


Abstract
The UDP-glucuronosyltransferases (UGTs) are a superfamily of membrane-bound enzymes whose active site is localized inside the endoplasmic reticulum. Glucuronidation using human liver microsomes has traditionally involved disruption of the membrane barrier, usually by detergent treatment, to attain maximal enzyme activity. The goals of the current work were to develop a universal method to glucuronidate xenobiotic substrates using microsomes, and to apply this method to sequential oxidation-glucuronidation reactions. Three assays of UGT catalytic activity estradiol-3-glucuronidation, acetaminophen-O-glucuronidation, and morphine-3-glucuronidation, which are relatively selective probes for human UGT1A1, 1A6, and 2B7 isoforms, respectively, were developed. Treatment of microsomes with the pore-forming peptide alamethicin (50 μg/mg protein) resulted in conjugation rates 2 to 3 times the rates observed with untreated microsomes. Addition of physiological concentrations of Mg2+ to the alamethicin-treated microsomes yielded rates that were 4 to 7 times the rates with untreated microsomes. Optimized assay conditions were found not to detrimentally affect cytochrome P450 activity as determined by effects on testosterone 6β-hydroxylation and 7-ethoxycoumarin deethylation. Formation of estradiol-3-glucuronide displayed atypical kinetics, and data best fit the Hill equation, yielding apparent kinetic parameters of Kmapp = 0.017 mM,Vmaxapp = 0.4 nmol/mg/min, and n = 1.8. Formation of acetaminophen-O-glucuronide also best fit the Hill equation, with Kmapp = 4 mM, Vmaxapp = 1.5 nmol/mg/min, and n = 1.4. Alternatively, morphine-3-glucuronide formation displayed Michaelis-Menten kinetics, with Kmapp = 2 mM andVmaxapp = 2.5 nmol/mg/min. Finally, alamethicin treatment of microsomes was found to be effective in facilitating the sequential oxidation-glucuronidation of 7-ethoxycoumarin.
Because a successful drug possesses a desirable pharmacokinetic profile in addition to pharmaceutical potency, drug metabolism plays an important role throughout the drug discovery and development processes. The ability to identify large numbers of biologically potent molecules has led to a need for efficient drug metabolism screening methods (Eddershaw and Dickins, 1999). Because hepatic cytochromes P450 (CYPs)2are responsible for a large percentage of the metabolism of drugs in vivo, human liver microsomes appear to be the in vitro drug metabolism screening system of choice (Rodrigues, 1994). Drug candidates can be screened for stability to CYP-mediated metabolism and for a favorable CYP inhibition profile using human liver microsomes (Crespi et al., 1998; Ito et al., 1998). Although UDP-glucuronosyltransferases (UGTs) are often involved in drug metabolism, candidates are not routinely examined for in vitro metabolic stability to conjugation or a favorable UGT inhibition profile. This is in part due to the lack of standard microsomal incubation conditions for the UGTs and the paucity of substrate specificity data (Burchell et al., 1995).
The UGTs are a superfamily of membrane-bound enzymes that catalyze the conjugation of endo- and xenobiotics with d-glucuronic acid. Conjugation can occur at hydroxyl, carboxylic acid, amino, and even carbon centers (Miners and Mackenzie, 1991). The resulting β-glucuronide metabolites possess increased aqueous solubility compared with the substrate, enhancing its biliary or renal excretion. Glucuronides excreted in the bile may undergo enterohepatic recycling via glucuronidase-catalyzed hydrolysis and subsequent intestinal reabsorption of the parent drug (Rowland and Tozer, 1995). Glucuronidation in this situation is primarily considered to be storage of parent drug. In general, glucuronide formation leads to termination of pharmacological potency. However, relevant examples exist of glucuronidation being a bioactivation event, which contributes to observed pharmacodynamic complexities (Mackenzie, 1995; Christrup, 1997)
In contrast to CYPs and the flavin-containing monooxygenases, the active site of the UGTs resides in the lumen of the endoplasmic reticulum (ER). Thus, a “latency” of activity occurs because the ER membrane provides a diffusional barrier for substrates, cofactors, and products (Meech and Mackenzie, 1997). In a microsomal incubation, disruption of this barrier is required to remove the latency and observe maximum enzyme activity. Most investigators use detergents for this purpose, empirically determining the optimum detergent type and concentration for the activity of interest. Therefore, assays with different substrates generally require different conditions (Lett et al., 1992). In addition, bell-shaped curves are often observed in detergent titrations (Fulceri et al., 1994), implying that detergents are affecting enzyme activity in addition to membrane permeability. For example, detergent treatment may affect the enzyme itself because it has been reported that detergent can make the UGT enzyme intrinsically more active (Trapnell et al., 1998).
The xenobiotic metabolizing UGTs exist in two subfamilies, designated 1A and 2B (Jedlitschky et al., 1999). Multiple members of these subfamilies exist in humans, and a nomenclature method has recently been adopted (Mackenzie et al., 1997). This nomenclature is directly analogous to the CYP nomenclature. Although specific probes have begun to elucidate the pattern of gene expression of these isoforms in different tissues, the lack of standard conditions for in vitro glucuronidation and the need for a complete battery of isoform-selective catalytic activities has hindered the development of a method to quantitatively predict drug-drug interactions. In humans, selective substrates for a few UGT isoforms have been identified through the use of expressed enzymes (Fig.1). For example, UGT1A1 is responsible for the formation of estradiol-3-glucuronide (E3G) and bilirubin glucuronides (Senafi et al., 1994). UGT1A6 is the hepatic isoform kinetically most important in the glucuronidation of acetaminophen (Bock et al., 1993). UGT2B7 is the enzyme responsible for morphine-3- and morphine-6-glucuronidation (Coffman et al., 1997).
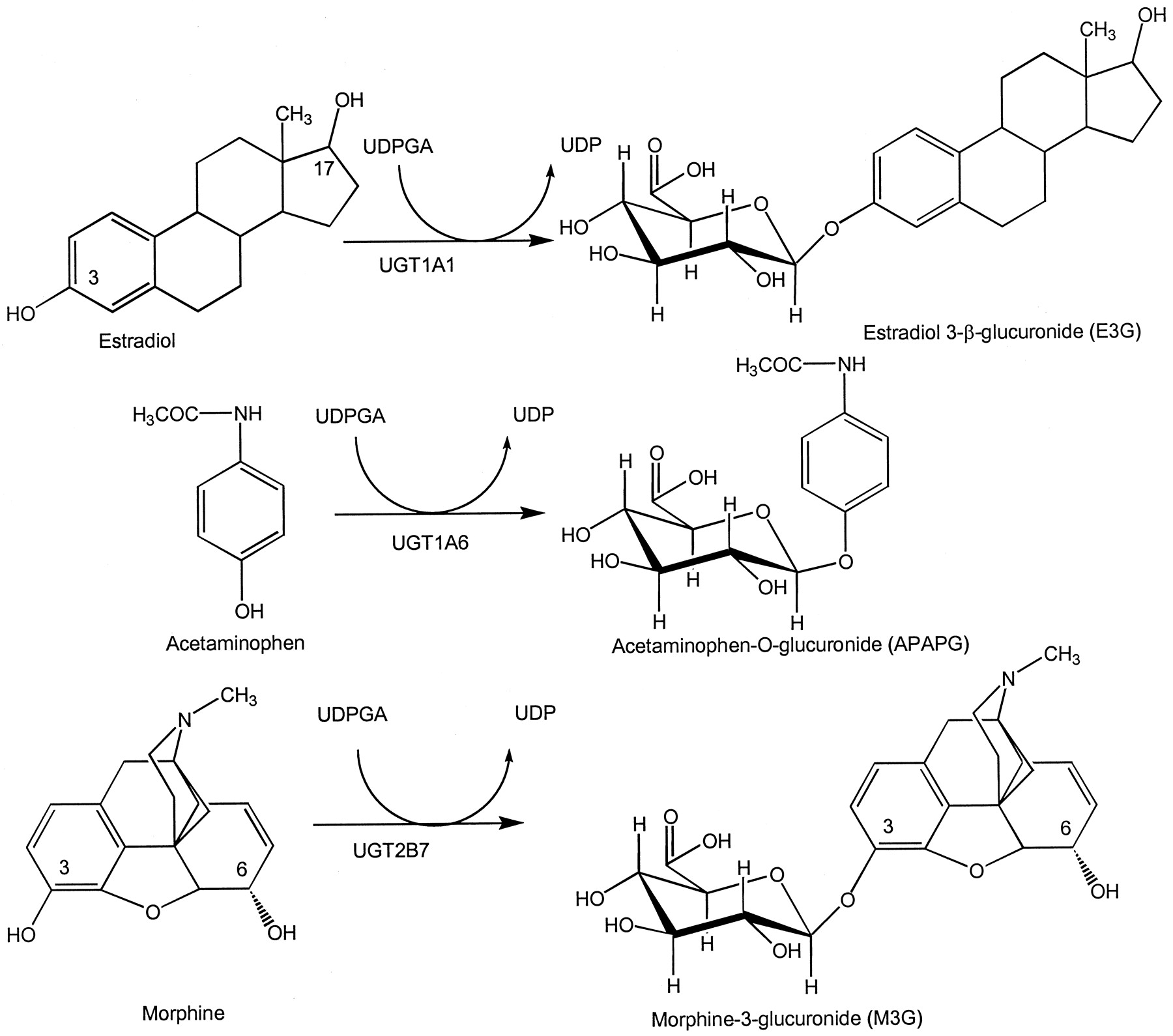
Structures of selective UGT substrates and metabolites used in these studies.
One of the goals of this study was to develop a universal in vitro method to study glucuronidation activity and kinetics in human liver microsomes, regardless of substrate. This requires a mechanism of obtaining maximal activity from microsomes that would be applicable to any UGT activity. A recent report (Little et al., 1997) indicated that the antibiotic fungal peptide alamethicin, which is known to insert into membranes and form well defined pores (He et al., 1996), could remove the latency of UGT activity for retinoic acid conjugation. This method was applied to develop assays and determine kinetic parameters for estradiol, acetaminophen, and morphine glucuronidation. Furthermore, this approach was then used to examine coupled oxidative metabolism and glucuronidation in microsomes.
Materials and Methods
Chemicals.
Uridine diphosphoglucuronic acid (UDPGA), saccharic acid-1,4-lactone, alamethicin, trifluoroacetic acid, polyoxyethylene(20) cetyl ether (Brij 58), testosterone, 6β-hydroxytestosterone, 11β-hydroxytestosterone, 7-ethoxycoumarin (7-EC), 7-hydroxycoumarin, α-naphthyl β-d-glucuronide (NG), acetaminophen-O-glucuronide (APAPG), β-estradiol, E3G, estradiol 17-glucuronide) (E17G), morphine-3-glucuronide (M3G), and morphine-6-glucuronide (M6G) were purchased from Sigma Chemical Co. (St. Louis, MO). HPLC grade acetonitrile was obtained from Burdick and Jackson (Muskegon, MI). Ammonium acetate was purchased from J.T. Baker (Phillipsburg, NJ). Acetaminophen was obtained from Kodak (Rochester, NY). Morphine was obtained from Research Biochemicals Inc. (Natick, MA). 7-Hydroxycoumarin glucuronide was obtained from Gentest Corp. (Woburn, MA) or Salford Ultrafine Chemicals and Research (Manchester, UK).
Liver Specimens.
Human liver samples designated HLA through HLT were obtained from the liver transplant unit at the Medical College of Wisconsin under a protocol approved by the Committee for the Conduct of Human Research. Microsomes from a mixture of nine (HL samples A, B, C, D, F, G, H, L, and M) or four (HL samples B, H, M, and P) human livers were prepared by differential centrifugation as described previously (van der Hoeven and Coon, 1974). These livers were originally chosen because they possessed average levels of CYP activities, contained no CYP2D6-deficient livers, and the donors were not known to have been exposed to any CYP inducers.
Statistical Analyses.
Statistics (mean, S.D., Student's t test) were performed using Microsoft Excel. The apparent kinetic parameters ofKm, Vmax, andn (where appropriate) were determined by nonlinear regression analysis using WinNonlin Standard for PC (version 1.5; Scientific Consulting, Inc., Cary, NC). The data were fit to the conventional Michaelis-Menten equation, or to the Hill equation when substrate activation was suspected (Ekins et al., 1998). The quality of fit to a particular model was determined by evaluation of criteria that included: 1) visual inspection of the Eadie-Hofstee plots of the data; 2) the sum of the squares of the residuals; and 3) the S.E. of the parameter estimates.
Acetaminophen Glucuronidation Assay.
Unless otherwise indicated, 0.5 mg of human liver microsomes, 0.1 M potassium phosphate buffer (pH 7.1), and 25 μg of alamethicin were mixed and placed on ice for 15 min. MgCl2 (1 mM in incubation), saccharolactone (5 mM in incubation), and acetaminophen (0.25–10 mM in incubation) were added, and the mixture was preincubated at 37°C for 3 min. To initiate the reaction, UDPGA (5 mM in incubation) was added to give a 200-μl final volume. Blank incubations were performed without UDPGA. Preliminary experiments indicated that the reaction was linear to 30-min incubation and up to 0.5 mg of protein. The reaction was stopped with 150 μl of ice-cold acetonitrile. After sitting on ice for 30 min, stopped reactions were centrifuged to pellet precipitated protein, and 30 μl of the supernatant were injected for HPLC analysis. Analysis was performed on a Shimadzu (Kyoto, Japan) HPLC system, equipped with two LC-10AD pumps, a SCL-10A system controller, a SIL-10A autoinjector, a SPD-10A UV/Vis detector, and a 3-μm C18 (3) Luna 100 × 4.6 mm column (Phenomenex, Torrance, CA). The mobile phase was 97% water/3% acetonitrile/0.05% trifluoroacetic acid at 1 ml/min. APAPG was detected at 254 nm and eluted at a retention time of 6.5 min. Metabolite formation was quantitated by comparing peak areas in incubations to a standard curve containing known amounts of metabolite. Standard curve correlation coefficients (r2) were ≥0.99.
Morphine Glucuronidation Assay.
Unless otherwise indicated, 0.3 mg of human liver microsomes, 0.1 M potassium phosphate buffer (pH 7.1), and 15 μg of alamethicin were mixed and placed on ice for 15 min. MgCl2 (1 mM in incubation), saccharolactone (5 mM in incubation), and morphine (0.1–5 mM in incubation) were added, and the mixture was preincubated at 37°C for 3 min. To initiate the reaction, UDPGA (5 mM in incubation) was added to give a 200-μl final volume. Blank incubations were performed without UDPGA. Preliminary experiments indicated that the reaction was linear to 30-min incubation and up to 0.3 mg of protein. The reaction was stopped with 150 μl of ice-cold acetonitrile. After sitting on ice for 30 min, stopped reactions were centrifuged to pellet precipitated protein, and 30 μl of the supernatant were injected for HPLC analysis. Analysis was performed as described above, except detection was performed with a Shimadzu RF-10A fluorescence detector. The glucuronide, M3G, was detected at an excitation wavelength of 210 nm, an emission wavelength of 350 nm, and eluted at a retention time of 7 min. The 6-O-glucuronide, M6G, coeluted with substrate under these conditions. Metabolite formation was quantitated by comparing peak areas in incubations to a standard curve containing known amounts of metabolite. Standard curve correlation coefficients (r2) were ≥0.99.
Estradiol Glucuronidation Assay.
In a typical incubation, 0.1 to 0.3 mg of human liver microsomes, 0.1 M potassium phosphate buffer (pH 7.1), and 5 to 15 μg of alamethicin (50 μg alamethicin/1 mg microsomal protein final concentration) were mixed and placed on ice for 15 min. MgCl2 (1 mM in incubation), saccharolactone (5 mM in incubation), and estradiol (0.5–100 μM final incubation concentration, added in 2 μl of methanol) were added, and the mixture was preincubated at 37°C for 3 min. To initiate the reaction, UDPGA (5 mM in incubation) was added to give a 200-μl final volume. Blank incubations were performed without UDPGA. Preliminary experiments indicated that the reaction was linear to 30-min incubation and up to 0.3 mg of protein. The reaction was stopped with 50 μl of ice-cold 25% (v/v) formic acid, and 2 nmol of NG was added. After sitting on ice for 30 min, stopped reactions were centrifuged to pellet precipitated protein, the supernatant was transferred to a 96-well plate, and 20 μl were injected for HPLC-mass spectrometry analysis. Analysis was performed on a Micromass Platform LCZ system (Manchester, UK) equipped with a Gilson 215 Liquid Handler, two Shimadzu LC-10AD pumps, a Shimadzu CTO-10AC column oven, and a 3-μm, 100 × 2 mm Prodigy ODS (3) HPLC column (Phenomenex, Torrance, CA). The mobile phase solution A was 10 mM ammonium acetate and solution B was 90% acetonitrile/10% water/10 mM ammonium acetate. Initial conditions were 85% A/15% B pumped at 0.25 ml/min with a 30°C column temperature. A linear gradient from 15 to 31% B between 0 and 8 min was used, followed by 1 min at 100% B, and a re-equilibration at 15% B. Analytes were detected as their [M−H]− ions using negative ion electrospray ionization. The source block temperature was 125°C, desolvation temperature was 400°C, the capillary voltage was −2.70 kV, and the cone voltage was −30 V. The internal standard NG, E3G, and E17G were detected by single ion recording monitoring atm/z 319, 447, and 447, and eluted at 5, 7.5, and 8 min, respectively. Metabolite formation was quantitated by comparing peak area ratios (metabolite/internal standard) in incubations to ratios obtained from a standard curve containing known amounts of metabolite. Standard curve correlation coefficients (r2) were ≥0.99.
Testosterone Assay.
In a typical incubation, 0.1 mg of human liver microsomes, 0.1 M potassium phosphate buffer (pH 7.1), or 0.1 M sodium phosphate buffer (pH 7.4), and 5 μg of alamethicin were mixed and placed on ice for 15 min. MgCl2 (1 mM in incubation), and testosterone (200 μM final incubation concentration, added in 2 μl of methanol) were added, and the mixture was preincubated at 37°C for 3 min. NADPH was added to give a final volume of 0.2 ml and a final concentration of 1 mM. Blank incubations were performed without NADPH. Reactions were allowed to proceed for 15 min before termination with 100 μl of acetonitrile. Internal standard was added (25 μl of a 0.1 mM solution of 11β-hydroxytestosterone), incubations were vortexed, and 50 μl were injected for HPLC analysis. Analysis was similar to the acetaminophen glucuronide assay described above, except the solvents were water (solvent A) and acetonitrile (B), and the analytes were detected at 242 nm. The flow rate was 1 ml/min, and the gradient was 30% B for 9 min, followed by a rapid increase to 90% B, 1 min at 90%, and a re-equilibration at 30% B. Under these conditions, 6β-hydroxytestosterone, 11β-hydroxytestosterone, and substrate elute at 5, 8.5, and 13 min, respectively. Metabolite formation was quantitated by comparing peak area ratios (metabolite/internal standard) in incubations to ratios obtained from a standard curve containing known amounts of metabolite. Standard curve correlation coefficients (r2) were ≥0.99.
Ethoxycoumarin Oxidation and Glucuronidation.
Typically, 0.1 to 0.3 mg of human liver microsomes, 0.1 M potassium phosphate buffer (pH 7.1) or 0.1 M sodium phosphate buffer (pH 7.4), and 50 μg of alamethicin/mg microsomes were mixed and placed on ice for 15 min. MgCl2 (1 mM in incubation), saccharolactone (5 mM in incubation), and ethoxycoumarin (250 μM final incubation concentration, added in 2 μl of methanol) were added, and the mixture was preincubated at 37°C for 3 min. To initiate the reaction, NADPH alone or with UDPGA was added to give a 200-μl final volume and final concentrations of 1 and 5 mM, respectively. Blank incubations were performed without cofactor. Reactions were terminated by the addition of 50 μl of 5% (v/v) HCl; 20 μl of 0.2 mM coumarin were added as internal standard, samples were centrifuged to pellet precipitated protein, and 30 μl of the supernatant was injected for HPLC analysis. Analysis was performed by HPLC/UV as described above for APAPG, except detection was at 320 nm. 7-Hydroxycoumarin glucuronide, 7-hydroxycoumarin, internal standard, and 7-EC standards eluted at 4.8, 7.5, 9.5, and 12 min. Metabolite formation was quantitated by comparing peak area ratios (metabolite/internal standard) in incubations to ratios obtained from a standard curve containing known amounts of metabolite. Standard curve correlation coefficients (r2) were ≥0.99.
Results
Optimization of Incubation Conditions.
Incubation conditions for glucuronidation by microsomes usually includes a detergent to disrupt the membrane barrier, a divalent metal ion, and optimal pH to obtain maximal glucuronidation activity. Using acetaminophen glucuronidation, in a mixture of liver microsomes from nine human livers, as a surrogate UGT activity, preliminary experiments indicated that enzyme activity increased with a Mg2+ concentration at least up to 10 mM, and that 0.1 mg polyoxyethylene(20) cetyl ether/mg microsomal protein was the optimal detergent treatment for this activity (data not shown). Combining these treatments gave an additive effect, yielding APAPG formation rates of 1.1 nmol/mg/min (Fig.2).

The effect of treatment of microsomes on acetaminophen glucuronidation.
Microsomes (0.5 mg) from a mixture of nine human livers were treated with polyoxyethylene(20) cetyl ether (0.1 mg/mg protein) and 10 mM MgCl2, or alamethicin (50 or 100 μg/mg protein, in methanol) as indicated, and incubated with 10 mM acetaminophen and 5 mM UDPGA for 30 min, as described under Materials and Methods. Control incubations were treated with an equivalent amount of methanol. Duplicate incubations were analyzed as described under Materials and Methods.
Conditions were sought that would most closely mimic the environment of the UGT found in vivo, specifically those found in the lumen of the ER, and would allow determination of enzyme kinetic parameters. The lumenal pH of the ER is reported to be approximately 7.1 (Kim et al., 1998), and the concentration of Mg2+ inside the ER is on the order of 1 mM (Berg et al., 1995; Sugiyama and Goldman, 1995). In addition to these conditions, a method of disrupting the ER membrane was desired that was nondetergent in nature, because detergents may have an artifactual effect on the native catalytic activity of the enzyme. It was recently shown that the antibiotic fungal peptide alamethicin, which is known to insert into membranes and form well-defined pores (He et al., 1996), removed the latency of UGT activity for the glucuronidation of retinoic acid (Little et al., 1997) and 7-hydroxy-4-trifluoromethylcoumarin (Patten et al., 1998) at approximately 50 μg alamethicin/mg microsomal protein. As shown in Fig. 2, alamethicin treatment alone (50 or 100 μg alamethicin/mg microsomal protein) was equally as effective as the conditions described above with 0.1 mg polyoxyethylene(20) cetyl ether/mg microsomes and 10 mM Mg2+ ion. Based on these results, 50 μg alamethicin/mg microsomes was chosen as standard incubation condition. In addition, the physiological conditions reflecting the lumen of the ER, 1 mM Mg2+, and pH 7.1, were used.
The effects of alamethicin and 1 mM Mg2+ on acetaminophen and morphine glucuronidation in the mixture of microsomes from human livers are shown in Fig. 3A. Relative to incubations with untreated microsomes (control), alamethicin enhanced the conjugation of both substrates 2- to 3-fold, and Mg2+ enhanced activity an additional 2- to 3-fold. This is similar to reports of a 2-fold increase in retinoic acid and 7-hydroxytrifluoromethylcoumarin conjugation on alamethicin pretreatment. Figure 3B demonstrates the effect of these standard assay conditions on estradiol conjugation. Again, a 2- to 3-fold increase in estradiol-3- and -17-glucuronidation is seen on alamethicin treatment. Addition of 1 mM Mg2+ to the alamethicin-treated microsomes yielded rates of glucuronidation that were 4 to 7 times the rates with untreated microsomes. These results demonstrate that alamethicin appears to increase enzyme activity in a substrate-independent fashion, presumably by facilitating entry of substrate and UDPGA into, and the diffusion of conjugate and UDP out of, the lumen of the microsomes.
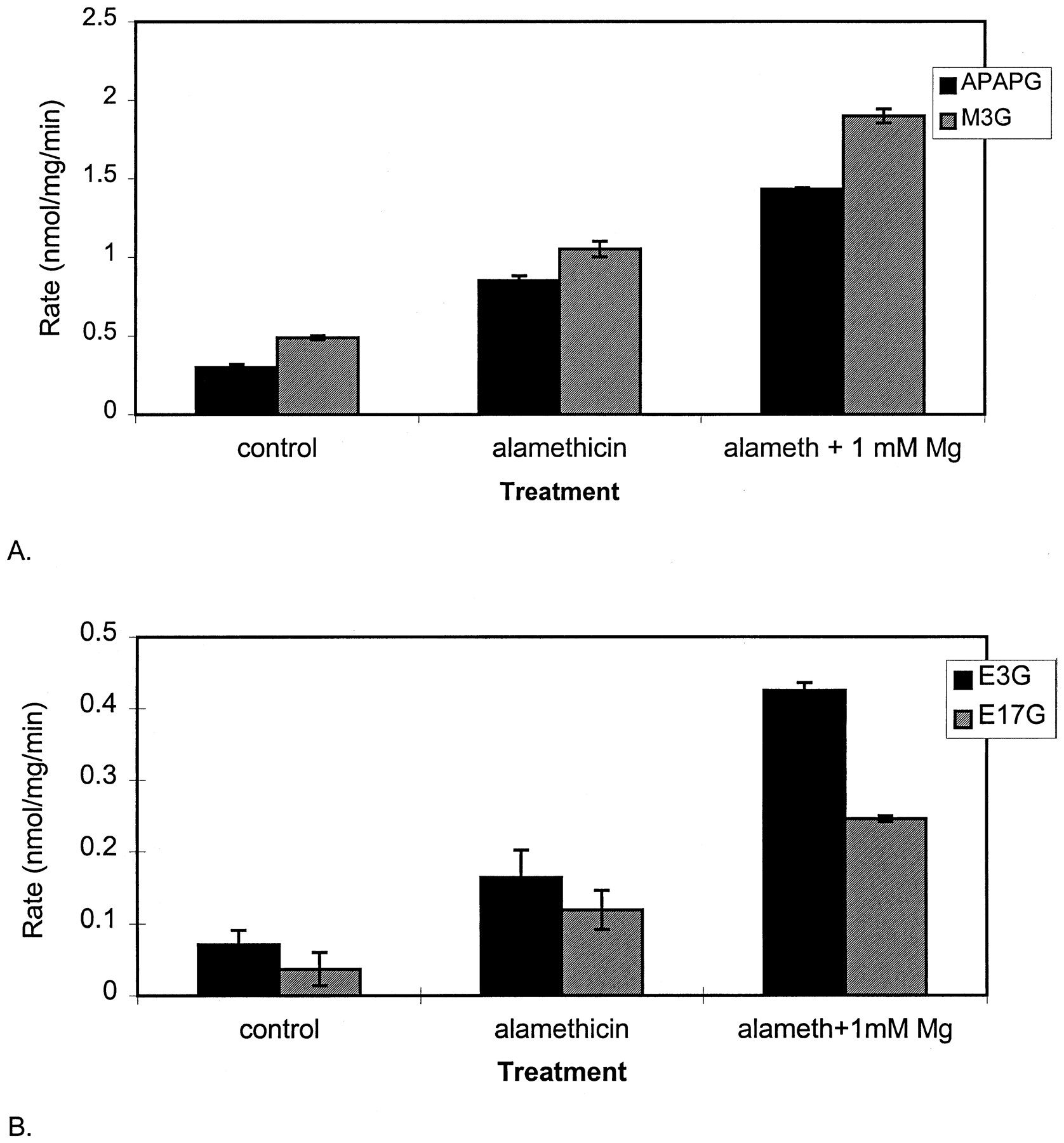
The effects of alamethicin and a physiological concentration of Mg2+ on the glucuronidation of estradiol, acetaminophen, and morphine.
Microsomes from a mixture of nine human livers were incubated with 100 μM estradiol, 10 mM acetaminophen, or 5 mM morphine in the presence of either alamethicin (50 μg/mg protein, in methanol), alamethicin and 1 mM MgCl2, or methanol control. Triplicate incubations were then performed and analyzed as described under Materials and Methods.
Effect of Alamethicin on CYP Activity.
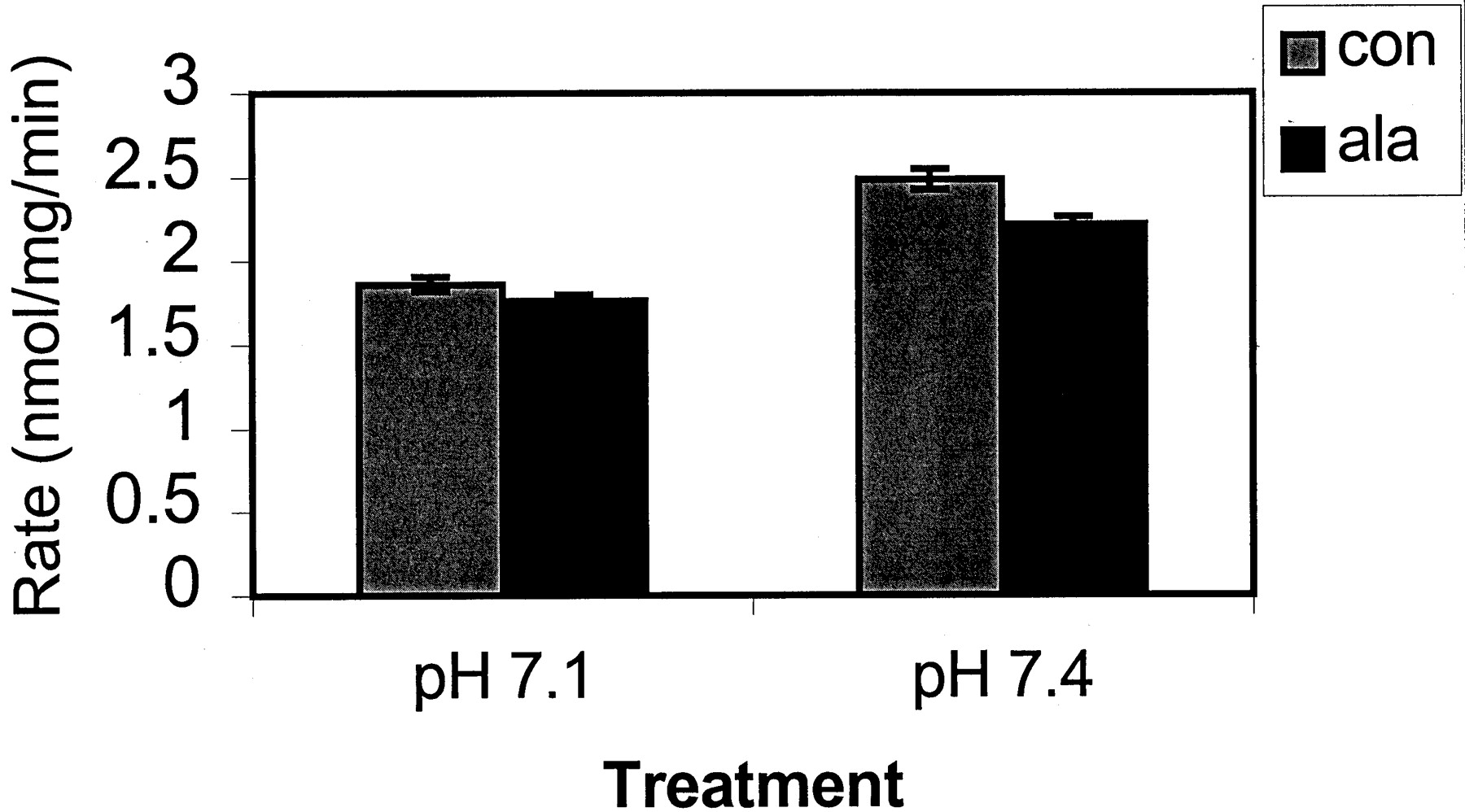
The data in Fig. 3 indicates that alamethicin has a UGT isoform-independent effect on glucuronidation, which probably results from pore formation without effects on UGT protein structure. To more directly examine the effect of alamethicin on the microsomal membrane and its potential disruptive and/or detergent-like effects, CYP3A4/5-catalyzed testosterone 6β-hydroxylation was examined in the presence and absence of alamethicin. This activity was chosen because members of the CYP3A subfamily are notoriously sensitive to membrane composition and structure (Halvorson et al., 1990; Imaoka et al., 1992), and because detergent-like effects could lead to an alteration in enzyme activity due to disruption of coenzyme interactions (Guengerich et al., 1998). Figure 4 shows that the presence of alamethicin did not have a significant effect on CYP3A4/5 activity compared with control (P > .05), at either pH 7.1 or at the more traditional CYP incubation pH of 7.4. These results indicate that the pore-forming peptide alamethicin does not significantly affect membrane structure or CYP-coenzyme interactions.

The effects of pH and alamethicin pretreatment on testosterone 6β-hydroxylation.
Microsomes from a mixture of nine human livers were pretreated either with 50 μg/mg alamethicin in methanol or with an equivalent amount of methanol. Treated microsomes were then incubated with 200 μM testosterone, 1 mM MgCl2, and 5 mM UDPGA at either pH 7.1 or 7.4. Triplicate incubations were analyzed as described underMaterials and Methods. Alamethicin treatment was not significantly different from control (P > .05).
Glucuronidation Kinetics In Vitro.
Using the standard incubation conditions described above, enzyme kinetic parameters for the formation of E3G, APAPG, and M3G were determined in human liver microsomes. Kinetic parameters are summarized in Table 1. As shown in Fig.5A, acetaminophen conjugation resulted in a hooked Eadie-Hofstee plot, consistent with allosterism or activation kinetics. These data were best fit to the Hill equation. However, theKm value (Table 1) determined agrees with that obtained previously (Bock et al., 1993). The formation of M3G was consistent with classical Michaelis-Menten kinetics at the substrate concentrations used in the experiment (Fig. 5B), and theKm value (Table 1) was in the expected range (Coffman et al., 1997). Interestingly, E3G formation resulted in a hooked Eadie-Hofstee plot and was best fit to the Hill equation (Fig.5C), whereas E17G formation in the same samples yielded classical Michaelis-Menten kinetics (Fig. 5D). The Kmvalue for E3G formation was somewhat lower than reported previously (Senafi et al., 1994; Ethell et al., 1998). To our knowledge, this is the first report of what appears to be homotropic activation in the glucuronidation of acetaminophen and estradiol. Interestingly, a recent report indicated that bilirubin conjugation in human hepatocytes exhibited activation, indicating that this may be more of a general phenomenon than previously recognized (Bruni and Chang, 1999).
Kinetic analysis of substrate glucuronidation in alamethicin-pretreated mixture of human liver microsomes

Eadie-Hofstee plots of acetaminophen, morphine, and estradiol glucuronidation kinetics.
Microsomes from a mixture of four human livers were incubated with alamethicin (50 μg/mg protein, in methanol), 1 mM MgCl2, 5 mM UDPGA, and a range of substrate concentrations. Incubations were then performed and analyzed as described under Materials and Methods. Kinetic constants determined after fitting the data to a model are shown in Table 1.
Coupled Oxidative-Conjugative Metabolism.
Because alamethicin does not seem to affect CYP activity (Fig. 4), this method provides an opportunity to examine coupled oxidation and glucuronidation in the same microsomal incubation. The effect of alamethicin on metabolite profiles from sequential oxidation-conjugation of 7-EC was determined. As shown in Table2, alamethicin treatment had no significant effect on ethoxycoumarin deethylation compared with control. This result is consistent with the findings in Fig. 4 with testosterone oxidation. Compared with NADPH alone, NADPH plus UDPGA treatment without alamethicin led to glucuronidation of half of the 7-hydroxycoumarin produced. However, in the presence of alamethicin, more than 80% of the 7-hydroxycoumarin product was subsequently conjugated under the conditions of the experiment, indicating that sequential phase I and phase II metabolism proceeds very efficiently in alamethicin-treated microsomes.
Examination of coupled oxidation and glucuronidation of 7-ethoxycoumarin by human liver microsomes
Discussion
The formation of glucuronides using hepatic microsomes have notoriously been problematic due to the lumenal localization of the UGT active site. Traditional methods of characterizing a conjugation reaction in vitro involved an initial detergent titration step to identify optimal conditions. However, detergent treatment may have artifactual effects on enzyme activity (Fulceri et al., 1994; Trapnell et al., 1998), and is known to have inhibitory effects on CYP activity (Guengerich et al., 1998). Other investigators have begun applying pore-forming peptides instead of detergents to incubations with UGTs and other lumenally localized enzymes. For instance,Staphylococcus aureus α-toxin (Vanstapel and Blanckaert, 1988; Bossuyt and Blanckaert, 1996) and filipin (Banhegyi et al., 1993) have been applied to permeabilize hepatocytes and microsomes to study UGT activity. Also, alamethicin pretreatment diminished the latency of uridine diphosphatase activity in the Golgi apparatus (Wang and Guidotti, 1998), and the latency of the following enzymatic activities found in the ER: gulonolactone oxidase (Puskas et al., 1998), β-glucuronidase (Banhegyi et al., 1996), and glucose-6-phosphatase (Banhegyi et al., 1997).
During the course of these studies, it was reported that the antibiotic fungal peptide alamethicin, which is known to insert into membranes and form well defined pores (He et al., 1996), could remove the latency of UGT activity for retinoic acid conjugation (Little et al., 1997). The results presented here indicate that alamethicin is allowing free diffusion of substrate, cofactor, and products without affecting the gross membrane structure and intrinsic enzyme catalytic activity. Namely, the alamethicin effects as shown in Fig. 3 are substrate-independent, whereas alterations in membrane and/or enzyme structure would more than likely demonstrate some isoform dependence. Also, we showed in Fig. 4 that alamethicin does not have a major effect on CYP3A4/5 catalytic activity, the 6β-hydroxylation of testosterone. CYP3A4 is an isoform that is notoriously sensitive to changes in the membrane structure, and any gross alterations in membrane structure would be expected to alter the activity of this CYP. Only a minimal effect was seen. In addition, the CYP-mediated 7-hydroxycoumarin formation was also unaffected by alamethicin treatment of human liver microsomes. Because of the evidence outlined above, alamethicin treatment appears to be a superior method of examining microsomal glucuronidations in vitro compared with detergent treatment.
The enhancement of glucuronide formation by Mg2+has been described as either an increase inVmax due to increasing enzyme catalysis, or by activating transporters responsible for cofactor access and product removal (Zakim et al., 1973). The data in Fig. 3 demonstrates that the enhancement seen with Mg2+ occurs in the presence of pores formed with alamethicin, when free diffusion of substrates and products occurs. Therefore, at least for the activities examined here, Mg2+ appears to exert its effects directly on the actual catalytic activity of the UGTs, increasing their catalytic activities.
Alamethicin also appears to have utility generating coupled oxidation-glucuronidation activity in microsomes (Table 2). Traditionally, this was examined only in intact cell systems, such as hepatocytes and liver slices (Rodrigues, 1994; Eddershaw and Dickins, 1999). These models were used for such studies because they contain active transporters, circumventing the issue of latency. Also, whole cell systems are energetically competent, which eliminates the need for cofactor supplementation. Now, with the addition of alamethicin, NADPH, and UDPGA, oxidation-glucuronidation occurs efficiently in microsomal incubations. In addition to direct glucuronidation reactions, these assay conditions for coupled oxidation and glucuronidation will have utility in the early determination of metabolic stability of drug candidates beyond the scope of simple microsomal oxidation, and in the generation of metabolite profiles without the need for intact cell systems.
Whereas the formation of M3G and E17G was consistent with Michaelis-Menten kinetics, E3G and APAPG formation catalyzed by UGT1A1 and 1A6, respectively, showed activation kinetics (Fig. 5). This appears to be the first report demonstrating UGT autoactivation in microsomal incubations. One obvious explanation for atypical E3G kinetics is that saturable protein binding is occurring at low substrate concentrations, resulting in a hooked Eadie-Hofstee plot. However, this appears not to be the case, because E17G formation in the same incubations demonstrated a linear Eadie-Hofstee plot and thus a classical Michaelis-Menten fit. Of interest is a recent report that indicated that bilirubin conjugation by UGT1A1 in hepatocytes also resulted in atypical kinetics that were fit to the Hill equation (Bruni and Chang, 1999). It should be noted that previous investigators have published acetaminophen kinetic plots suggestive of atypical kinetics, but fit the data using a Michaelis-Menten model (Bock et al., 1993). Estradiol kinetics also have been fit previously to the Michaelis-Menten model (Senafi et al., 1994; Ethell et al., 1998), but it is unclear if the regioselectivity of estradiol glucuronidation was determined in those studies.
Autoactivation kinetics have been reported for some substrates of CYP3A4 and, to a lesser extent, other CYPs, but the mechanism of this activation has not yet been fully elucidated (Ekins et al., 1998).Korzekwa and coworkers (1998) proposed that atypical CYP kinetics is due to multiple substrates present in the active site of a single enzyme. This is consistent with the relatively open and accessible active sites proposed for xenobiotic-metabolizing enzymes, which have likely evolved in this fashion to handle the broad spectrum of environmental chemicals and drugs they may encounter. The UGTs and CYPs are the major xenobiotic-metabolizing enzyme superfamilies, and thus the activation kinetics observed with both families probably result from evolutionary pressure for the need to process a diverse and unexpected milieu of substrates.
In conclusion, the pore forming peptide alamethicin was used to develop a standard set of incubation conditions that were applied to multiple UGTs and substrates. These conditions were shown to have minimal effects on the microsomal membrane and on CYP catalytic activity, and were used in a sequential oxidation-glucuronidation reaction. In vitro glucuronidation rates have recently been used in the rank ordering of drug candidates for selection of a new drug entity with improved pharmacokinetic parameters (Bouska et al., 1997). This was one of the few reports where microsomal glucuronidation was used as a drug candidate selection method. Perhaps the work described above will allow in vitro glucuronidation methods to be more universally applied to the drug discovery and development processes.
Acknowledgments
We thank Barbara J. Ring, Dr. Michael L. Schrag, and Cliff Fisher for many helpful discussions.
Footnotes
-
Send reprint requests to: Dr. Steven A. Wrighton, Drop 0730, Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, IN 46285. E-mail: WRIGHTON_STEVEN{at}Lilly.Com
-
↵1 Present address: Pfizer Central Research, Eastern Point Rd., Groton, CT 06340.
- Abbreviations used are::
- CYP
- cytochrome P450
- APAPG
- acetaminophen-O-glucuronide
- 7-EC
- 7-ethoxycoumarin
- E3G
- estradiol-3-glucuronide
- E17G
- estradiol 17-glucuronide
- M3G
- morphine-3-glucuronide
- M6G
- morphine-6-glucuronide
- NG
- α-naphthyl β-d-glucuronide
- UDPGA
- uridine diphosphoglucuronic acid
- UGT
- UDP-glucuronosyltransferase
- ER
- endoplasmic reticulum
- Received October 25, 1999.
- Accepted February 14, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}