydroxylation by intact human liver microsomes, as the expression level of CYP2C19 is low relative to CYP2C9 (Lasker et al., 1998). If CYP2C19 is as active as CYP2C9, then CYP2C19 would be an important determinant of tolbutamide disposition in individuals with elevated hepatic levels of this CYP and in individuals expressing CYP2C9 allelic variants that are catalytically deficient toward this drug (Sullivan-Klose et al., 1996).
Assessing the contribution of CYP2C19 to hepatic tolbutamide hydroxylation has been challenging for several reasons. First, CYP2C19 possesses a Km value for tolbutamide (Lasker et al., 1998; Venkatakrishnan et al., 1998) similar to that of CYP2C9, thereby obviating kinetic approaches for determining the contribution of the former enzyme to this microsomal drug-metabolizing reaction. Second, the development of monospecific CYP2C19 antibodies for use in immunoinhibition studies has been hampered by the extensive degree of sequence homology (92%) found between this CYP2C protein and CYP2C9 (Romkes et al., 1991). As polyclonal antibodies produced against purified CYP2C19 recognize not only the corresponding immunogen but also CYP2C9 (and CYP2C8), the inhibition of microsomal tolbutamide hydroxylation elicited by these antibodies (95%) overestimates the CYP2C19 contribution (Lasker et al., 1998). Chemical inhibitors, such as sulfaphenazole, have also been used to determine the contribution of CYP2Cs to this reaction (Newton et al., 1995). However, sulfaphenazole exhibits greater specificity toward CYP2C9 than CYP2C19. Correlation analyses between CYP2C19 and/or CYP2C9 enzyme levels and tolbutamide hydroxylase activity in human liver microsomes have also yielded equivocal results (Forrester et al., 1992; Hall et al., 1994; Inoue et al., 1997; Lasker et al., 1998). Moreover, the use of such correlation analyses for determining the metabolic specificity of CYP2C19 and/or CYP2C9 toward tolbutamide suffers from inherent limitations, including the inability to reveal strong relationships when allelic variants exist at a relatively high frequency, but contribute only slightly to tolbutamide hydroxylation.
We used an immunochemical-based approach to assess the respective roles of CYP2C19 and CYP2C9 in hepatic tolbutamide metabolism. Solid-phase adsorption was used to derive a monospecific anti-CYP2C9 antibody. Recombinant CYP2C19 was used as the back adsorbent, and the resultant anti-CYP2C9 preparation reacted specifically with CYP2C9 on Western blots. Using this specific inhibitory probe, it was established that CYP2C9 catalyzes the majority of tolbutamide hydroxylation in human liver microsomes. However, CYP2C19 was also found to contribute to metabolism of tolbutamide, particularly in subjects expressing high levels of this hepatic CYP2C enzyme.
Experimental Procedures
Human Liver Specimens.
Liver samples were obtained from organ donors through the Liver Tissue Procurement and Distribution System (LTPADS, University of Minnesota, Minneapolis, MN). None of the subjects had a reported history of alcohol or drug abuse. The livers were removed within 12 h of death, frozen in liquid nitrogen, and stored at −80°C until used for microsomal preparation (Raucy and Lasker, 1991). Protein and CYP contents were determined using the bicinchoninic acid procedure (Smith et al., 1985) and the method of Omura and Sato (1964), respectively.
Expression and Purification of Recombinant CYP2C Enzymes.
CYP2C8, CYP2C9, and CYP2C19 were expressed in Escherichia coli as described previously (Richardson et al., 1995). The recombinant CYP enzymes were purified from E. coli membranes as follows. A 3.0-liter culture of E. coli expressing CYP2C8, CYP2C9, or CYP2C19 was centrifuged at 8000g for 10 min, and the pellet was resuspended in 450 ml of 100 mM Tris buffer (pH 7.8) containing 0.5 mM EDTA and 20% glycerol by stirring for 10 min at 4°C. Lysozyme was added to a final concentration of 0.2 mg/ml, the solution was diluted 1:1 with distilled water, stirred for an additional 30 min at 4°C, and then centrifuged at 8000gfor 10 min. The pellet was resuspended in 90 ml of 10 mM Tris-HCl buffer (pH 7.3) containing 14 mM magnesium acetate, 60 mM potassium acetate, and 0.1 mM dithiothreitol (DTT), and then frozen at −80°C. After thawing and the addition of phenylmethylsulfonyl fluoride to a final concentration of 0.5 mM, the sample was subjected to 10 cycles of sonication at 100W for 30 s with a 60-s cooling period on ice between cycles. The sample was again centrifuged at 8000gfor 10 min, and the pellet was resuspended in 2.0 ml of 10 mM KPO4 buffer (pH 7.4) containing 0.1 mM EDTA and 20% glycerol. The bacterial membranes were then solubilized by dropwise addition of 10% Lubrol PX to a final concentration of 0.5% while stirring at 4°C for 60 min. The clarified solution was centrifuged at 150,000g for 60 min, and the supernatant was applied to a 1.5 × 10 cm column of hydroxylapatite (Hypatite C; Clarkson Chromatography Products, Inc., South Williamsport, PA) that had been equilibrated with 10 mM KPO4 buffer (pH 7.4) containing 0.5% Lubrol PX, 1 mM DTT, 0.1 mM EDTA, and 20% glycerol. Bound proteins were eluted from the column by washing with 12 volumes of equilibration buffer, followed by 12 volumes each of equilibration buffer containing 25 and 50 mM KPO4buffer (pH 7.4). Lubrol PX was removed from the bound CYP sample by washing the column with 50 volumes of the same buffer used for equilibration, but with the detergent omitted. CYP2C proteins were then eluted from the hydroxylapatite resin with 300 mM KPO4 buffer (pH 7.4) containing 0.5% cholate, 1 mM DTT, 1 mM EDTA, and 20% glycerol (Raucy and Lasker, 1991). The final CYP2C8, CYP2C9, and CYP2C19 enzyme preparations were exhaustively dialyzed against 100 mM KPO4 buffer (pH 7.4) containing 1 mM DTT, 1 mM EDTA, and 20% glycerol to remove cholate, and concentrated by ultrafiltration through a Filtron Omega 30k membrane (Filtron Technology Corp., Northborough, MA). Cytochromeb5 (b5; specific content = 41.5 nmol/mg protein) and NADPH:P-450 oxidoreductase (P-450 reductase; specific activity = 40,000 U or 11.9 nmol/mg protein) were isolated from human liver microsomes as described elsewhere (Raucy and Lasker, 1991). One nanomole of P-450 reductase was considered equivalent to 3,370 U of activity; 1 U was defined as that amount catalyzing the reduction of 1 nmol of ferrocytochrome c/min at 30°C in 300 mM potassium phosphate buffer (pH 7.7).
Catalytic Activities.
Hydroxylation of S-mephenytoin, diclofenac, and tolbutamide by human liver microsomes and purified recombinant CYP2C enzymes were determined under conditions where product formation was directly proportional to both CYP concentration and time of incubation. Reactions with microsomes contained an amount of protein equivalent to 25 to 150 pmol of aggregate CYP, whereas those with reconstituted systems contained 5 to 25 pmol of purified CYP2C enzyme, 50 to 250 pmol of P-450 reductase, 1.5 to 7.5 μg ofl-α-dilauroylphosphatidylcholine (DLPC), and 20 to 100 pmol of b5. Other reaction components are given below. For antibody inhibition studies, microsomes or reconstituted CYP enzymes were first incubated with anti-CYP2C9 or preimmune IgG (see below) for 3 min at 37°C, and then for 10 min at room temperature, followed by the addition of the remaining reaction components. Antibody titration curves contained increasing amounts of either polyspecific anti-CYP2C9 or monospecific anti-CYP2C9 IgG (0–20 mg of IgG/nmol CYP). The amount of IgG added to the incubation mixtures was maintained at a constant level by the addition of preimmune IgG.
S-Mephenytoin 4′-hydroxylation was determined using a combination of previously published methods (Meier et al., 1985;Shimada et al., 1985, 1986; Lasker et al., 1998). Briefly, incubation mixtures (0.25 ml) contained human liver microsomes or reconstituted CYP2Cs, 100 μM [14C]S-mephenytoin (3.8 μCi/mmol), and 1.0 mM NADPH. Reactions with microsomes and reconstituted CYPs were performed in 100 mM KPO4buffer (pH 7.4) and 50 mM HEPES buffer (pH 7.4), respectively. Incubations were terminated after 30 min by the addition of 200 μl of 1 N HCl, followed by extraction with 3 ml of ethyl acetate. The organic extracts were dried under nitrogen gas at room temperature, and dissolved in 25 μl of acetonitrile. Formation of 4-hydroxymephenytoin was assessed by injecting the resolubilized residues onto a Versapack C18 column (4.6 × 300 mm; Alltech, Deerfield, IL) with 26% (v/v) aqueous acetonitrile as the mobile phase. The flow rate used was 1.0 ml/min, and radioactivity in the column eluates was detected with a Radiomatic 150TR radio-HPLC detector (scintillant flow rate of 2.5 ml/min). Under these conditions, 4′-hydroxymephenytoin and mephenytoin exhibited retention times of 7.9 and 19.5 min, respectively.
Diclofenac 4′-hydroxylation was assessed essentially as described byLeemann et al. (1993). Incubation mixtures (1.0 ml) contained human liver microsomes or reconstituted CYP2Cs, 10 μM diclofenac, and 1.0 mM NADPH. Reactions with microsomes and reconstituted CYPs were performed in 100 mM KPO4 buffer (pH 7.4) and 50 mM HEPES buffer (pH 7.4), respectively. Incubations were terminated after 30 min by the addition of 200 μl of 1 N HCl containing 100 μM chlorpropamide (used as the internal standard), followed by extraction with 3 ml of ethyl acetate. The organic extracts were dried under nitrogen gas at room temperature, and dissolved in 25 μl of acetonitrile. Formation of 4′-hydroxydiclofenac was assessed by injecting the resolubilized residues onto a Versapack C18 column (4.6 × 300 mm; Alltech) developed with a linear gradient of 45 to 59% aqueous acetonitrile containing 0.025% phosphoric acid. The flow rate used was 1.0 ml/min, and eluates were continuously monitored at 282 nm. These HPLC conditions gave retention times for 4′-hydroxydiclofenac and diclofenac of 7.5 and 13.3 min, respectively.
Tolbutamide hydroxylation was assessed according to a previously published procedure (Knodell et al., 1987) but with the following modifications. Incubation mixtures (1.0 ml) contained human liver microsomes (protein equivalent to 50 pmol) or 50 pmol of reconstituted CYP2Cs, 100 mM potassium phosphate buffer (pH 7.4), 0.25 mM [14C]tolbutamide (μCi/mmol), and 1.0 mM NADPH. When sulfaphenazole was included, the final concentration ranged from 50 to 100 μM. Reactions were terminated after 60 min at 37°C with 200 μl of 1 N HCl, followed by extraction with 3 ml of ethyl acetate. The ethyl acetate extracts were dried with nitrogen gas at room temperature, and the residue was resolubilized in 25 μl of acetonitrile. OHT formation was assessed by injecting the resolubilized residues onto a Versapack C18 column (4.6 × 300 mm; Alltech) with 40% acetonitrile containing 0.03% phosphoric acid (pH 2.6) as the mobile phase. The flow rate used was 1.0 ml/min, and radioactivity in the column eluates was detected with a Radiomatic 150TR radio-HPLC detector (scintillant flow rate of 2.5 ml/min). Under these conditions, OHT and tolbutamide exhibited retention times of 3.8 and 7.9 min, respectively.
Immunochemical Methods.
Polyclonal antibodies to CYP2C9 were raised in male New Zealand White rabbits, and the IgG fractions were derived from sera using caprylic acid/ammonium sulfate precipitation (Lasker et al., 1998). These polyspecific CYP2C9 antibodies (anti-CYP2C9-Ps), which exhibited cross-reactivity with CYP2C8 as well as CYP2C19 (Lasker et al., 1998), were subsequently made monospecific for CYP2C9 by repeated passage over a column of cross-linked agarose to which purified recombinant CYP2C19 had been covalently coupled. The immunosorbent resin was prepared by reacting 12 ml of activated Affi-Gel 10 agarose resin (Bio-Rad Labs, Richmond, CA) with 20 ml of 250 mM sodium bicarbonate buffer (pH 8.6) containing 1% Lubrol PX, 1 mM EDTA, and 200 nmol recombinant CYP2C19 for 3 h at ambient temperature according to the manufacturer's instructions. The column was regenerated between anti-CYP2C9 applications by washing with 20 ml of 100 mM glycine-HCl buffer (pH 3.1), followed by rapid neutralization with 50 ml of 100 mM KPO4 buffer (pH 7.4). Preimmune (control) IgG was prepared from rabbit sera obtained before immunization.
Protein blotting of microsomal proteins and purified recombinant enzymes to nitrocellulose and subsequent immunochemical staining with anti-CYP2C9 antibodies was performed using a strepavidin peroxidase-based system as described elsewhere (Lasker et al., 1998). CYP2C9 and CYP2C19 levels in the human liver samples were quantified on Western blots by staining with polyspecific anti-CYP2C9 and polyspecific anti-CYP2C19, respectively. The blots were scanned with a Microtek ScanMaker II HR flat bed scanner interfaced to a computer, and immunoreactive areas were measured using ImageQuant software (Molecular Dynamics, Sunnyvale, CA). All immunochemical staining was performed under conditions where the peroxidase reaction density was directly proportional to the amount of protein applied to the original polyacrylamide gels.
Statistical Analysis.
Data was analyzed using repeated-measure ANOVA or by Student'st test. Levels of significance were set at P≤ .05.
Materials.
[14C]tolbutamide and [14C]S-mephenytoin were purchased from Amersham (Arlington Heights, IL). UnlabeledS-mephenytoin was obtained from Salford Ultrafine Chemicals (Manchester, England). Unlabeled tolbutamide, diclofenac, sulfaphenazole, cytochrome c, and Lubrol PX were obtained from Sigma Chemical Co. (St. Louis, MO), whereas NADPH was purchased from Boehringer-Mannheim (Indianapolis, IN). Hydroxylapatite (Hypatite C) was obtained from Clarkson Chromatography Products, Inc. (South Williamsport, PA). All other chemicals used were of the highest grade commercially available.
Results
Preparation and Characterization of Monospecific CYP2C9 Antibodies.
Rabbit polyclonal antibodies to human CYP2C9 were found to recognize CYP2C9 as well as CYP2C8 and CYP2C19 on immunoblots containing purified human liver CYP2C enzymes and hepatic microsomes (Lasker et al., 1998). Preliminary small-scale experiments indicated that monospecific CYP2C9 antibodies (anti-CYP2C9-Ms) could be derived from anti-CYP2C9-Ps by back-adsorption against heterologously expressed CYP2C19. We expanded this procedure to derive CYP2C9 IgG preparations that were not only monospecific on Western blots but were also inhibitory toward only CYP2C9-catalyzed reactions. To obtain anti-CYP2C9-M, we used recombinant CYP2C19-based immunoadsorption. Expressed CYP2C19 (Richardson et al., 1995) was purified using a single chromatographic step, hydroxylapatite chromatography. A yield of 43% (200 of the 470 nmol originally processed) was achieved. The entire preparation of purified CYP2C19 was then covalently linked to Affigel 10, giving a solid-phase immunoadsorbent containing 16.7 nmol hemoprotein/ml agarose resin. Back-adsorption of 30 mg of anti-CYP2C9-P using this immunosorbent resin resulted in a yield of 15.4 mg of anti-CYP2C9-M.
As shown in Fig. 1A, anti-CYP2C9-P recognized CYP2C9 (lane 2), and also purified recombinant CYP2C8 and CYP2C19 (lanes 1 and 3). Moreover, the polyspecific antibody reacted with the same CYP2C proteins in human liver microsomes (lanes 4–11). However, after back-adsorption against CYP2C19, the anti-CYP2C9-M preparation recognized only recombinant CYP2C9 (Fig. 1B, lane 2) and the corresponding enzyme in microsomes (Fig. 1B, lanes 4–11). Cross-reactivity of anti-CYP2C9-M with CYP2C8 and CYP2C19 was completely eliminated by immunoadsorption (Fig. 1B, lanes 1 and 3). The figure also reveals that subjects E, G, I, and F expressed negligible levels of hepatic CYP2C19 (Fig. 1A, lanes 8–11).

Immunoreactivity of anti-CYP2C9-Ps and anti-CYP2C9-Ms with recombinant CYP2C enzymes and human liver microsomes.
Recombinant human CYP2C enzymes and liver microsomes were subjected to SDS-polyacrylamide gel electrophoresis, followed by electrophoretic transfer to nitrocellulose filters. A, filter immunostained with anti-CYP2C9-P IgGs. B, membrane stained with anti-CYP2C9-M IgGs. Lane 1: recombinant CYP2C8 (25 pmol); lane 2: recombinant CYP2C9 (25 pmol); lane 3: recombinant CYP2C19 (25 pmol); lanes 4 to 11: liver microsomes (10 μg) from subjects A, B, C, D, E, G, I, and F, respectively. As shown (see Table 2), the latter four subjects are CYP2C19−. The arrowheads denote the immunoreactive bands corresponding to CYP2C9, CYP2C8, and CYP2C19 (top to bottom).
The inhibitory properties of anti-CYP2C9-M IgG were assessed with two substrates, diclofenac and S-mephenytoin, which are specifically metabolized by CYP2C9 and CYP2C19, respectively. Immunoinhibition reactions were performed with either recombinant CYPs or human liver microsomes derived from subject H. As shown in Fig.2A, diclofenac 4′-hydroxylation mediated by recombinant CYP2C9 was inhibited to a similar extent by both anti-CYP2C9-P and anti-CYP2C9-M, with maximal inhibition (90%) achieved at an IgG/CYP ratio of 20 mg/nmol. Comparable results were obtained using human liver microsomes, where both anti-CYP2C9 IgG preparations were found to maximally inhibit (≥75%) diclofenac 4′-hydroxylation at an IgG/CYP ratio of 5 mg/nmol (Fig. 2B).
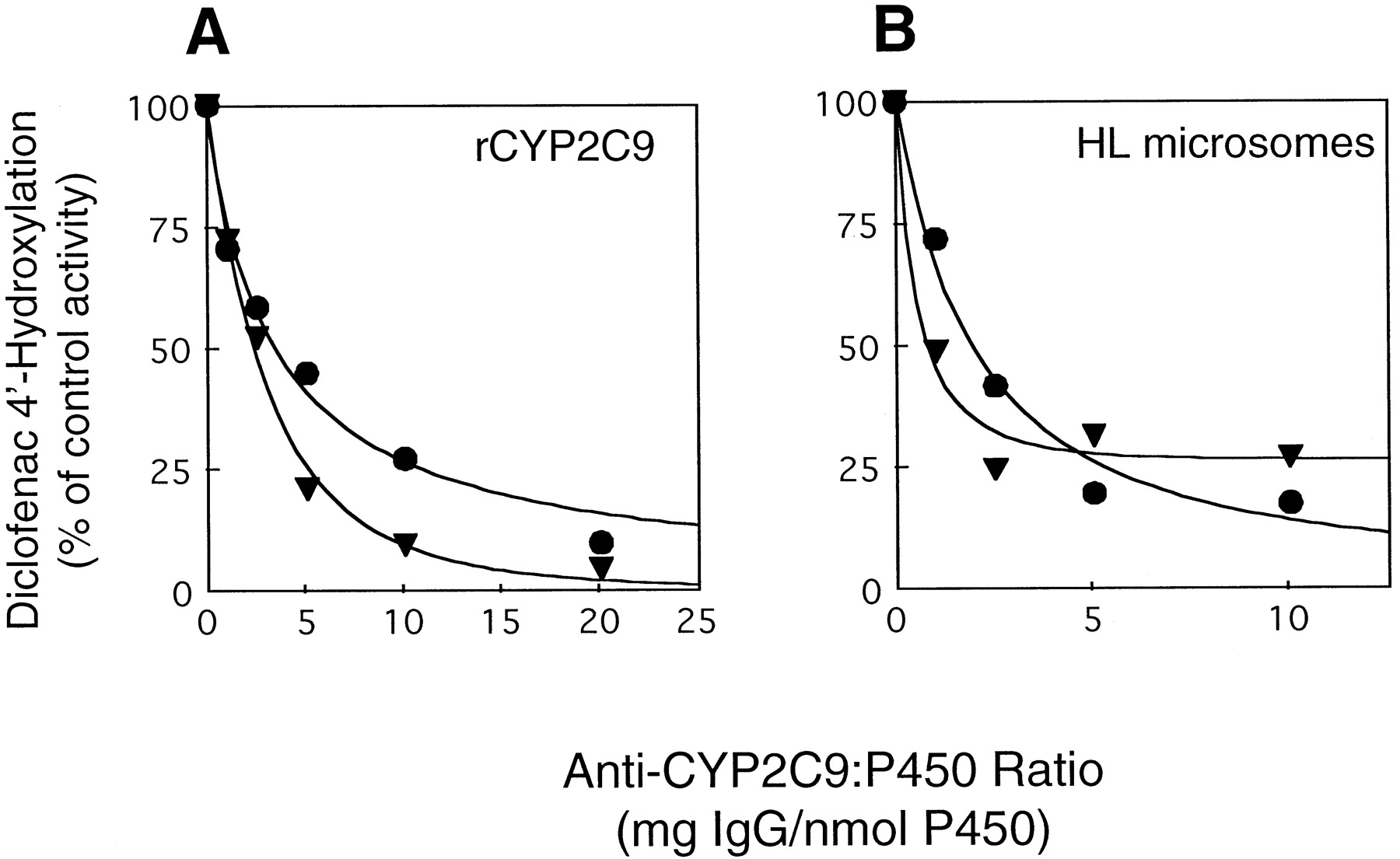
The effect of CYP2C9 antibodies on diclofenac 4′-hydroxylase activity.
A, incubation mixtures contained a CYP2C9 reconstituted system (5 pmol of recombinant CYP2C9, 50 pmol of P-450-reductase, 1.5 μg of DLPC, and 20 pmol of b5), 50 mM HEPES buffer, (pH 7.4), 10 μM diclofenac, 1 mM NADPH, and increasing amounts (0–200 μg) of preimmune, anti-CYP2C9-P (▾), or anti-CYP2C9-M (●) IgG. The total amount of IgG added (anti-CYP2C9 plus preimmune IgG) was maintained at 200 μg. B, incubation mixtures contained liver microsomes from subject H (Table 2) (25 pmol of CYP), 100 mM KPO4 buffer (pH 7.4), 10 μM diclofenac, 1 mM NADPH, and increasing amounts (0–250 μg) of preimmune, anti-CYP2C9-P (▾), or anti-CYP2C9-M (●) IgG. Diclofenac 4′-hydroxylation was determined as described in Experimental Procedures, except that before reaction initiation, the CYP2C9-reconstituted system and microsomes were preincubated with antibodies for 3 min at 37°C, followed by 10 min at 25°C. Rates of 4′-hydroxydiclofenac formation in the presence of preimmune IgG were 4.69 nmol/min/nmol CYP with recombinant CYP2C9 and 0.78 nmol/min/nmol CYP with liver microsomes.
In contrast to the above observations, only anti-CYP2C9-P significantly inhibited S-mephenytoin 4′-hydroxylation catalyzed by recombinant CYP2C19 (Fig. 3A). The back-adsorbed CYP2C9 antibody preparation gave negligible nonspecific inhibition (<15%) of this CYP2C19-promoted activity, regardless of the IgG/CYP ratio used. Similarly, when hepatic microsomes from subject H were used as the catalyst (Fig. 3B), anti-CYP2C9-P markedly inhibited 4-hydroxymephenytoin formation (≥75%) at IgG/CYP ratios ranging from 2.5 to 10 mg/nmol, whereas the anti-CYP2C9-M preparation was much less inhibitory (≤25%) at these same IgG/CYP ratios. Similar results were obtained with other human liver samples examined (Table1). As shown in Table 1, anti-CYP2C9-P completely inhibited microsomal S-mephenytoin 4-hydroxylation whereas anti-CYP2C9-M produced only a mean of 8.5 ± 5.3% inhibition (n = 4). Conversely, both anti-CYP2C9-P and anti-CYP2C9-M markedly inhibited (89 ± 6 and 89.3 ± 2%, respectively) diclofenac 4′-hydroxylation in the liver samples (n = 4) (Table 1).
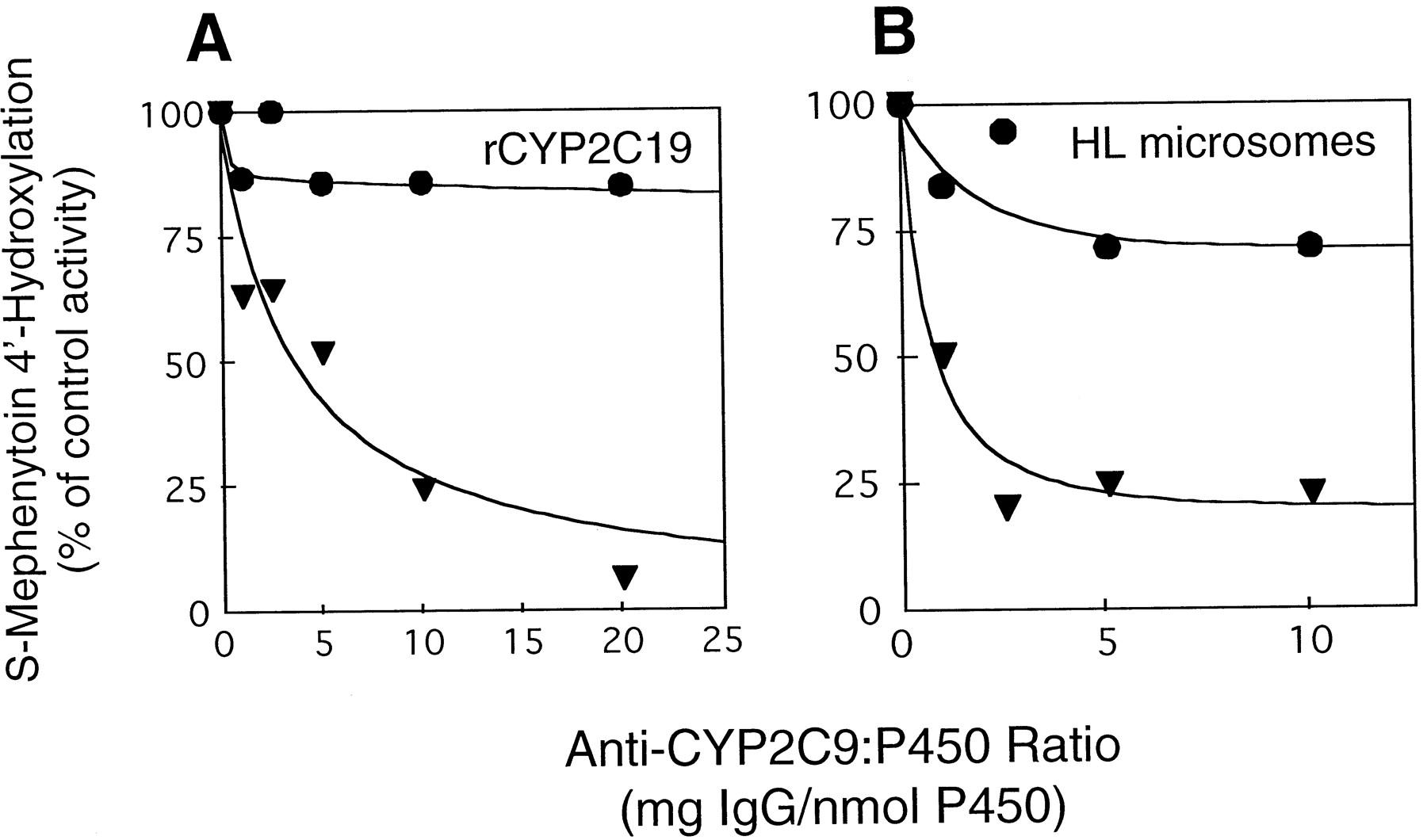
The effect of CYP2C9 antibodies on S-mephenytoin 4′-hydroxylase activity.
A, incubation mixtures contained a CYP2C19 reconstituted system (10 pmol of recombinant CYP2C19, 50 pmol of P-450 reductase, 3 μg of DLPC, and 40 pmol of b5), 50 mM HEPES buffer, (pH 7.4), 100 μM [14C]S-mephenytoin, 1 mM NADPH, and increasing amounts (0–200 μg) of preimmune IgG, anti-CYP2C9-P (▾), or anti-CYP2C9-M (●). The total amount of IgG added (anti-CYP2C9 plus preimmune) was maintained at 200 μg. B, incubation mixtures contained liver microsomes from subject H (25 pmol of CYP) 100 mM KPO4 buffer, 100 μM [14C]S-mephenytoin, 1 mM NADPH and increasing amounts (0–250 μg) of preimmune IgG, anti-CYP2C9-P (▾) or anti-CYP2C9-M (●). S-Mephenytoin 4′-hydroxylation was determined as described in Experimental Procedures, except that before reaction initiation, the CYP2C19-reconstituted system and microsomes were preincubated with antibodies for 3 min at 37°C, followed by 10 min at 25°C. Rates of hydroxymephenytoin formation in the presence of preimmune IgG were 3.6 nmol/min/nmol CYP for recombinant CYP2C19 and 0.18 nmol/min/nmol CYP for microsomes from subject H.
Effect of CYP2C9 antibodies on diclofenac and S-mephenytoin hydroxylation by human liver microsomes
CYP2C Enzymes Catalyzing Tolbutamide Hydroxylation.
We recently reported that tolbutamide hydroxylation in human liver microsomes was catalyzed by both CYP2C9 and CYP2C19 (Lasker et al., 1998). However, the lack of monospecific CYP2C antibodies prevented us from quantitatively assessing the contribution of CYP2C9 and CYP2C19 to overall microsomal metabolism of this drug. We thus examined the utility of anti-CYP2C9-M preparations to differentiate the respective roles of these CYP2C enzymes in tolbutamide hydroxylation by human liver samples that either contained CYP2C19 (CYP2C19+) or were deficient in the enzyme (CYP2C19−) (Table2). Incubation of CYP2C19+microsomes with optimal amounts of anti-CYP2C9-M produced 78 ± 6% inhibition of tolbutamide hydroxylase activity, whereas the same amount of anti-CYP2C9-P gave complete (100%) inhibition (Fig.4). With CYP2C19− microsomes, the inhibition of tolbutamide metabolism observed with anti-CYP2C9-M increased to 93 ± 4%. CYP2C9 immunoquantitation performed with anti-CYP2C9-M revealed a correlation (r = 0.72,P < .02) between CYP2C9 content and anti-CYP2C9-M inhibited rates of tolbutamide hydroxylation in the seven human samples examined. A somewhat weaker but still significant (r = 0.66; P < .05) correlation was noted between microsomal CYP2C9 levels and anti-CYP2C9-P inhibited rates of tolbutamide metabolism, which was similar to the correlation found between levels of this CYP enzyme and uninhibited rates of OHT formation.
CYP2C9 and CYP2C19 content in human liver microsomes
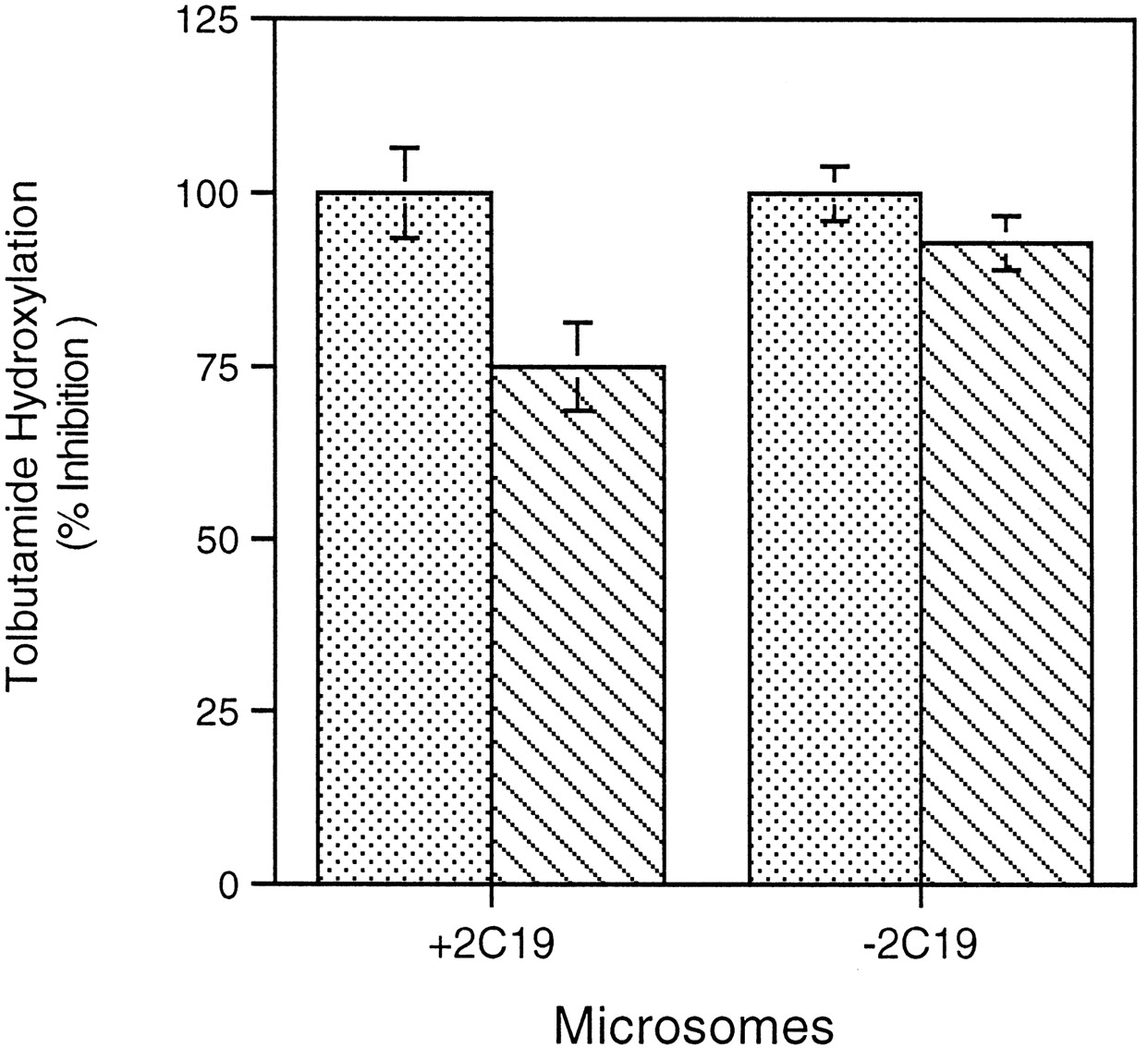
Effect of CYP2C9 antibodies on tolbutamide hydroxylation by human liver microsomes that contain or lack CYP2C19.
Tolbutamide hydroxylation was determined in incubation mixtures containing liver microsomes (50 pmol of CYP), 100 mM KPO4buffer (pH 7.4), 250 μM [14C]tolbutamide, 0.5 mM NADPH, and 250 μg of preimmune (control), anti-CYP2C9-P, or anti-CYP2C9-M IgG. Four CYP2C19+ microsomal preparations were compared with four CYP2C19− microsomal preparations (see Table 2). OHT formation was assessed as described in Experimental Procedures. The percent inhibition obtained in the presence of polyspecific anti-CYP2C9 and monospecific anti-CYP2C9 are represented by dotted bars and diagonal-hatched bars, respectively, and denote the mean ± S.E. from the four different subjects. Control rates of OHT formation (determined in the presence of preimmune IgG) were 0.365 ± 0.09 (P < .05) and 0.164 ± 0.12 (P < .05) nmol/min/nmol CYP for CYP2C19+ microsomes and CYP2C19− microsomes, respectively.
To confirm participation of a CYP other than CYP2C9 in microsomal tolbutamide metabolism, we examined the effects of sulfaphenazole, a specific CYP2C9 chemical inhibitor (Newton et al., 1995). Tolbutamide hydroxylation by recombinant CYP2C9 was inhibited by 75% in the presence of 50 μM sulfaphenazole, whereas this compound had negligible (4%) effects on the CYP2C19-mediated reaction. With CYP2C19+ microsomes, sulfaphenazole inhibited rates of OHT formation by an average of 57% (n = 2), whereas with CYP2C19− microsomes inhibition (65%, n = 2) was somewhat greater. Sulfaphenazole at 100 μM produced inhibition that was similar in CYP2C19− and CYP2C19+ microsomes, indicating a loss of specificity with increasing concentrations of the inhibitor.
Discussion
In this study, we demonstrated that both CYP2C9 and CYP2C19 participate in metabolism of the antidiabetic agent tolbutamide in human liver. Despite the marked sequence similarity between these CYP2C enzymes, we used an immunochemical approach to estimate the contribution of each CYP toward microsomal tolbutamide hydroxylation. After eliminating the cross-reactivity of CYP2C9 antibodies with CYP2C19 (and CYP2C8), use of the monospecific anti-CYP2C9 preparation indicated that CYP2C9 catalyzed from 78 to 93% of the total tolbutamide hydroxylation occurring in human hepatic microsomes, and the remaining 7 to 22% could be attributed to CYP2C19 catalysis. These findings were subsequently validated in experiments with the specific CYP2C9 chemical inhibitor, sulfaphenazole, which also indicated that CYP2C9 was the major tolbutamide hydroxylase in microsomes from human liver.
Tolbutamide has been used extensively as a probe of CYP function, a fact that may partly stem from the polymorphism purported to underlie its oxidative metabolism (Scott and Poffenbarger, 1979). Whereas the majority of studies have found that tolbutamide is specifically hydroxylated by CYP2C9, recent reports have suggested that this specificity may extend to another member of the human CYP2C subfamily, CYP2C19 (Richardson et al., 1995; Lasker et al., 1998; Venkatakrishnan et al., 1998). Indeed, we found that rates of tolbutamide hydroxylation mediated by native and recombinant CYP2C19 were essentially equivalent to those of purified CYP2C9 and the corresponding recombinant CYP (Lasker et al., 1998). However, as catalysis of a given drug-metabolizing reaction by a purified and/or recombinant CYP enzyme in reconstituted systems does not imply that it contributes extensively to the reaction in intact liver microsomes (Guengerich et al., 1998), we attempted to discriminate CYP2C19-catalyzed tolbutamide hydroxylation from that mediated by CYP2C9. A kinetic approach to this problem proved unsuccessful as the Michaelis constants for tolbutamide derived for these two CYP2C enzymes were similar, and different human liver specimens exhibited monophasic rather than biphasic tolbutamide hydroxylation kinetics (Lasker et al., 1998). Furthermore, an immunochemical approach was precluded due to the extensive cross-reactivity of the CYP2C9 and CYP2C19 antibody preparations used in our previous study. In fact, the marked (94–96%) inhibition of microsomal tolbutamide hydroxylation elicited by the latter IgG preparation grossly overestimated the contribution of CYP2C19 to the reaction.
Herein, we refined the immunochemical-based methodology to discern the respective roles of CYP2C9 and CYP2C19 in hepatic tolbutamide metabolism. In contrast to our previous efforts, we derived a monospecific CYP2C9 antibody from its polyspecific precursor by back-adsorption of the IgG against purified recombinant CYP2C19 coupled to a solid-phase support. Exposure of anti-CYP2C9-P to this immunoadsorbent gave an antibody preparation that: 1) reacted on protein blots with only CYP2C9 and no longer recognized either CYP2C19 or CYP2C8 (compare lanes 1–3 in Fig. 1, A and B); and 2) inhibited CYP2C9-catalyzed but not CYP2C19-catalyzed reactions. For instance, diclofenac 4′-hydroxylation mediated by either recombinant CYP2C9 or intact liver microsomes was inhibited to essentially the same extent by both monospecific and polyspecific CYP2C9 antibodies (Fig. 2, A and B). In contrast, the inhibition of CYP2C19-mediatedS-mephenytoin 4′-hydroxylation noted with anti-CYP2C9-P (Fig. 3A) was minimal but nonspecific after back-adsorption of this polyspecific CYP antibody against CYP2C19 (Fig. 3B). To the best of our knowledge, this is the first report of a specific inhibitory CYP2C polyclonal antibody that was originally raised to the corresponding native CYP apoprotein.
Using anti-CYP2C9-M as a specific immunochemical probe for CYP2C9-mediated catalysis demonstrated that this human CYP enzyme was the predominant tolbutamide hydroxylase in human liver microsomes, confirming our earlier observations (Lasker et al., 1998). Indeed, antibodies to CYP2C9, even after back-adsorption, retained the capacity to markedly inhibit (78–93%) microsomal OHT formation (Fig. 4). A significant correlation (r = 0.72; P < .02) was established between hepatic CYP2C9 content and anti-CYP2C9-M inhibitable rates of tolbutamide hydroxylation in the seven subjects studied. Nevertheless, several lines of evidence presented here indicated that CYP2C19 was also involved in hepatic tolbutamide metabolism. First, the capacity of anti-CYP2C9-M to inhibit tolbutamide metabolism was reduced in hepatic microsomes containing CYP2C19 (78 ± 6% inhibition) compared with microsomes deficient in CYP2C19 (93 ± 4% inhibition) (Fig. 4). Similarly, less average inhibition of tolbutamide hydroxylase activity with CYP2C19+ liver microsomes compared with CYP2C19− microsomes (Table 2) was also observed with sulfaphenazole, a known chemical inhibitor of CYP2C9-mediated reactions (Newton et al., 1995 and references therein). Evidence that sulfaphenazole, like anti-CYP2C9-M, was a specific probe for CYP-mediated tolbutamide metabolism was indicated by its capacity to inhibit OHT formation only by recombinant CYP2C9 and not by CYP2C19 (Table 2). Despite this specificity, sulfaphenazole proved considerably less efficacious than anti-CYP2C9-M as an inhibitor of microsomal tolbutamide metabolism (concentrations higher than 50 μM failed to elicit additional decreases in metabolic rates), and the results obtained with this chemical inhibitor were not as clear-cut as those obtained with the immunochemical reagents. Finally, no clear relationship (r = 0.62; P = .14) was found between hepatic CYP2C19 levels and tolbutamide hydroxylase activity, which was expected because of the low hepatic CYP2C19 content relative to that of CYP2C9 in the subjects studied (seeResults).
In conclusion, our results indicate that CYP2C9 and CYP2C19 function as the major and minor tolbutamide hydroxylases, respectively, in human liver. When considered together with abundance of hepatic CYP2C9 relative to that of CYP2C19 (Inoue et al., 1997; Lasker et al., 1998), our findings imply that the latter CYP typically plays a nominal role in the metabolism of this oral antidiabetic agent. However, CYP2C19 involvement in hepatic tolbutamide disposition may be much greater in subjects who express CYP2C9 and CYP2C19 at comparable levels or in individuals who express a catalytically deficient CYP2C9 enzyme, such as the CYP2C9V1 allelic variant (Sullivan-Klose et al., 1996). This CYP2C9 variant exhibits a marked decrease in both tolbutamide and S-warfarin hydroxylase activity compared with the wild-type CYP2C9 as a result of an increasedKm and decreasedVmax for these therapeutic substrates. Clearly, more studies of this type are required to better differentiate the contribution of individual CYP2C enzymes to the oxidative metabolism of other pharmaceutical agents.
Footnotes
-
Send reprint requests to: Dr. Judy L. Raucy, The La Jolla Institute for Experimental Medicine, 505 Coast Blvd. S., La Jolla, CA 92037. E-mail: jraucy{at}agi.org
-
↵1 This work was presented in partial fulfillment of the requirements for a doctoral degree (M.R.W.) in Toxicology at the University of New Mexico.
-
↵2 Present address: The Scripps Research Institute, Dept. of Molecular and Experimental Medicine, La Jolla, CA 92037.
-
Supported by National Institutes of Health Grants GM49511 (J.L.R., J.M.L.), AA07842 (J.M.L.), GM31001 (E.F.J.), and DK62274 (Liver Transplant, Procurement and Distribution System).
- Abbreviations used are::
- CYP
- cytochrome P-450
- b5
- cytochromeb5
- P-450 reductase
- NADPH:P-450 oxidoreductase
- anti-CYP2C9-P
- polyspecific CYP2C9 antibodies
- anti-CYP2C9-M
- monospecific CYP2C9 antibodies
- DLPC
- l-α-dilauroylphosphatidylcholine
- DTT
- dithiothreitol
- OHT
- hydroxytolbutamide
- CYP2C19+, microsomes containing CYP2C19
- CYP2C19−, CYP2C19-deficient microsomes
- Received July 27, 1999.
- Accepted November 11, 1999.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}