


Abstract
Drug-drug interactions between tolbutamide and sulfonamides have extensively been reported. We attempted to predict the in vivo interaction between tolbutamide and sulfonamides from the in vitro metabolic inhibition studies. The inhibition constant (Ki) was derived from the inhibitory effects of eight sulfonamides (sulfaphenazole, sulfadiazine, sulfamethizole, sulfisoxazole, sulfamethoxazole, sulfapyridine, sulfadimethoxine, and sulfamonomethoxine) on tolbutamide metabolism. We found that the inhibitory effect of sulfaphenazole was greatest among the eight sulfonamides examined. Furthermore, the contribution of each P450 enzyme to tolbutamide metabolism was investigated by using recombinant P450 enzymes. Although cytochrome P450 (CYP) 2C8, 2C9, and 2C19 metabolized tolbutamide, the main enzyme involved was CYP2C9. TheKi values of several sulfonamides were comparable between human liver microsomes and recombinant CYP2C9. The maximum unbound plasma concentration of sulfonamides in the portal vein was calculated from literature data on the pharmacokinetics of sulfonamides. Using the Ki values obtained from in vitro inhibition studies, the degree of increase in tolbutamide area under the plasma concentration-time curve (AUC) was predicted. About 4.8- and 1.6-fold increases in tolbutamide AUC were predicted by coadministration of sulfaphenazole and sulfamethizole, respectively, which agreed well with the reported increases in humans. Furthermore, the increase in tolbutamide AUC by coadministration of sulfadiazine, sulfisoxazole, and sulfamethizole was predicted to be 1.5- to 2.6-fold, although the corresponding in vivo effects have not been reported. It is concluded that some of these sulfonamides have to be carefully coadministered with CYP2C9 substrates such as tolbutamide although coadministration of sulfaphenazole needs the greatest care.
Drug-drug interaction may cause serious side effects by raising the blood concentration of a drug whose metabolism is inhibited by coadministered drug. It is, therefore, important to predict any change in drug disposition caused by a drug-drug interaction. There are many reports of the prediction of in vivo drug disposition in humans based on animal experiments (Iwatsubo et al., 1996, 1997). However, because of species differences in metabolic enzymes, not only the metabolic activity but also metabolic pathway of drugs in animals may be different from those in humans. Therefore, predicting in vivo drug disposition in humans from animal data may sometimes lead to incorrect results. Recently, human liver samples and recombinant enzymes have become readily available, and prediction based on in vitro experiments is becoming more important (Iwatsubo et al., 1997; Ito et al., 1998a).
In this study, in vivo drug-drug interactions involving tolbutamide metabolism in humans were predicted based on in vitro studies using human liver microsomes and recombinant cytochrome P450 (CYP)1enzymes. The aromatic methyl group of tolbutamide (1-butyl-3-p-tolylsulfonylurea), an antidiabetic drug, is hydroxylated mainly by CYP2C9 in the liver, giving hydroxytolbutamide (Thomas and Ikeda, 1966). Hydroxytolbutamide is further oxidized by alcohol dehydrogenase and excreted (Thomas and Ikeda, 1966; Scott and Poffenbarger, 1979). About 80% of total tolbutamide elimination is accounted for by this single and sequential metabolic pathway. It is, therefore, reasonable to believe that coadministered P450 inhibitors may cause drug-drug interactions with tolbutamide due to metabolic inhibition.
It has been reported that sulfaphenazole, an antibacterial drug used to treat tuberculosis, etc., increases the area under the plasma concentration-time curve (AUC) of tolbutamide 5-fold in humans (Veronese et al., 1990b), and can provoke a hypoglycemic attack (Christensen et al., 1963). This was followed by a report that sulfaphenazole is a specific inhibitor of CYP2C9, and competitively inhibits the metabolism of tolbutamide (Pond et al., 1977;Miners et al., 1982, 1995; Back et al., 1988; Brian et al., 1989;Bourrie et al., 1996). Moreover, some sulfonamides have been reported to inhibit the metabolism of tolbutamide and to increase the blood concentrations of tolbutamide in both humans and animals (Hansen and Christensen, 1977; Thiessen and Rowland, 1977; Sugita et al., 1984a,b; Veronese et al., 1990b).
We have proposed a method for the quantitative prediction of in vivo drug-drug interactions from in vitro data (Sugiyama et al., 1996; Ito et al., 1998a,b). Interaction between tolbutamide and sulfaphenazole has been successfully predicted using the pharmacokinetic data on both drugs reported in literature (Ito et al., 1998b). In this study, we used sulfaphenazole and seven other commercially available sulfonamides (sulfadiazine, sulfamethizole, sulfisoxazole, sulfamethoxazole, sulfapyridine, sulfadimethoxine, and sulfamonomethoxine) to investigate the inhibitory effects on tolbutamide hydroxylation in a series of in vitro experiments. Furthermore, the contribution of each P450 enzyme to in vivo tolbutamide metabolism was estimated using physiological amounts of recombinant P450 enzymes. Taking the pharmacokinetic feature of each sulfonamide into consideration, we predicted the degree of increase in tolbutamide AUC caused by coadministration of sulfonamides from the in vitro experiments using microsomes obtained from human livers and CYP2C9-expressed lymphoblastoid cells.
Materials and Methods
Chemicals and Reagents.
Tolbutamide, diethylether, sulfamonomethoxine, sulfadimethoxine, sulfamethoxazole, magnesium chloride, hydrochloric acid, and dipotassium hydrogenphosphate were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Hydroxytolbutamide was kindly provided by Daiichi Pure Chemicals, Co. Ltd. (Tokyo, Japan). NADP, glucose 6-phosphate, and glucose 6-phosphate dehydrogenase were obtained from Boehringer Mannheim (Mannheim, Germany). Sulfaphenazole, sulfisoxazole, sulfadiazine, and sulfapyridine were purchased from Sigma Chemical Co. (St. Louis, MO). Methanol of HPLC grade was purchased from Wako Pure Chemical Industries, Ltd. All other chemicals were of reagent grade.
Human Liver Microsomes and Recombinant CYP Enzymes.
Human liver microsomes obtained from ten donors (six males and four females; 31–57 years old; H-19, H-35, H-36, H-38, H-50, H-51, H-56, H-57, H-66, H-67) were generous gifts among 26 different microsomes prepared from human livers stored in the human liver bank of SRI International (Menlo Park, CA). Microsomal preparations of recombinant human CYP enzymes expressed by the human B lymphoblastoid cell line, AHH-1 (recombinant microsomes) were a gift from Gentest Corp. (Woburn, MA). The level of CYP2C9 in each microsome was assayed by immunoblotting as described previously (Imaoka et al., 1996).
Metabolic Assay of Tolbutamide Hydroxylation by Human Liver Microsome or Recombinant CYP2C9.
Tolbutamide was incubated with reaction mixture (1 ml) consisting of 0.2 mg of human liver microsomal protein or 3.5 pmol of recombinant CYP2C9 and NADPH-generating system (1 mM NADP, 10 mM glucose 6-phosphate, 0.1 U/ml glucose 6-phosphate dehydrogenase, and 5 mM MgCl2) in 100 mM potassium phosphate buffer (pH 7.4). Reactions were initiated by adding 100 μl of the NADPH-generating system (preincubated for 5 min). After incubation at 37°C in a shaking water bath for 75 min (human liver microsomes) or 60 min (recombinant CYP2C9), incubations were terminated by adding 100 μl of 2 M hydrochloric acid. After terminating the reaction, 2 ml of diethylether and 100 μl of chlorpropamide solution (internal standard; 6 μg/ml) were added. Incubation mixtures were centrifuged for 10 min (3000 rpm). The supernatant fraction from each incubation was transferred to a 15-ml test tube and evaporated to dryness under a mild stream of N2 gas. Residues were resuspended in 100 μl of the HPLC mobile phase, and 50 μl was used for analysis. The chromatograph was fitted with a TOSOH ODS-80TM column, which was eluted with 0.05% phosphoric acid/methanol (60:40) at a flow rate of 1 ml/min. Peaks were monitored by ultraviolet detection at 235 nm (Csillag et al., 1989; St-Hilaire and Belanger, 1989; Ho and Moody, 1992). Retention times for tolbutamide, hydroxytolbutamide, and chlorpropamide were 31.0, 7.9, and 21.0 min, respectively. The metabolic rate by human liver microsomes and recombinant CYP enzymes was linear for at least 120 and 60 min, respectively. All of the experiments were performed in triplicate unless otherwise indicated.
Enzyme Kinetics of Tolbutamide Hydroxylation by Human Liver Microsomes or Recombinant CYP2C9.
Using four human liver microsomes (H-38, H-56, H-66, H-67) or recombinant CYP2C9 (Lot 2, Lot 27), enzyme kinetics for tolbutamide hydroxylation was investigated. Concentration of tolbutamide was set at 50, 75, 100, 125, 250, 500, 750, and 1000 μM. The initial formation rate of hydroxytolbutamide was plotted against concentration of substrate. Km and maximum metabolic rate (Vmax) values were estimated by the nonlinear least-squares regression method “MULTI” (Yamaoka et al., 1981).
Correlation between Tolbutamide Hydroxylating Activity and 2C9 Content of Human Liver Microsomes.
Metabolic assays for tolbutamide hydroxylation were carried out using ten human liver microsomes (H-19, H-35, H-36, H-38, H-50, H-51, H-56, H-57, H-66, H-67) containing various amounts of CYP2C9. The initial concentration of tolbutamide was set at 100 μM and 1 mM. We investigated the correlation between CYP2C9 content of each microsome and the metabolic activity.
Identification of the CYP Enzyme Involved in Tolbutamide Hydroxylation.
Metabolic assays for tolbutamide hydroxylation were carried out using nine enzymes of P450 (1A1, 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4). The initial concentration of tolbutamide was set at 300 μM. Reaction mixtures were incubated for 60 min. To estimate the contribution of each enzyme in in vivo tolbutamide metabolism, we used average amounts of enzyme contained in 0.2 mg of human liver microsomal protein (Shimada et al., 1994). The amounts of each enzyme used were 8.4 pmol of CYP1A1 or 1A2, 0.2 pmol of 2B6, 3.5 pmol of 2C8, 2C9, or 2C19, 1.0 pmol of 2D6, 4.4 pmol of 2E1, or 19.2 pmol of 3A4.
Inhibition Study.
Inhibitory effects of eight sulfonamides on tolbutamide metabolism were investigated using H-67 microsome. The concentration of tolbutamide was set at 100 μM. The inhibition constant (Ki) was obtained by fitting the inhibition curve to the following equation using the nonlinear least-squares regression method “MULTI”:
Prediction of Increase in AUC of Tolbutamide from In Vitro Metabolic Data.
In the case of competitive or noncompetitive inhibition, the ratio of intrinsic metabolic clearance (CLint) in the presence and absence of the inhibitor can be described as follows when the substrate concentration is much lower thanKm:
Results
Enzyme Kinetics of Tolbutamide Hydroxylation by Human Liver Microsomes.
Tolbutamide metabolism to hydroxytolbutamide was studied using four human liver microsomes that contained different amounts of CYP2C9. The hydroxylation by all microsomes followed Michaelis-Menten kinetics. Figure 1 shows typical Eadie-Hofstee plots for two microsomal samples. Km andVmax values obtained by nonlinear least-squares regression method are summarized in Table1.

Eadie-Hofstee plots for tolbutamide hydroxylation by human liver microsomes H-38 (a) and H-67 (b).
Reaction mixture contained 0.2 mg of microsomal protein, and was incubated for 75 min.
Computer derived Michaelis-Menten parameters for hydroxylation of tolbutamide by human liver microsomes and recombinant CYP2C9
Interindividual Difference in Tolbutamide Hydroxylation by Human Liver Microsomes.
Good correlations were observed between CYP2C9 content and tolbutamide hydroxylating activity of ten human liver microsomes (Fig.2). The correlation coefficient wasr = 0.955 and r = 0.904 when tolbutamide concentration was set at 1 mM and 100 μM, respectively.
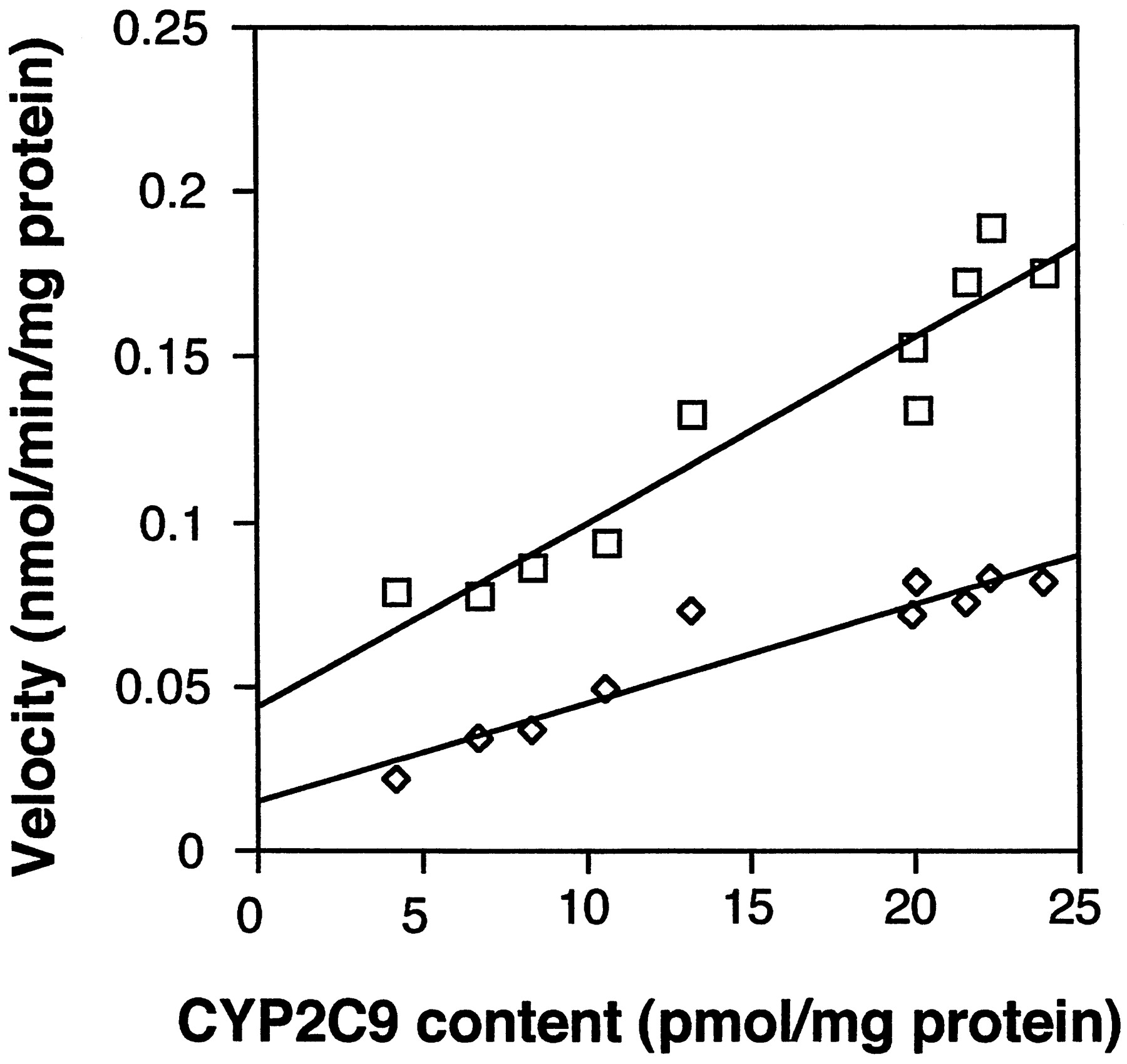
CYP2C9 content dependence of tolbutamide hydroxylation.
The metabolic rates of tolbutamide by ten microsomes are plotted against CYP2C9 content in each microsome. The concentrations of tolbutamide were 1 mM (■; r = 0.955) and 100 μM (⋄; r = 0.904). Reaction mixtures contained 0.2 mg of human liver microsomes and were incubated for 75 min.
Contribution of Each CYP Enzyme to Tolbutamide Hydroxylation.
The CYP enzymes expressed in human B lymphoblastoid cells did not metabolize tolbutamide except for CYP2C8, 2C9, and 2C19 (Fig.3). The tolbutamide hydroxylation activity of CYP2C8 was less than one-third that of CYP2C9. When the same amounts of CYP2C enzymes were used for the assay, CYP2C19 had an even weaker activity, only about one-fifth that of CYP2C9. We also used the physiological amount of CYP2C19 (0.27 pmol; average amount of ten human liver microsomes used in our assay) for this assay, but 0.27 pmol of CYP2C19 did not produce hydroxytolbutamide more than the detection limit. From these data, it was shown that the major enzyme for tolbutamide hydroxylation is CYP2C9.

The contribution of each P450 enzyme in tolbutamide hydroxylation.
Reaction mixture contained 8.4 pmol of recombinant CYP1A1 or 1A2; 0.2 pmol of 2B6; 3.5 pmol of 2C8, 2C9, or 2C19; 1.0 pmol of 2D6; 4.4 pmol of 2E1; or 19.2 pmol of 3A4. Each recombinant enzyme was incubated with tolbutamide (300 μM) for 60 min. Each point and vertical bar represents the mean ± S.E.
Enzyme Kinetics of Tolbutamide Hydroxylation by Recombinant CYP2C9.
Tolbutamide hydroxylation by two lots of recombinant CYP2C9 that was expressed by human B lymphoblastoid cells followed Michaelis-Menten kinetics (Fig. 4).Km and Vmaxvalues obtained by the nonlinear least-squares regression method were approximately 130 to 190 μM and 8.3 to 9.1 pmol/min/pmol enzyme, respectively, which were comparable with those obtained by human liver microsomes (Table 1).

Tolbutamide hydroxylation by recombinant CYP2C9.
Reaction mixture contained 3.5 pmol of recombinant CYP2C9, and was incubated for 60 min. Each point and vertical bar represents the mean ± S.E. (a) and (b), Lot 2; (c) and (d), Lot 27.
Inhibitory Effects of Sulfonamides.
All of the eight sulfonamides showed inhibitory effects on tolbutamide hydroxylation in both human liver microsomes (Fig.5) and recombinant CYP2C9 (Fig.6). The extent of inhibition, however, differed among the sulfonamides. The most potent inhibitor was sulfaphenazole whose Ki value in human liver microsomes was about 0.3 μM, which was much smaller than that of other sulfonamides (Table 2). Among the drugs investigated, sulfadiazine and sulfamethizole were relatively potent inhibitors with Kivalues of about 50 μM. The Ki values of sulfaphenazole and these two drugs in the recombinant CYP2C9 were comparable with those in human liver microsomes (Table3).
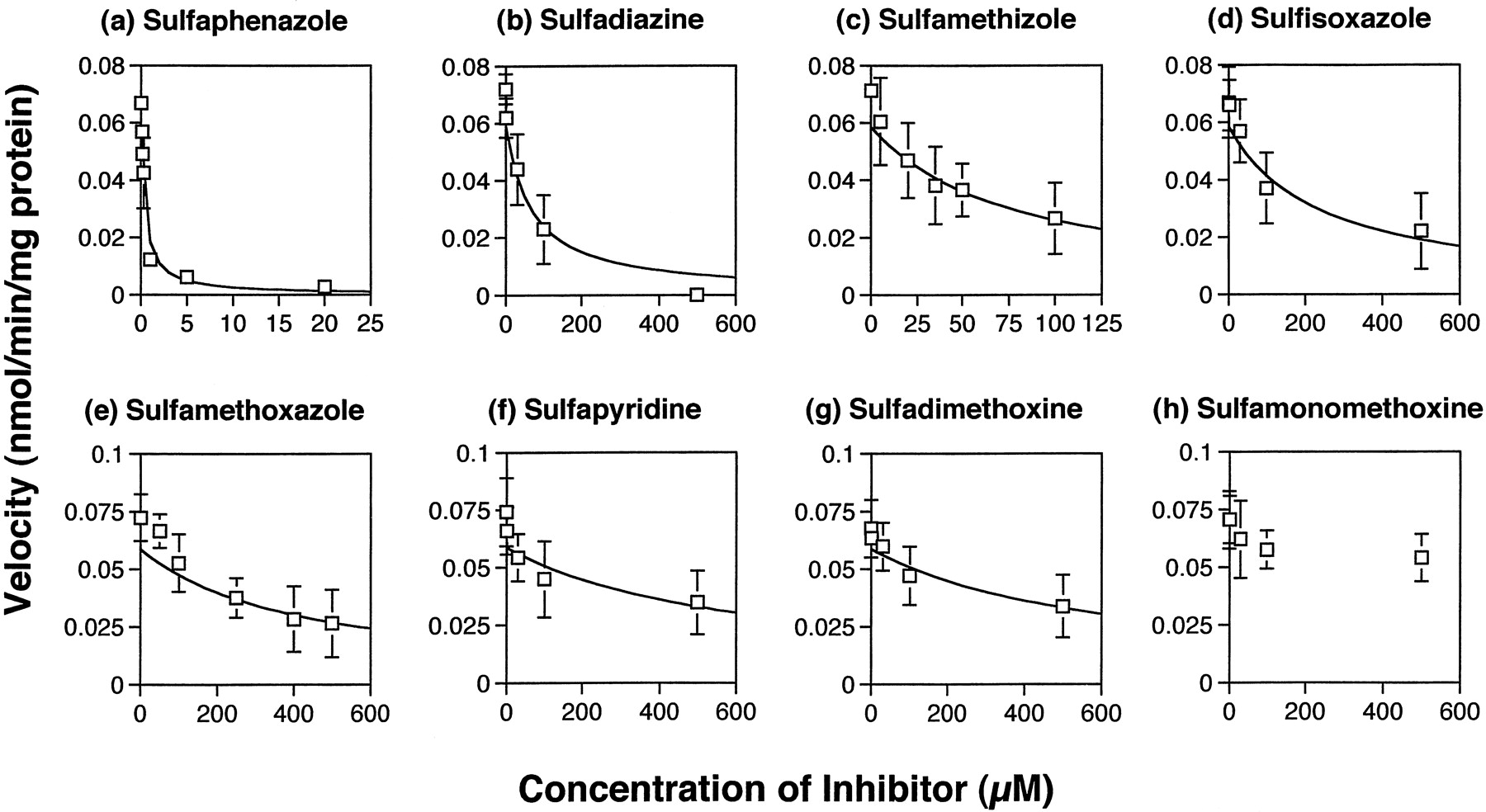
Inhibitory effects of sulfonamides on tolbutamide metabolism by human liver microsome (H-67).
Reaction mixture contained 100 μM tolbutamide.Ki values for the sulfonamides are summarized in Table 2. Each point and vertical bar represents the mean ± S.E. Curves represent the fitting lines (eq. 1). a, sulfaphenazole (n = 6); b, sulfadiazine (n = 6); c, sulfamethizole (n = 4); d, sulfisoxazole (n = 3); e, sulfamethoxazole (n = 4); f, sulfapyridine (n = 6); g, sulfadimethoxine (n = 6); h, sulfamonomethoxine (n = 3).

Inhibitory effects of sulfonamides on tolbutamide metabolism by recombinant CYP2C9 (Lot 27).
Reaction mixture contained 100 μM tolbutamide.Ki values for the sulfonamides are summarized in Table 3. Each point and vertical bar represents the mean ± S.E. Curves represent the fitting lines (eq. 1). a, sulfaphenazole; b, sulfadiazine; c, sulfamethizole.
The Ki values for the inhibition of tolbutamide metabolism by various sulfonamides in human liver microsomes
The Ki values for the inhibition of tolbutamide metabolism by various sulfonamides in recombinant CYP2C9 (Lot 27)
Inhibition Type of Sulfonamides.
The slope of the Eadie-Hofstee plot for tolbutamide hydroxylation by human liver microsomes was decreased by both sulfaphenazole and sulfamethizole with no substantial change in x-intercept (Fig. 7). This showed that inhibition type of sulfonamides was competitive. Furthermore, tolbutamide hydroxylation activities of human liver microsome (H-67) after a 10- and 40-min preincubation with 3 μM sulfaphenazole were equal to the control value without preincubation.

Eadie-Hofstee plot for tolbutamide metabolism by human liver microsome (H-67) in the presence of 0.35 μM sulfaphenazole (■), 50 μM sulfamethizole(○), and vehicle (⋄).
Each point and vertical bar represents the mean ± S.E. Reaction mixture was incubated for 75 min.
Prediction of AUC Increase.
Iu (maximum unbound concentration of inhibitor in the portal vein) was calculated from literature data on an average dose in clinical practice, plasma unbound fraction, maximum systemic concentration, absorption rate constant, and fraction absorbed of each sulfonamide (Table 4). The calculated degrees of increase in tolbutamide AUC caused by coadministration of sulfonamides are summarized in Table5. The most potent inhibitor was sulfaphenazole, which was estimated to increase tolbutamide AUC about 5-fold. Tolbutamide AUC was estimated to be increased around or more than 2-fold by three sulfonamides that have relatively smallKi values, i.e., sulfamethizole, sulfadiazine, and sulfisoxazole. AUC increase by other sulfonamides was predicted to be less than 1.5-fold.
Summary of pharmacokinetic parameters of sulfonamides
Prediction of in vivo interaction between tolbutamide and sulfonamides
Discussion
The purpose of this study was to predict in vivo drug-drug interactions in humans quantitatively from in vitro experiments. If metabolism of tolbutamide is inhibited, the elevated concentration may cause side effects of tolbutamide such as hypoglycemia attack. Because tolbutamide is metabolized by a single pathway (Thomas and Ikeda, 1966), its inhibition will have serious effects.
Tolbutamide hydroxylation in human liver microsomes followed Michaelis-Menten kinetics (Fig. 1). Km andVmax values obtained in the present study (Table 1) were consistent with those reported by Doecke et al. (1991;Km = 85.6 μM) and by Miners et al. (1988;Km = 120 μM,Vmax = 0.273 nmol/min/mg).
In metabolic studies using ten human liver microsomes containing various amounts of CYP2C9, significant correlation was observed between CYP2C9 content and initial velocity of tolbutamide hydroxylation (Fig.2). This suggests that hydroxylation of tolbutamide is mainly mediated by CYP2C9, which supports the previous reports (Knodell et al., 1987;Miners et al., 1988; Back and Orme, 1989; Brian et al., 1989; Veronese et al., 1990a,1993). Furthermore, estimation of the contribution of each enzyme using recombinant CYP enzymes also showed that CYP2C9 is a major enzyme in tolbutamide hydroxylation (Fig. 3). Figure 3 shows that tolbutamide is slightly metabolized also by the other CYP2C enzymes. Some groups have reported that CYP2C8 is also involved in hydroxylation of tolbutamide (Relling et al., 1990; Srivastava et al., 1991; Veronese et al., 1993). However, the average level of CYP2C8 in human liver microsomes is reported to be less than one-third that of CYP2C9 (Imaoka et al., 1996), suggesting little contribution of CYP2C8 in tolbutamide hydroxylation. Nevertheless, the correlation between tolbutamide hydroxylation activity and CYP2C9 content does not intercept at the origin (Fig. 2), suggesting that part of tolbutamide hydroxylation (e.g., about 25% of total hydroxylation of 100 μM tolbutamide by microsomes with average content of CYP2C9, i.e., about 15 pmol/mg protein) may be mediated by some other enzymes. It cannot be ruled out that such enzymes as CYP2A6, CYP3A5, or CYP4A9/11 etc., the recombinant systems of which have not been investigated in Fig. 3, may be partly involved in tolbutamide hydroxylation.
The Km and Vmaxvalues (per picomole of CYP2C9) for tolbutamide hydroxylation by recombinant CYP2C9 were comparable with those obtained using human liver microsomes (Table 1). This finding indicates that recombinant CYP2C9 can be used as an alternative to human liver microsomes in prediction of in vivo drug interactions of tolbutamide.
The inhibition study using human liver microsomes showed that sulfonamides had various Ki values for tolbutamide hydroxylation (Table 2). Sulfaphenazole was the most potent inhibitor among all the sulfonamides examined. From the Dixon plot analysis using human liver microsomes, theKi values of sulfaphenazole, sulfamethizole, and sulfamethoxazole for tolbutamide hydroxylation are reported to be 0.3, 35, and 254 μM, respectively (Back et al., 1988), which are consistent with our results. TheKi values of sulfonamides, except for sulfaphenazole, showed that they are less potent inhibitors. This experimental result could explain why there are fewer reports of elevation of tolbutamide concentration or hypoglycemia due to these other sulfonamides. In addition, tolbutamide hydroxylation by recombinant CYP2C9 was also inhibited by sulfonamides (Table 3).Ki values obtained in this study were comparable with those obtained in the liver microsomal study.
The reason why only sulfaphenazole has such a potent inhibitory effect is unknown. One hypothesis is that sulfaphenazole could inhibit tolbutamide hydroxylation by a mechanism-based inhibition (Ito et al., 1998b). However, metabolic assay was performed after a 10- and 40-min preincubation of human liver microsomes with sulfaphenazole and the inhibitory effect exhibited no significant differences, suggesting that the mechanism-based inhibition is not involved in the inhibition of CYP2C9 by sulfaphenazole. Also, an Eadie-Hofstee plot analysis using a fixed concentration of inhibitor showed changes inKm values with minor changes inVmax values (Fig. 7), indicating a competitive inhibition at least at this concentration of inhibitor. TheKi values for sulfaphenazole and sulfamethizole estimated from Fig. 7 (0.22 and 39 μM, respectively) were not so different from those obtained from Fig. 5, assuming a competitive inhibition (0.31 and 53 μM, respectively; Table 5), suggesting that a competitive inhibition also takes place at other concentrations of inhibitor.
The AUC of tolbutamide (500 mg p.o.) was predicted to increase about 5-fold by coadministration of sulfaphenazole 500 mg, which is equal to the reported increase (Veronese et al., 1990b) (Table 5). Furthermore, the AUC of tolbutamide (750 mg p.o.) is reported to increase by 1.6 times when sulfamethizole (1 g) is coadministered (Lumholtz et al., 1975), which was also predicted well by our method. Our prediction is based on avoiding false negatives, so that the portal vein concentration of the inhibitor may be overestimated. Even under this condition, the predicted increase in plasma concentration of tolbutamide is still not very high except for sulfaphenazole, suggesting that the risk of hypoglycemia is limited when sulfonamides other than sulfaphenazole are coadministered with tolbutamide. However, it should be kept in mind that a fewfold increase in the AUC of tolbutamide may occur by coadministration of other sulfonamides (sulfadiazine, sulfisoxazole, sulfamethizole). Furthermore, predictions in the present study were based on plasma concentrations of sulfonamides after single administration of their typical therapeutic dose. Concentrations in the clinical situation, especially in the case of repeated administration of higher doses of the sulfonamides, may be higher than estimated in this study, which may result in greater degrees of in vivo interactions.
In this study, we systematically evaluated the inhibitory effects of sulfonamides on tolbutamide metabolism mediated by CYP2C9. This evaluation may also be applied to other drugs that are metabolized by CYP2C9. Therefore, attention should be paid in coadministration of sulfonamides with relatively small Kivalues (sulfaphenazole, sulfadiazine, sulfamethizole, and sulfisoxazole) and CYP2C9 substrates with narrow therapeutic ranges such as phenytoin (an antiepileptic) and warfarin (an anticoagulant).
Footnotes
-
Send reprint requests to: Yuichi Sugiyama, Graduate School of Pharmaceutical Sciences, University of Tokyo, 7–3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail:sugiyama{at}seizai.f.u-tokyo.ac.jp
- Abbreviations used are::
- CYP
- cytochrome P450
- AUC
- area under the plasma concentration-time curve
- Fa
- fraction absorbed from the intestinal tract
- fp
- unbound fraction in plasma
- Imax
- maximum plasma concentration in circulating blood
- Iu
- unbound concentration of inhibitor
- ka
- absorption rate constant
- Vmax
- maximum metabolic rate
- Received July 26, 1999.
- Accepted January 10, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}