


Abstract
HIV protease inhibitors have proven remarkably effective in treating HIV-1 infection. However, some tissues such as the brain and testes (sanctuary sites) are possibly protected from exposure to HIV protease inhibitors due to drug entry being limited by the membrane efflux transporter P-glycoprotein, located in the capillary endothelium. Intravenous administration of the novel and potent P-glycoprotein inhibitor LY-335979 to mice (1–50 mg/kg) increased brain and testes concentration of [14C]nelfinavir, up to 37- and 4-fold, respectively, in a dose-dependent fashion. Similar effects in brain levels were also observed with 14C-labeled amprenavir, indinavir, and saquinavir. Because [14C]nelfinavir plasma drug levels were only modestly increased by LY-335979, the increase in brain/plasma and testes/plasma ratios of 14- to 17- and 2- to 5-fold, respectively, was due to increased tissue penetration. Less potent P-glycoprotein inhibitors like valspodar (PSC-833), cyclosporin A, and ketoconazole, as well as quinidine and verapamil, had modest or little effect on brain/plasma ratios but increased plasma nelfinavir concentrations due to inhibition of CYP3A-mediated metabolism. Collectively, these findings provide “proof-of-concept” for increasing HIV protease inhibitor distribution into pharmacologic sanctuary sites by targeted inhibition of P-glycoprotein using selective and potent agents and suggest a new therapeutic strategy to reduce HIV-1 viral replication.
Highly active antiviral therapy involving HIV protease inhibitors has dramatically improved the clinical management of HIV-1 infection, however, significant problems remain. Thus, despite nondetectable plasma viral RNA levels, low-level active replication of the virus is still present in the central nervous system, which is often associated with progressive loss of cognitive and motor function characteristic of the AIDS dementia complex (Kolson et al., 1998). This may occur because effective levels of antiviral agent are not achieved in the central nervous system within the limits of clinical toxicity (Groothuis and Levy, 1997). A similar pharmacologic sanctuary site appears to be the testes, contributing to sexual transmission of the infection (Zhang et al., 1998). A common characteristic of the blood-brain and blood-testes barriers is the presence of a membrane efflux transporter, termed P-glycoprotein (P-gp),1 in the capillary endothelial cells of these tissues (Thiebaut et al., 1987;Cordon-Cardo et al., 1989). Expression of this transporter is polarized to the lumenal surface of the endothelial cell so that uptake of a drug substrate is countered by efficient back-efflux into the circulating blood, which limits drug entry into the tissue. Recently, we have shown that HIV protease inhibitors are excellent substrates for this transporter (Kim RB et al., 1998); this has been confirmed by others (Kim AE et al., 1998; Lee et al., 1998). Moreover, in themdr1a(−/−) “knockout” mouse, lacking P-gp expression, the brain levels of saquinavir, indinavir, and nelfinavir were 7- to 35-fold higher than in syngeneic wild-type [mdr1a(+/+)] animals (Kim RB et al., 1998).
P-gp is also a major mechanistic contributor to the pleiotropic multidrug resistance phenomenon associated with the chronic use of many anticancer agents such as the anthracyclines, epidophyllotoxins, and vinca alkaloids (Bellamy, 1996; Ambudkar et al., 1999). That is, intracellular drug concentrations are markedly reduced in resistant tumor cells due to overexpression of this efflux transporter. In vitro, such resistance may be reversed by inhibitors of P-gp function (Ford and Hait, 1990; Ford, 1996) and a similar strategy using the cyclosporine derivative valspodar (PSC-833) is currently under clinical investigation, with some success being reported (Boote et al., 1996;Sonneveld et al., 1996).
This study was designed to determine whether this approach could be extended to the treatment of HIV infection by determining whether pharmacologic modulation of P-gp activity would alter the distribution of HIV protease inhibitors into the brain and testes. Demonstration of such a “proof of concept” would provide support for initiating clinical investigation of this strategy to enhance the in vivo efficacy of an important class of drugs in the treatment of HIV-1 infection.
Materials and Methods
Inhibition of Digoxin Transport by P-gp In Vitro.
Caco-2 cells were grown and cultured on 0.4-μm polycarbonate membrane filters (Transwell; Costar Corp., Cambridge, MA) as previously described (Kim RB et al., 1998). Transport of [3H]digoxin (15 Ci/mmol; DuPont-New England Nuclear, Boston, MA) was determined by its addition to either the basal or apical side of the polarized cell monolayer and by measuring the transport of radioactivity into the other compartment over 4 h, in the absence or presence of putative inhibitor in both compartments. The extent of inhibition was determined from the following equation, where i and a are the percentages of digoxin transport in the presence and absence of inhibitor, according to the direction of transport (Kim et al., 1999):
Inhibition of CYP3A-Mediated Metabolism of Nifedipine In Vitro.
CYP3A activity was determined using human liver microsomes prepared from sample HL110, as previously described (Wandel et al., 1999), and based on the conversion of nifedipine to its dihydropyridine metabolite. Briefly, the incubation mixture consisted of microsomes equivalent to 100 pmol of total cytochrome P450, 1.5 mM NADPH, 20 μM nifedipine, and 0.5 to 40 μM putative inhibitor dissolved in either dimethyl sulfoxide (2%) or acetonitrile (1%) in a total volume of 500 μl of 0.1 M phosphate buffer (pH 7.4). Incubations were performed at 37°C, and the reaction was stopped after 10 min by the addition of 1 ml CH2Cl2. Dihydropyridine metabolite production was measured using HPLC, and the IC50 value for inhibition was estimated in the same fashion as described with respect to analogous studies with P-gp.
Comparative Tissue Distribution in mdr1a(+/+) and (−/−) Mice.
Male mdr1a(−/−) mice (FVB/TacfBR-[KO]mdr1aN7), 6 to 12 weeks of age and genetically matched male mdr1a(+/+) mice (FVB/MTtacfBR) weighing 20 to 30 g were obtained from Taconic (Germantown, NY). The animals were cared for in accordance with the U.S. Public Health Service policy for the Care and Use of Laboratory Animals, and the experimental studies were approved by the Vanderbilt University Animal Care Committee. The tissue distribution of [14C]nelfinavir was determined following i.v. injection (5 mg/kg) of an ethanol (20%)/saline (0.9%) solution over 5 min into a tail vein; the total volume injected was 4 μl/g (Kim RB et al., 1998). At specific times after drug administration and following anesthesia with isoflurane (Isoflo; Abbott Laboratories, Abbott Park, IL), blood was removed by orbital bleeding, and the animal was sacrificed. Subsequently, tissues were harvested, weighed, and homogenized with 4% BSA solution. Total radioactivity was determined after the addition of 100 μl of plasma or 500 μl of tissue homogenate to vials containing 4 ml of scintillation fluid (Scintiverse BD; Fisher Scientific Co., Fairlawn, NJ).
The effect of P-gp inhibitors was investigated by pretreatment with equally divided doses given by i.v. tail vein injection, 30 min before and concurrently with administration of nelfinavir. Inhibitors investigated included freshly prepared LY-335979 (2 × 0.5–25 mg/kg), verapamil (2 × 6.25 mg/kg; Sigma-Aldrich), and quinidine (2 × 25 mg/kg; Sigma-Aldrich), each dissolved in 20% ethanol/0.9% saline; valspodar (2 × 0.5–25 mg/kg; Novartis Pharma AG, Basel, Switzerland); and cyclosporine A (2 × 0.5–25 mg/kg; Novartis Pharma AG) dissolved in 10% ethanol/60% propylene glycol/30% water; nelfinavir (2 × 25 mg/kg; Agouron Pharmaceuticals Inc., San Diego, CA), ritonavir (2 × 12.5 mg/kg; Abbott Laboratories, Abbott Park, IL), saquinavir (2 × 25 mg/kg; Roche Products Ltd.), and indinavir (2 × 25 mg/kg; Merck Research Laboratories), each dissolved in 10% ethanol/40% propylene glycol/50% 0.9% saline; and ketoconazole (2 × 25 mg/kg; Sigma-Aldrich) dissolved in 25% 0.2 N HCl. All drugs were injected in a total volume of 4 μl/g, and appropriate vehicle solutions were used in control studies.
Similar tissue distribution studies were also performed with [14C]amprenavir (49.2 mCi/mmol; Glaxo Wellcome Inc., Research Triangle Park, NC), [14C]indinavir, and [14C]saquinavir following administration of LY-335979 (2 × 25 mg/kg). At least three mice were studied at each time point, and differences in radioactivity between treated and control groups were analyzed by a two-sided Student's ttest with P < .05 as the limit of statistical significance.
HPLC Analysis of [14C]Nelfinavir Tissue Levels.
Plasma and brain samples obtained at 2 h following administration of nelfinavir along with LY-335979 (50 mg/kg) were extracted and analyzed by HPLC to determine the chemical nature of the measured radioactivity. Briefly, 0.3 ml of acetonitrile was added to 0.25 ml of plasma to which 10 μg of unlabeled nelfinavir had been added. After vortexing, the supernatant was removed and evaporated at room temperature under a stream of nitrogen; the residue was reconstituted with 200 μl of methanol. Brain homogenate (1:10 dilution) was similarly prepared except that 0.5 ml of acetonitrile was added to 0.5 ml of homogenate. Subsequently, 100 μl of the reconstituted extraction was analyzed by HPLC using a 4.6-m × 150-mm, 5 μm PrimeSphere C18 column (Phenomenex, Torrance, CA) and a mobile phase (1 ml/min) consisting of 25 mM ammonium phosphate (pH 4.5) to which acetonitrile was added as a 20 to 70% linear gradient over 25 min. UV detection at 220 nm was used to monitor the elution of unlabeled nelfinavir, and fractions of the eluate were collected every minute for the first 15 min and then every 30 s for the remainder of the separation. Scintillation fluid (5 ml; Scintiverse BD) was added to the collected eluate, which was then analyzed by liquid scintillation counting. Under these chromatographic conditions, nelfinavir had a retention time of 21.4 min, which was comparable with the value of 21.6 min reported by others using the same HPLC method and conditions (Wu et al., 1999). In the latter case, various identified metabolites of nelfinavir had shorter retention times than the unchanged drug (M-11, 16.1 min; M-8, 17.2 min; M-10, 18.2 min; and M-3, 19.3 min).
Statistical Analysis.
Mean ± S.E. values were compared using Student's ttest or the Mann-Whitney U test, and P < .05 was taken as the minimum level of significance.
Results
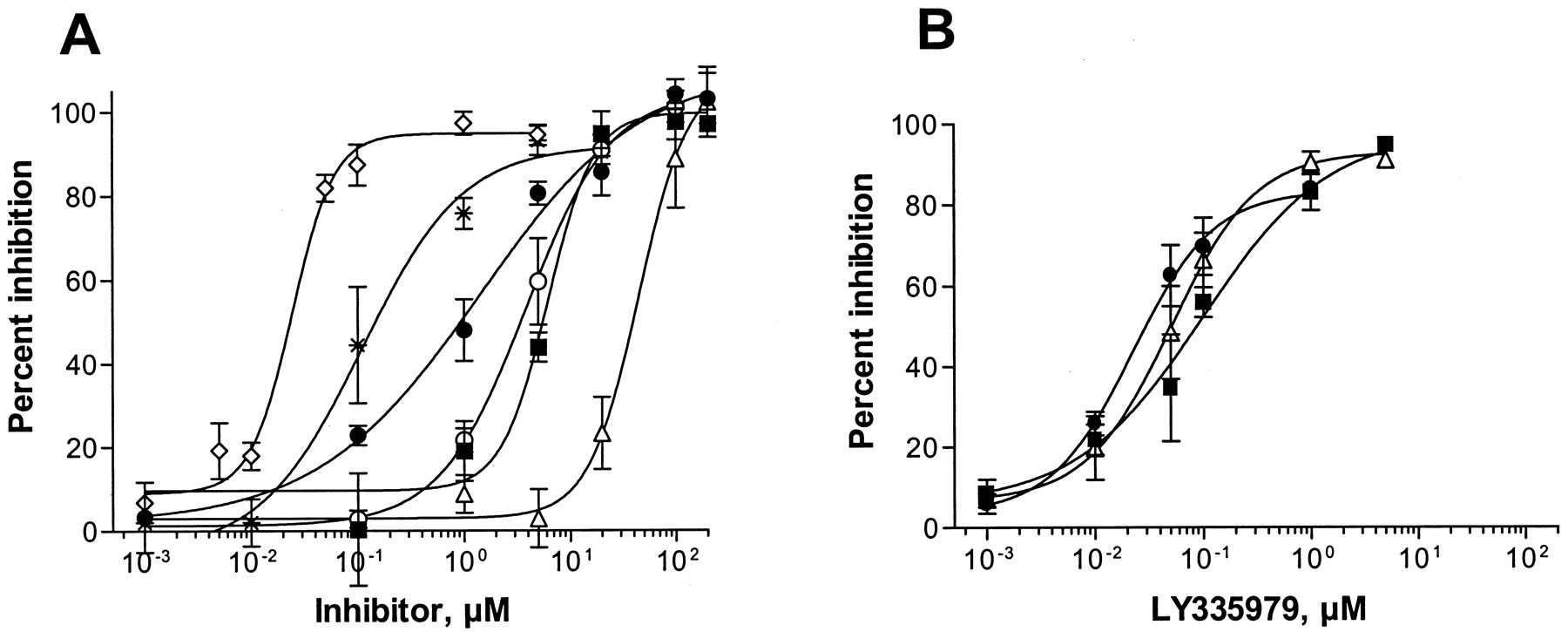
The IC50 values for drugs previously shown to inhibit P-gp transport such as ketoconazole (1.2 μM), cyclosporin A (1.3 μM), verapamil (2.1 μM), and quinidine (2.2 μM) were in the low micromolar range, using the translocation of digoxin across Caco-2 cells as a model system for P-gp-mediated transport. Nelfinavir exhibited comparable inhibitory potency (1.4 μM); however, ritonavir (3.8 μM) and saquinavir (6.5 μM) were somewhat less potent, and the IC50 value for indinavir (44 μM) was about an order of magnitude greater than those for the other HIV protease inhibitors (Fig. 1A). As expected, valspodar was found to be a much more potent inhibitor of digoxin transport (IC50 = 0.11 μM) than its structurally related analog cyclosporin A and, in turn, the inhibitory potency of LY-335979 was an additional 5-fold higher (IC50 = 0.024 μM) (Fig. 1A). LY-335979 was also found to be a potent inhibitor of the translocation of HIV protease inhibitor across Caco-2 cells with IC50 values of 0.02 μM for nelfinavir, 0.05 μM for indinavir, and 0.08 μM for saquinavir (Fig. 1B).

Interaction of HIV protease inhibitors with P-glycoprotein.
A, inhibition of P-gp-mediated [3H]digoxin (5 μM) transport across a Caco-2 cell culture monolayer (n = 3 or more) at varying concentrations of LY-335979 (⋄), valspodar (*), nelfinavir (●), ritonavir (○), saquinavir (▪), and indinavir (▵). Comparable data for ketoconazole, cyclosporine A, verapamil, and quinidine were within the boundaries of nelfinavir and ritonavir and are not shown for reasons of clarity. B, inhibition of14C-labeled nelfinavir (●), saquinavir (▪), and indinavir (▵) transport (5 μM) across the Caco-2 cell monolayer in the presence of varying concentrations of LY-335979.
All of the HIV protease inhibitors were able to inhibit the CYP3A-mediated metabolism of nifedipine, with ritonavir (IC50 = 0.32 μM) and indinavir (0.67 μM) being considerably more potent than nelfinavir (3.8 μM) and saquinavir (6.5 μM). Ketoconazole (0.15 μM) was also a potent CYP3A inhibitor, whereas quinidine (50 μM) was 300-fold less potent. In addition, the IC50 values for cyclosporin A (3 μM) was almost 3-fold smaller than its analog valspodar (10 μM), and LY-335979 had a similar IC50 value of 5 μM. We have previously described the ratio of the IC50 (CYP3A) to the IC50(P-gp) as an index of the relative selectivity of a drug to inhibit the efflux transporter in contrast to drug metabolism (Wandel et al., 1999). This ratio was the highest for LY-335979 at 208, compared with a value of 91 for valspodar; the ratios for all the other drugs were substantially lower (<3).
Pretreatment with 25 mg/kg LY-335979, 30 min before and concurrently with [14C]nelfinavir, markedly altered the disposition of total radioactivity in mdr1a(+/+) mice. The brain concentration-time profile in particular was especially affected (Fig. 2). In untreated mice, radioactivity in the plasma was more than 17 times greater than that in brain with a mean brain/plasma concentration ratio of 0.06, based on the relative area-under-the-concentration-time curves. LY-335979 increased brain levels by 25-fold in contrast to those in the plasma, which only changed 1.8-fold. Subsequent studies, based on tissue distribution measured 2 h after nelfinavir administration, showed that these changes were dose-dependent (Fig.3; Table1). At lower LY-335979 doses between 12.5 and 25 mg/kg, 10- to 15-fold higher brain levels could be achieved with no effect on nelfinavir plasma concentrations. Comparison of these findings with those in mdr1a(−/−) mice administered LY-335979 indicated that, if all of the effects were accounted for by P-gp inhibition, the transporter was inhibited by about 75% following a total dose of 50 mg/kg LY-335979 (Fig. 3; Table 1). Similarly, results were obtained in the testes with nelfinavir where P-gp activity appeared to be inhibited by over 90% (Table 1) compared withmdr1a(−/−) mice given LY-335979. HPLC-radiochromatography of the radioactivity present in plasma and brain samples obtained 2 h after nelfinavir and LY-335979 administration indicated the presence of a single peak with an identical retention time to nelfinavir. Changes in tissue/plasma distribution were also noted after i.v. administration of [14C]saquinavir, [14C] indinavir, and [14C]amprenavir following pretreatment with 50 mg/kg LY-335979; however, these were less marked than with nelfinavir (Fig. 4). In addition, the tissue/plasma ratio of HIV-1 protease inhibitors in tissues not expressing significant amounts of P-gp, such as spleen and heart, were not significantly altered by LY-335979 administration.

Tissue levels of [14C]nelfinavir (5 mg/kg) in mdr1a wild-type mice given 25 mg/kg LY-335979 [plasma (○), brain (●)] or vehicle [plasma (▵), brain (▴)] 30 min before and simultaneously with [14C]nelfinavir.
Each point represents three mice, except at 2 h (n= 5).
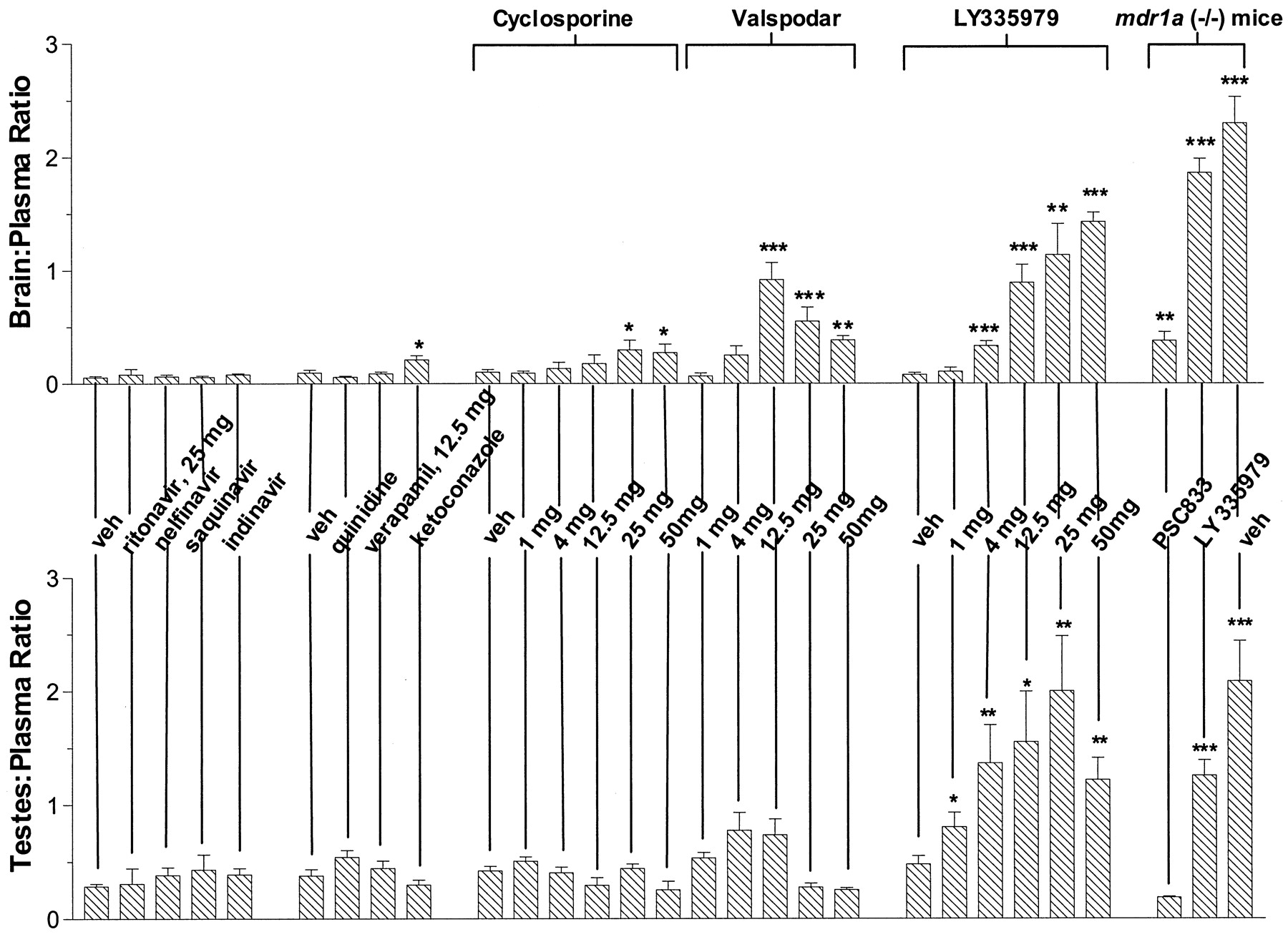
Effect of P-gp inhibitors (dose shown is per kilogram of body weight) on brain/plasma and testes/plasma distribution ratios of [14C]nelfinavir in wild-type mdr1a(+/+) and mdr1a(−/−) mice.
Actual values are shown in Table 1. Statistical differences compared with vehicle-treated mdr1a wild-type mice were assessed using a two-sided Student's t test, or a Mann-WhitneyU test dependent on the variance, with P < .05 as the limit of significance (*P < .05, **P < .01, ***P < .001).
Tissue levels of radioactivity (nanograms per gram of tissue) in wild-type and mdr1a(−/−) mice at 2 h after i.v. injection of [14C]nelfinavir (5 mg/kg)
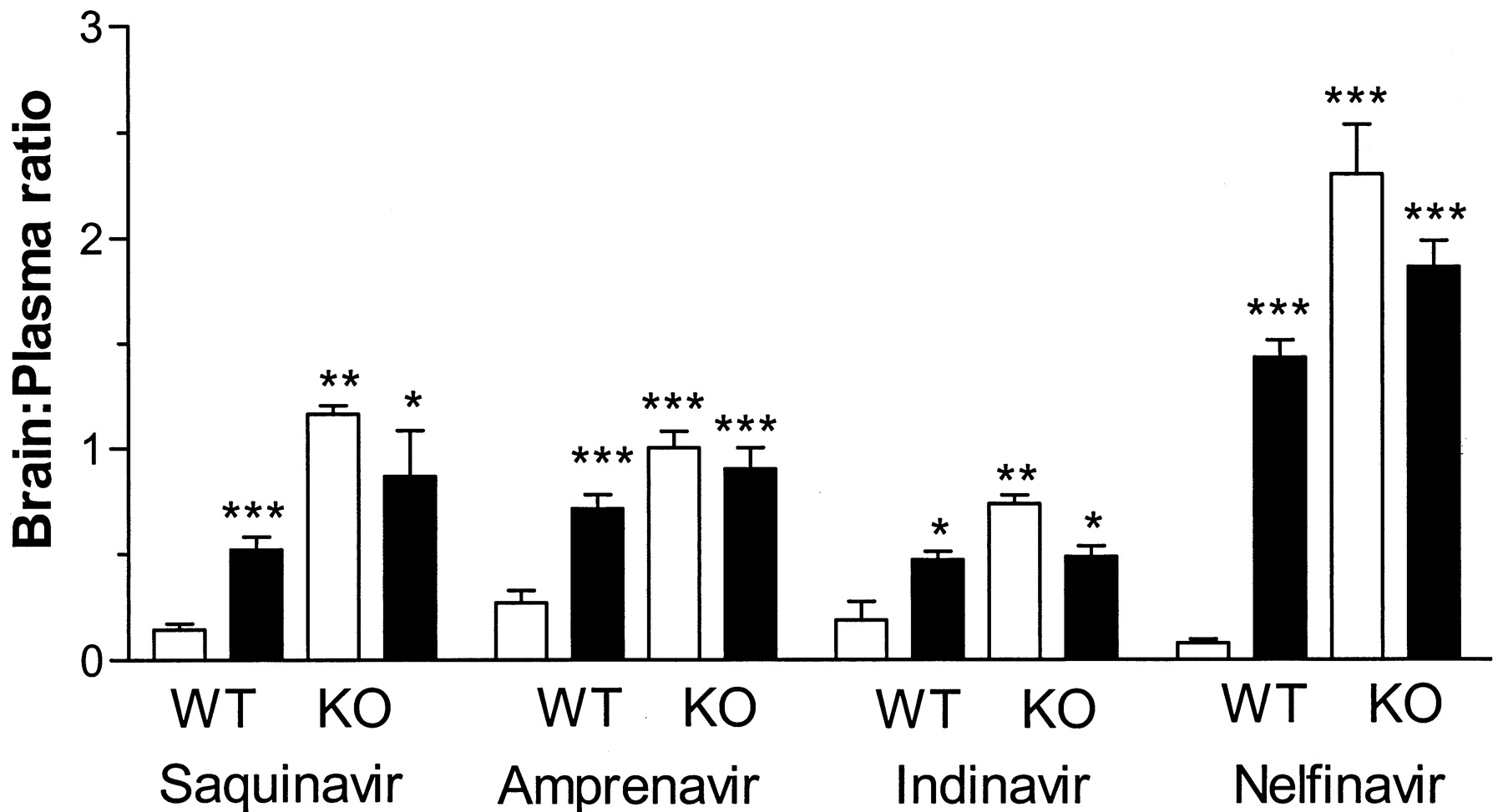
Brain/plasma distribution ratios determined 2 h after radiolabeled HIV-protease inhibitor administration (mean ± S.E., n = 3 or greater) in vehicle control mice (open column) and LY335959 (50 mg/kg) treated (filled column) mdr1a(+/+) wild type (WT) and mdr1a(−/−) knockout (KO) mice.
Statistical differences in radioactivity compared with untreatedmdr1a wild-type mice were assessed using a two-sided Student's t test, or Mann-Whitney U test dependent on the variance, with P < .05 as the limit of significance (*P < .05, **P < .01, ***P < .001).
Valspodar, like LY-335979, increased nelfinavir levels in brain and testes in a dose-dependent fashion, and at the highest dose level brain and testes concentrations were similar to those observed inmdr1a(−/−) mice (Table 1). However, in large part, this appeared attributable to the valspodar-induced increase in the plasma concentrations of the HIV protease inhibitor, with the brain/plasma ratio of nelfinavir being maximally increased (13-fold) following pretreatment with 12.5 mg/kg valspodar, which was comparable with the change after the same dose of LY-335979. At higher doses, valspodar increased plasma nelfinavir levels disproportionately to those in brain and testes so that the tissue/plasma ratios declined. In contrast to the brain, valspodar had only a modest effect on the testes/plasma ratio of nelfinavir and, again its value was reduced from its maximum at doses above 12.5 mg/kg. More modest, although statistically significant, increases in brain levels were produced by cyclosporin A, ketoconazole, and ritonavir administration, but again, these largely reflected increased nelfinavir plasma concentrations rather than a selective alteration in tissue distribution, secondary to inhibition of P-gp. Finally, neither quinidine, verapamil, nelfinavir, saquinavir, nor indinavir produced significant changes in the disposition of nelfinavir at the doses studied (Fig. 3; Table 1).
Discussion
HIV-1 enters the brain relatively early after primary infection (Kolson et al., 1998) and a similar distribution pattern probably occurs with other organs like the testes. Reduction of the viral load in such organs has, however, proven to be difficult in comparison to the plasma, possibly, in part, because most of the current HIV antiviral agents have difficulty in penetrating into the tissues and achieving sufficiently high concentrations to prevent replication (Groothuis and Levy, 1997). Drug properties like plasma protein binding may be contributory to this phenomenon, but it is increasingly recognized that these pharmacologic sanctuary sites possess a functional barrier to drug entry. For example, recent studies have demonstrated that HIV protease inhibitors are excellent substrates for the membrane efflux pump P-gp (Kim AE et al., 1998; Kim RB et al., 1998; Lee et al., 1998) that is localized to the lumenal surface of capillary endothelial cells of the brain and testes (Thiebaut et al., 1987; Cordon-Cardo et al., 1989), and limits drug distribution into these tissues. The major finding of this study is that the functional activity of this transporter can be pharmacologically inhibited and that such modulation results in significantly enhanced HIV protease inhibitor concentrations in both the brain and testes. Importantly, a highly potent and specific inhibitor of P-gp, such as LY-335979 (Dantzig et al., 1996, 1999), can achieve this effect by increasing the distribution of the antiviral drug into these tissues disproportionately to any change in plasma concentration, an observation consistent with a targeted impairment of P-gp-mediated efflux in the capillary endothelial cells.
P-gp inhibitors such as LY-335979 did not equally affect the tissue distribution of the HIV protease inhibitors (Fig. 4). This could reflect differences in inhibitory potency; however, in the Caco-2 system. LY-335979 was approximately equipotent with respect to inhibition of the individual HIV-1 protease inhibitors' transport (Fig. 1B). Moreover, previous studies comparing HIV protease inhibitor distribution between mdr1a(+/+) and mdr1a(−/−) mice showed that the brain/plasma ratio of nelfinavir was increased to a far greater extent than that of indinavir and saquinavir (Kim RB et al., 1998). These observations suggest that the relative roles of P-gp-mediated efflux versus uptake by passive and/or active transport in determining the brain entry of HIV protease inhibitors varies between drugs.
It is also apparent that the magnitude of the effect of P-gp inhibition is tissue-dependent, that is, the brain/plasma ratio increased to a greater extent than the testes/plasma value, whereas in tissues lacking significant expression such as heart and spleen, tissue/plasma ratios were unaffected. A possible explanation for this difference may be the greater openness of the intercellular junction between the capillary endothelium of the testes than in the brain (Holash et al., 1993). Moreover, at the highest LY-335979 doses, the brain and testes concentrations of nelfinavir were equal to or higher than the plasma level of the drug, suggesting that therapeutic effects would be substantially enhanced relative to the noninhibited situation. In fact, a similar finding of enhanced amprenavir central nervous system entry has been noted after administration of the potent P-gp inhibitor GF-120908 (Polli et al., 1999).
The experimental findings also illustrate major problems that have confounded attempts to inhibit P-gp activity in the clinical setting. The first of these is that drugs previously investigated, such as quinidine, verapamil, and cyclosporin A, have the potential to interact with drug-metabolizing enzymes (Thummel and Wilkinson, 1998), in particular, members of the cytochrome P450–3A subfamily (CYP3A). Not only is there considerable overlap in the substrates of these two proteins, but inhibitors of P-gp are also frequently inhibitors of CYP3A, and vice versa (Wacher et al., 1995; Kim et al., 1999). Accordingly, a dual interaction occurs whereby reduced P-gp function is also associated with increased plasma levels of the CYP3A substrate. It is, therefore, not surprising that such well established CYP3A inhibitors such as ketoconazole, HIV protease inhibitors, and cyclosporin A (Thummel and Wilkinson, 1998) resulted in higher plasma concentrations of nelfinavir, because CYP3A is importantly involved in the metabolism of this and other HIV protease inhibitors, especially ritonavir (Lin, 1997; Wu et al., 1999). Moreover, such an interaction also resulted in enhanced drug levels in brain and testes; in fact, administration of valspodar, a known CYP3A inhibitor (Fischer et al., 1998), produced pronounced increases in plasma concentrations that contributed significantly to the elevated tissue level. Because of this type of interaction, substantial dosage reduction of cytotoxic cancer chemotherapeutic agents has been required in the clinical investigations of valspodar as a reversing agent in patients exhibiting multidrug resistance (Boote et al., 1996; Sonneveld et al., 1996).
Although many P-gp inhibitors impair CYP3A-mediated metabolism (Thummel and Wilkinson, 1998), this is not an absolute relationship. In fact, the two characteristics appear to be independently determined so that some CYP3A inhibitors do not cause significant impairment of P-gp function and, more importantly, the reverse situation is possible, i.e., effective transporter inhibition with minimal effect on CYP3A (Wandel et al., 1999). Because of this, it has been suggested that relative selectivity with respect to P-gp inhibition versus CYP3A inhibition can be assessed from the ratio of the IC50 values for the two interactions (Wandel et al., 1999). Based on this index, it is apparent that valspodar and LY-335979 are significantly more selective inhibitors of P-gp compared with CYP3A than are the other drugs studied. Moreover, the greater selectivity of LY-335979 relative to valspodar is due almost entirely to its considerably greater potency as a P-gp inhibitor, because the effectiveness of both agents to inhibit CYP3A activity is about the same. Similar findings on the effect of LY-335979 on CYP3A have been recently reported (Dantzig et al., 1999). This selectivity would account for the relatively small LY-335979-induced changes in the plasma level of nelfinavir associated with effective P-gp inhibition.
The second problem associated with the previous investigation and clinical use of P-gp modulators has been their limited potency. The observed minimal effects of quinidine, verapamil, ketoconazole, and cyclosporin A on the tissue/plasma ratios of nelfinavir are consistent with such low potency as demonstrated by their IC50 values relative to digoxin translocation across Caco-2 cells. By contrast, LY-335979 produced over 75 and 90% inhibition of P-gp transport at the brain and testes, respectively. It is also noteworthy that the HIV protease inhibitors were not particularly potent inhibitors of P-gp-mediated digoxin transport, in vitro; their IC50 values being 10- to 400-fold greater than that of valspodar or LY-335979. Moreover, no change in the tissue/plasma distribution ratio of nelfinavir was observed when another HIV protease inhibitor was coadministered (Fig. 4). This would suggest that any enhanced tissue bioavailability and efficacy resulting from the combined use of several HIV protease inhibitors, e.g., ritonavir and saquinavir, in HIV-infected patients (Piketty et al., 1999), does not involve inhibition of P-gp, as recently speculated (Drewe et al., 1999).
Another issue of selectivity by the currently available P-gp modulators would appear to be related to the inhibition of P-gp itself versus other transporters. A growing number of uptake and efflux transporters have been identified and characterized in various different cells/tissues within the body (Ambudkar and Gottesman, 1998), and overlapping inhibition of these transporters appears to occur. For example, a number of P-gp inhibitors such as quinidine, verapamil, ketoconazole, and valspodar also impair drug uptake by human organic anion transporting polypeptide (OATP) (Cvetkovic et al., 1999), but generally at higher concentrations than those required for inhibition of P-gp. There is significant substrate overlap between members of the OATP drug uptake transport family and the drug efflux transporter P-gp, including digoxin, fexofenadine, and steroid hormones such as cortisol (Bossuyt et al., 1996a,b; Noe et al., 1997; Cvetkovic et al., 1999;Kusuhara et al., 1999). In fact, an OATP type of transporter (Oatp2) is present in the brain (Noe et al., 1997; Kusuhara et al., 1999) and more recently localized to the capillary endothelial cells that make up the blood-brain barrier (Gao et al., 1999). Therefore, it is not unreasonable to suggest that the observed reduction in the tissue/plasma ratio of nelfinavir at the higher doses of valspodar inmdr1a(−/−) mice reflects inhibition of drug uptake transport system(s), because a reduction in the brain/plasma ratio could not occur in mdr1a(−/−) mice if P-gp was the only target of inhibition. A similar effect with valspodar has also been observed with another P-gp substrate, digoxin (Mayer et al., 1997).
In summary, findings from this study suggest selective increases in brain and testes levels of HIV protease inhibitors can be achieved by targeted pharmacologic inhibition of P-gp. However, to achieve the desired inhibition of P-gp at these sites, the inhibitor must be both highly potent and selective for the transporter. In the case of nelfinavir and LY-335979, the enhancement is substantial, suggesting that if a similar situation occurs in humans this could potentially increase the efficacy of the antiviral and reduce active HIV-1 replication in tissues that normally are relatively protected from the effects of the antiviral.
Acknowledgments
We acknowledge the assistance and cooperation of the following individuals and companies in supplying 14C-labeled and other drugs: nelfinavir, Dr. Bhasker Shetty, Agouron Pharmaceuticals Inc.; amprenavir, Dr. Joe L. Woolley, GlaxoWellcome Inc.; saquinavir, Dr. Hugh Wiltshire, Roche Products Ltd.; indinavir, Dr. Alan S. Nies, Merck Research Laboratories; ritonavir, Dr. John M. Leonard, Abbott Laboratories: valspodar, Dr. Sean Wells, Novartis Pharm AG; and LY-335979, Dr. Anne H. Dantzig, Lilly Research Laboratories.
Footnotes
-
Send reprint requests to: Richard B. Kim, M.D., 572 MRB1 Division of Clinical Pharmacology, Vanderbilt University School of Medicine, Nashville, TN 37232-6602. E-mail:richard.kim{at}mcmail.vanderbilt.edu
-
This work was supported in part by U.S. Public Health Service Grants GM31304, GM54724, CA68485; the Deutsche Forschungsgemeinschaft (to C.W.); and a grant in-aid from Lilly Research Laboratories.
- Abbreviations used are::
- P-gp
- P-glycoprotein
- MDR
- multidrug resistance
- OATP
- human organic anion transporting polypeptide
- LY-335979
- (2R)-anti-5-{3-[4-(10,11-difluoromethanodibenzosuber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinilone trihydrochloride
- Received November 18, 1999.
- Accepted February 28, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}