


Abstract
The anticonvulsant phenytoin (5,5-diphenylhydantoin) provokes a skin rash in 5 to 10% of patients, which heralds the start of an idiosyncratic reaction that may result from covalent modification of normal self proteins by reactive drug metabolites. Phenytoin is metabolized by cytochrome P450 (P450) enzymes primarily to 5-(p-hydroxyphenyl-),5-phenylhydantoin (HPPH), which may be further metabolized to a catechol that spontaneously oxidizes to semiquinone and quinone species that covalently modify proteins. The aim of this study was to determine which P450s catalyze HPPH metabolism to the catechol, proposed to be the final enzymatic step in phenytoin bioactivation. Recombinant human P450s were coexpressed with NADPH-cytochrome P450 reductase in Escherichia coli. Novel bicistronic expression vectors were constructed for P450 2C19 and the three major variants of P450 2C9, i.e., 2C9*1, 2C9*2, and 2C9*3. HPPH metabolism and covalent adduct formation were assessed in parallel. P450 2C19 was the most effective catalyst of HPPH oxidation to the catechol metabolite and was also associated with the highest levels of covalent adduct formation. P450 3A4, 3A5, 3A7, 2C9*1, and 2C9*2 also catalyzed bioactivation of HPPH, but to a lesser extent. Fluorographic analysis showed that the major targets of adduct formation in bacterial membranes were the catalytic P450 forms, as suggested from experiments with human liver microsomes. These results suggest that P450 2C19 and other forms from the 2C and 3A subfamilies may be targets as well as catalysts of drug-protein adduct formation from phenytoin.
The anticonvulsant phenytoin (5,5-diphenylhydantoin) has been associated with a number of toxic effects, some of which may result from primary and secondary metabolites, rather than the parent drug (Spielberg et al., 1981). In particular, drug-induced hypersensitivity reactions (Haruda, 1997), which occur in 5 to 10% of patients started on phenytoin therapy (Hyson and Sadler, 1996), may be due to haptenation of normal tissue proteins by phenytoin metabolites, leading to initiation of an inappropriate immune response directed at normal or haptenated proteins (Park et al., 1987). Patients experiencing hypersensitivity reactions show serum antibodies that react with cytochrome P450 (P450)2-related epitopes, specifically antigenic determinants on rat P450 3A and 2C forms and human P450 5 and P450 8 peptides (Leeder et al., 1996, 1998).
The major pathway of phenytoin clearance is hydroxylation by P450 enzymes to its phenol metabolite, 5-(p-hydroxyphenyl-),5-phenylhydantoin (HPPH). HPPH can be further oxidized to the catechol, which may then undergo methylation by catechol O-methyl transferase.
Several enzymes have been proposed to play a role in phenytoin bioactivation, namely, prostaglandin synthetase (by cooxidation) (Kubow and Wells, 1989), myeloperoxidase (Uetrecht and Zahid, 1988; Mays et al., 1995), and P450 (Roy and Snodgrass, 1988). Other mechanisms of activation, such as oxidation by superoxide, have been considered. However, to date, a link between the mechanism behind drug activation and the antigen toward which the autoimmune response is directed has not been elucidated.
We have recently shown that metabolism of phenytoin to its catechol derivative is followed by spontaneous oxidation to semiquinone and quinone species capable of forming covalent adducts with proteins in human liver microsomes (Munns et al., 1997). The most probable target of covalent adduct formation might be expected to be the P450 responsible for catechol formation, because further oxidation could occur in the active site and the reactive species could then preferentially react with neighboring nucleophilic amino acid residues. In support of this contention, mechanism-based inactivation of P450 was observed in parallel with catechol formation (Munns et al., 1997) in rat liver microsomes. Also, in liver microsomes incubated with radiolabeled phenytoin and HPPH, proteins in the molecular weight range associated with P450 forms became specifically radiolabeled under conditions supporting P450 catalysis (Munns et al., 1997).
P450 2C9 and 2C19 have been established to be the catalysts of phenytoin metabolism to HPPH in human liver microsomes (Veronese et al., 1991; Bajpai et al., 1996). However, the P450 species responsible for the further metabolism of HPPH have not yet been characterized. It was of interest to determine which P450 forms could catalyze further oxidative biotransformation of HPPH to the derivative, which could be critical in the haptenation of tissue proteins (including the P450 species responsible for catechol production). In particular, we were interested to determine whether P450 2C or 3A forms were involved in HPPH oxidation because patient antibodies reacted with P450 3A- and 2C-related epitopes (Leeder et al., 1996). The hypothesis underlying this work was that adduct formation may alter the antigenic processing of these P450s in such a way as to lead to a breakdown in tolerance to these forms or related P450s, such as thromboxane synthase or prostacyclin synthase, forms that have been identified as putative autoantigens in anticonvulsant-induced hypersensitivity (Leeder et al., 1998).
Experimental Procedures
Materials.
The bicistronic bacterial expression plasmids for P450s 1A1, 1A2, 1B1, 2D6, 2E1, 3A4, 3A5, and 3A7 were constructed as described previously (Gillam et al., 1997; Parikh et al., 1997; Shimada et al., 1998). The human NADPH P450 (hNPR) expression vector, pCW′/hNPR, containing the reductase cDNA common to all bicistronic plasmids used here, but containing no P450 cDNA, was obtained from Professor F. P. Guengerich (Vanderbilt University, Nashville, TN) as were the yeast and bacterial expression vectors for the P450 2C9 variant designated 2C10 (reported to contain codons for the following residues: Arg 144, pAAH5/MP8 (Brian et al., 1989) and pCW′/2C1029 (Sandhu et al., 1993). The bacterial expression plasmid for P450 2C19 was a generous gift from Dr. T. Baba of Shionogi and Co. (Osaka, Japan). Professor D. J. Birkett (Flinders University, Adelaide, Australia) donated the mammalian expression plasmids for P450 2C9 (p91023/2C9V2A (2C9*1; designated as the wild type sequence, encoding a protein with Arg at position 144, Tyr at 358, Ile at 359, and Gly at 417), p91023/2C9V2 (2C9*2; encoding a protein with Cys at 144, Tyr at 358, Ile at 359, and Gly at 417), p91023/2C9V1 (2C9*3; encoding a protein with Arg at position 144, Tyr at 358, Leu at 359, and Gly at 417), and p91023/2C9V2ABC (variant 2C10; described as encoding a protein with Arg at position 144, Cys at 358, Ile at 359, and Asp at 417) (Veronese et al., 1993).
Rabbit anti-human P450 3A4 and goat anti-rabbit P450 2C3 were generous gifts from Professor F. P. Guengerich (Vanderbilt University) and Professor M. E. McManus (University of Queensland, Australia), respectively. Rabbit antiserum raised against a P450 2C10-specific peptide (residues 413–424 LDEGDNFKKSKY) was provided by Hermann Esselmann (Medischine Hochschule, Hannover, Germany). Liver samples were obtained from organ donors under procedures approved by local institutional ethics committees, snap-frozen in liquid nitrogen, and stored at −70°C until use. Microsomes were prepared as described previously (Guengerich, 1994).
Restriction endonucleases, DNA ligase, and other enzymes for molecular biology were obtained from New England Biolabs. [14C]HPPH and [14C]catechol were isolated as previously described (Munns et al., 1997). All other reagents were purchased from local suppliers at the highest quality available.
Construction of Bacterial Expression Plasmids.
Expression plasmids for P450 2C9 were generated by the cloning strategy outlined in Fig. 1. Briefly, wild type and variant 2C9 sequences, excised from p91023 as EcoRI fragments, were cloned into pCW′/3A5#1 (Gillam et al., 1995) in place of the internal and C-terminal EcoRI fragments of P450 3A5. The residual P450 3A5 5′ sequence was then removed by replacing thePflMI fragment containing the 3A5 and 2C9 N-terminal sequences with that derived from the expression plasmid for P450 2C10 (Sandhu et al., 1993). This step served to introduce the N-terminal modifications found necessary for expression of P450 2C10. Bicistronic expression constructs were then prepared by subcloning the modified P450 2C9 sequences as NdeI-SalI fragments into the cognate sites of the bicistronic expression vector for P450 2C10. Constructs were checked for retention of 3′EcoRI site, present in all P450 2C9 fragments but absent from the P450 2C10 vector.

Cloning strategy for expression of P450 2C9 variants.
The bicistronic expression vector for P450 2C19 was constructed by inserting the HindIII fragment from pCW′/1A2/hNPR (Parikh et al., 1997) containing the reductase cDNA into the cognate site in pCW′/2C19 (S. Kirita, J. Aoyama, Y. Yamaguchi, K. Kadono, K. Ohno, and T. Baba, unpublished data). All constructs were confirmed by restriction mapping.
Expression of Recombinant Human P450 Enzymes and Quantification of Recombinant Proteins.
Recombinant P450 forms were expressed in bacteria in bicistronic format with hNPR as previously described (Gillam et al., 1997; Parikh et al., 1997; Shimada et al., 1998). Briefly, cultures were grown at 30°C in modified terrific broth containing 1 mM isopropyl-1-thio-β-d-galactopyranoside, 0.5 mM δ-aminolevulinic acid, 1 mM thiamine, 0.2 g/liter bactopeptone, and 100 μg/ml of ampicillin. Cells were harvested and membranes prepared according to established procedures (Gillam et al., 1993). Hemoprotein expression was quantified by difference spectroscopy. Fe2+ versus Fe2+ · CO difference spectra were recorded on a Beckman DU-640 UV-visible spectrophotometer. NADPH-P450 reductase expression was assayed according to Guengerich (1994) and yields (nanomoles per liter of culture) were calculated using a specific activity of 3200 nmol of cytochrome c reduced/min/nmol of reductase.
Analysis of Enzyme Activity.
Examination of phenytoin and HPPH metabolism to stable and reactive metabolites was carried out generally as described previously (Munns et al., 1997) but using membranes from bacteria transformed with bicistronic constructs or liver microsomes. In some experiments, bacterial membranes were supplemented to P450/reductase ratios of 1:2 or 1:5 with additional reductase using membranes from bacteria transformed with the reductase expression vector pCW′/hNPR. Substrates were added from ethanolic stocks, and the organic solvent content was maintained at or below 1% (v/v) in all cases. Sulfaphenazole was added from an ethanolic stock and troleandomycin was added from dimethyl sulfoxide. Where inhibitors were used, the total concentration of organic solvent was maintained at 1% and controls (no inhibitor) contained the same proportions of all solvents. HPPH and the catechol metabolite were quantified by HPLC analysis, and covalent adduct formation was measured as described previously (Munns et al., 1997).
Other Methods.
Protein concentrations were quantified by the Lowry method as described by Guengerich (1994). SDS-polyacrylamide gel electrophoresis was performed according to Laemmli (1970) in 8.3% gels. Immunoblotting was carried out as described previously using a 1:2000 dilution of rabbit anti-human P450 3A4 antiserum and a 1:5000 dilution of goat anti-rabbit P450 2C3 IgG. Blots were visualized using horseradish peroxidase-coupled secondary antibodies and 4-chloronaphthol as the chromogenic substrate. Fluorographs were prepared as described (Munns et al., 1997). Manual DNA sequencing was performed using the dideoxy method and Sequenase version 2.0 (Amersham, Castle Hill, Australia) according to the manufacturer's instructions. Automatic sequencing was performed at the Australian Genome Research Facility (St. Lucia, Australia) using the Big Dye Terminator system (Perkin Elmer Applied Biosystems, Scoresby, Australia).
Results
The isoform specificity of catechol formation was examined using the form-selective inhibitors, troleandomycin (P450 3A) and sulfaphenazole (P450 2C), in a human liver microsomal preparation (HL27) previously shown to catalyze covalent adduct formation and to contain average levels of P450 3A and 2C immunoreactivity (data not shown). Although troleandomycin showed little inhibition of HPPH production from phenytoin, further metabolism of HPPH to catechol was significantly affected. Sulfaphenazole inhibited both pathways (Table1) at concentrations selective for P450 2C forms. Covalent adduct formation, using 5.2 μM [14C]HPPH as substrate, was reduced by comparable concentrations of sulfaphenazole to 44 and 0% (12 and 120 μM sulfaphenazole, respectively) and troleandomycin to 42 and ∼48% (12 and 120 μM troleandomycin, respectively) of control values, in the same liver preparation.
Form-selective inhibition of phenytoin and HPPH metabolism in human liver microsomes
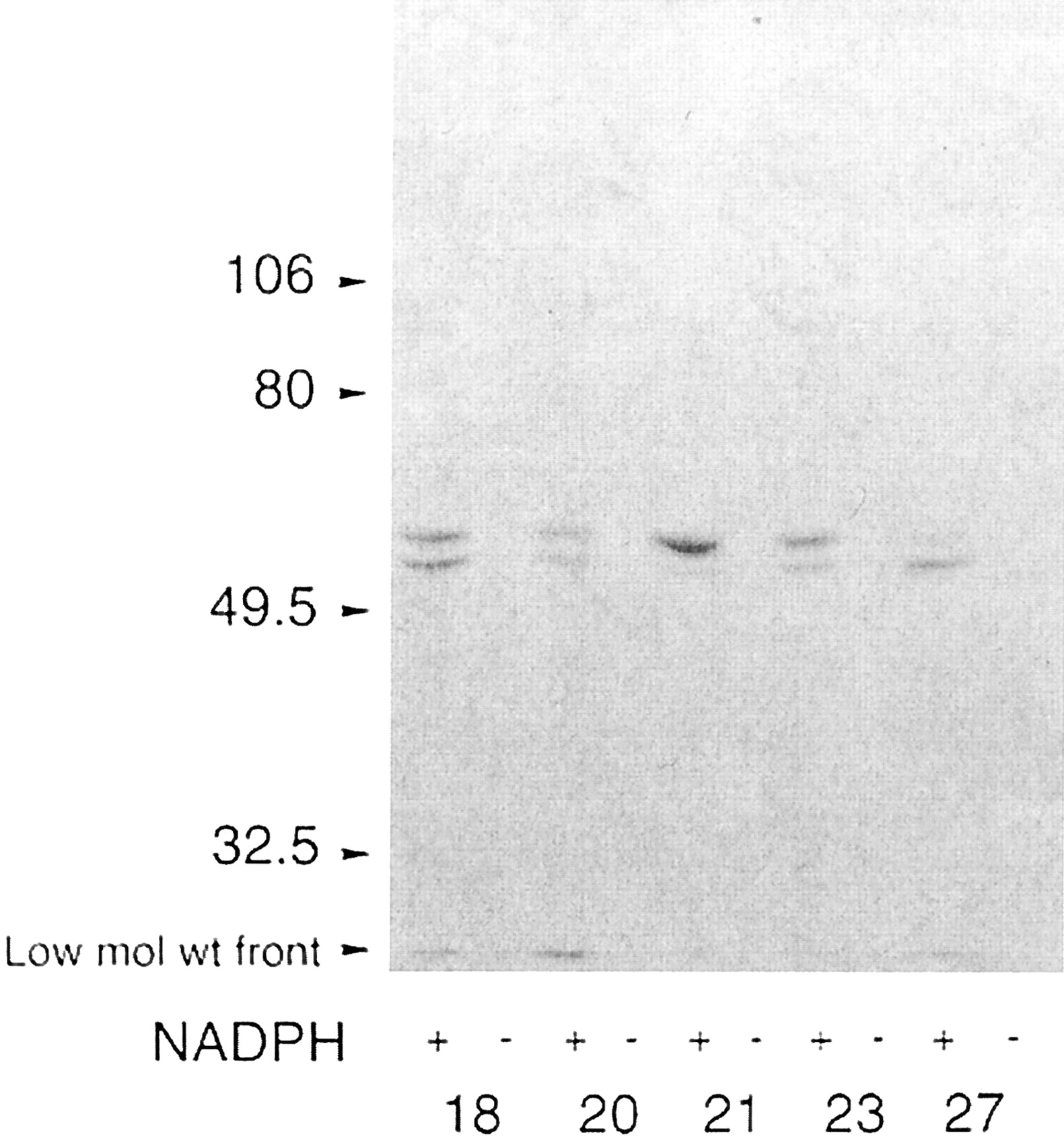
Incubation of human liver microsomes with [14C]HPPH showed that two bands of molecular weight within the range expected for P450 enzymes became modified in five different livers, consistent with covalent modification of at least two specific catalysts of HPPH bioactivation (Fig.2).

Fluorograph of covalent adduct formation in human liver microsomes.
Liver microsomes prepared from five different individuals (designated HL18, 20, 21, 23, and 27) were incubated with 8.7 μM [14C]HPPH at a P450 concentration of 0.75 μM for 2 h at 37°C. Aliquots of incubation mixtures were subjected to electrophoresis and fluorography directly. Each lane contains 37.5 pmol of P450. Arrows indicate the relative migration of molecular weight markers of the indicated sizes.
P450 forms were coexpressed with NADPH P450 reductase in bacterial membranes so as to facilitate examination of the relative ability of different P450 forms in phenytoin and HPPH metabolism and covalent adduct formation. Novel bicistronic expression constructs were prepared for the wild type and two major variant forms of P450 2C9 using the same strategy as used previously for the variant designated P450 2C10. During the course of the plasmid construction we observed an anomaly with the 2C10 sequence. The reported sequence of P450 2C10 at codon 358 (numbering corresponds to the full-length native sequence) contains anNsiI site; however, restriction analysis withNsiI failed to show any digestion at this site using either the 2C10 sequence present in the bacterial expression vector pCW′/2C1029 (Sandhu et al., 1993) or the original yeast expression vector pAAH5/MP8 (Brian et al., 1989). NsiI digestion was seen with the vector p91023/2C9V2ABC containing a 2C10 sequence constructed by site-directed mutagenesis (Veronese et al., 1993). DNA sequencing revealed that the sequence at codon 358 was wild type Tyr rather than the reported Cys codon. Sequencing also confirmed that the Asp codon reported uniquely in the P450 2C10 was present at position 417. In addition, immunoblotting with an antipeptide antibody raised to a 2C10-specific peptide centered on residue 417 showed that P450 recombinant proteins derived from the P450 2C10 sequence were markedly more immunoreactive than the other P450 2C9 variant proteins (data not shown). Expression levels for recombinant P450 2C forms are shown in Table 2.
Yields of P450 2C forms and hNPR expressed using novel expression vectors
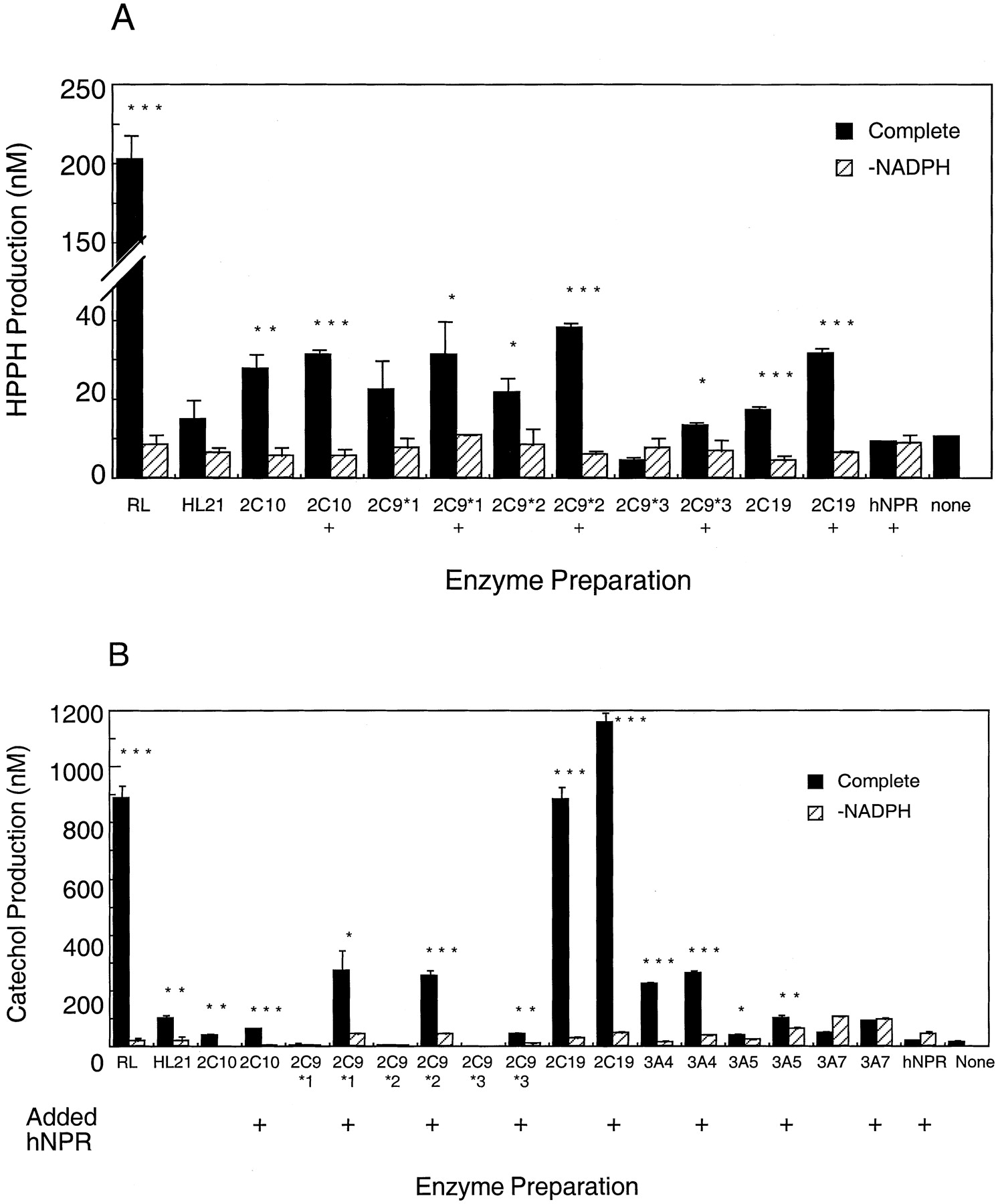
The activity of recombinant P450 2C and other forms was assessed toward phenytoin and HPPH. The effect of supplementing reductase levels was examined because this had been seen previously to enhance nifedipine oxidase and midazolam hydroxylase activities for recombinant P450 3A forms expressed in bacterial membranes (results not shown). Although rates for P450 2C forms were substantially enhanced by the presence of additional reductase, less effect was seen on P450 3A forms. All recombinant P450 2C forms showed activity toward phenytoin, which could be enhanced by supplementation of NPR to an NPR/P450 ratio of 5:1 (Fig.3A). No other P450 forms tested (P450s 1A1, 1A2, 1B1, 2D6, 2E1, 3A4, 3A5, 3A7) showed significant phenytoin hydroxylase activity (results not shown). In contrast, both P450 2C and 3A forms participated in catechol formation from HPPH (Fig. 3B); no other forms tested catalyzed catechol formation at any significant level (data not shown). A marked effect of reductase supplementation was seen with P450 2C9 variants. Incubations with P450 3A forms were supplemented to an NPR/P450 ratio of 2:1 as found optimal previously.
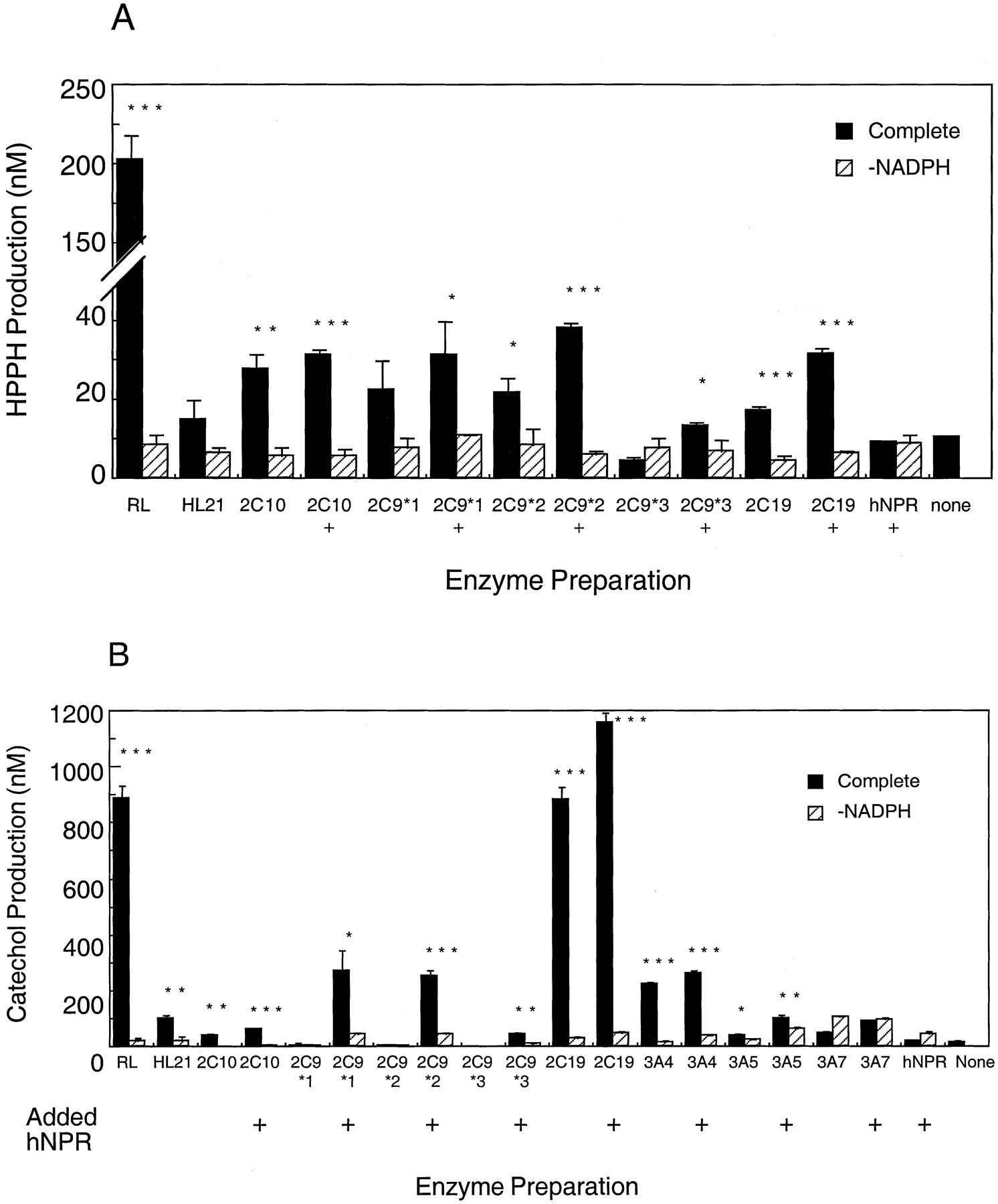
HPPH and catechol production by recombinant human P450s and human and rat liver microsomes.
Liver microsomes or recombinant P450 enzymes were incubated with 150 μM phenytoin (A) or HPPH (B) at a P450 concentration of 0.1 μM for 2 h at 37°C. HL and RL denote experiments using human and rat liver microsomes, respectively. +, indicates incubations where extra hNPR was added to the incubations in the forms of membranes for cells expressing hNPR alone to a reductase concentration of five (P450 2C forms) or two (P450 3A forms) times the P450 concentration. hNPR and none denote incubations with membranes isolated from cells expressing hNPR alone, and no recombinant proteins, respectively. HPPH metabolism was also checked using bacterial membranes derived from cells transformed with the bicistronic expression vectors for P450s 1A1, 1A2, 1B1, 2E1, and 2D6; none of these forms showed any significant activity. Data represent the means ± S.D. of triplicate determinations. Asterisks indicate significant increase over -NADPH controls at the following levels (Student's t test) *P < .05; **P < .01; ***P < .001.
Drug-protein adduct formation was examined in incubations of recombinant P450s or microsomes with [14C]HPPH. Figure 4 shows that relative adduct formation was greatest with P450 2C19 among the membranes containing recombinant P450s; however, significant activity was seen for all P450 2C and 3A forms with the exception of the 2C10 variant. The proteins modified by drug metabolites were examined by fluorography. Figure5 shows that adduct formation could be observed with proteins in bacterial membranes containing P450s 3A4, 3A5, 3A7, and 2C19, that comigrated approximately with the proteins modified in microsomes. Adduct formation was enhanced in NADPH-replete samples versus NADPH-deficient controls but was less intense in recombinant preparations than human liver preparations. Some background radiolabel could be seen in controls, consistent with low level contamination of [14C]HPPH with [14C]catechol (Munns et al., 1997).

Covalent binding of radiolabel derived from [14C]HPPH to bacterial membranes containing recombinant P450 forms or to liver microsomes.
Drug protein adduct formation by P450 2C and 3A4 forms and liver microsomes. Liver microsomes or recombinant P450 enzymes were incubated with 5.6 μM [14C]HPPH at a P450 concentration of 0.75 μM (liver microsomes) or 0.25 μM (recombinant P450s in bacterial membranes) for 2 h at 37°C. HL and RL denote experiments using human and rat liver microsomes, respectively. pCW′ and hNPR signify incubations where membranes were used from bacteria transformed with the vector pCW′ alone and the vector pCW′/hNPR, respectively. Protein and reductase concentrations in these incubations were 3 mg/ml (equivalent to the highest concentration of protein in the other incubations with bacterial membranes) and 1.25 μM, respectively. +, Indicates incubations where extra hNPR was added in the form of membranes from cells expressing hNPR alone to a reductase concentration five times (2C forms) or two times (3A forms) the P450 concentration. Data represent the means ± S.D. of triplicate determinations. Asterisks indicate significantly higher radiolabel binding in complete incubations compared with -NADPH controls at the following levels (Student's t test): *P < .05; **P < .01; ***P < .001.

Fluorographic analysis of the distribution of protein adducts from [14C]HPPH in bacterial membranes containing recombinant P450 forms or in liver microsomes.
Liver microsomes or recombinant P450 enzymes were incubated with 25.4 μM [14C]HPPH at a P450 concentration of 0.75 μM (liver microsomes) or 0.25 μM (recombinant P450s in bacterial membranes) for 2 h at 37°C. For experiments with bacterial membranes, hNPR was supplemented in the form of membranes from cells expressing hNPR alone to a reductase concentration of 1.25 μM (2C forms) or 0.5 μM (3A forms). Aliquots of incubation mixtures were subjected to electrophoresis and fluorography directly. Each lane contains 37.5 pmol of P450 (microsomes) or 12.5 pmol of P450 (membranes).
Discussion
Previous studies in this laboratory have demonstrated that human liver P450 enzymes can metabolize phenytoin to reactive products that form drug-protein adducts, and have implicated the catechol secondary metabolite in this bioactivation process. P450 2C forms have been established to be the major catalysts of phenytoin metabolism to HPPH in human liver microsomes (Veronese et al., 1991; Bajpai et al., 1996); however, the P450 species responsible for the further metabolism of HPPH had not been characterized at the start of this study. It was of interest, therefore, to determine which P450 forms could catalyze further oxidative biotransformation of HPPH, in particular, to the catechol derivative that might ultimately lead to generation of a reactive metabolite in the proximity of the P450 concerned (Munns et al., 1997). The issue of whether P450 2C or 3A forms may be involved was particularly relevant because epitopes related to these P450s have been shown to be antigenic in patients suffering phenytoin hypersensitivity (Leeder et al., 1996).
To compare the activity of different P450 2C and 3A forms, recombinant P450s were coexpressed with hNPR in bicistronic format in E. coli. Novel expression constructs were developed for the wild type and two principal allelic variants of P450 2C9, using the expression plasmid for the 2C10 variant as a cloning vector. The P450 2C10 variant has been reported to differ from all other 2C9 variant sequences at two positions, containing codons for Cys rather than Tyr at codon 358 and Asp rather than Gly at codon 417 (Umbenhauer et al., 1987). In the course of this work, it became obvious that the 2C10 expression plasmid differed at codon 358 from the reported sequence, and DNA sequencing revealed the presence of the wild type Tyr codon rather than the reported Cys codon. Neither the Cys mutation nor the Asp allele have ever been detected in any population study (Stubbins et al., 1996;Bhasker et al., 1997). The results presented here support the contention that the reported Cys codon was the result of a sequencing error; however, the source of the apparent Asp mutation remains to be established, because the original cDNA was confirmed to have this variant sequence.
The activity of the recombinant P450 2C and other forms was examined with phenytoin and HPPH as substrates. For P450 2C forms, activity was enhanced in the presence of a 5-fold excess of hNPR. Previously we have found hNPR supplementation to an hNPR/P450 ratio of 2:1 to enhance prototypical P450 3A activities (results not shown), and this result was also confirmed using the activities examined here. These studies suggest that recombinant hNPR and P450 proteins can couple effectively, to at least some degree, in mixed membranes without the requirement for coexpression and translation within the same membrane population or the addition of detergents or other measures to facilitate membrane mixing.
Initial studies in human liver microsomes supported the contention that both P450 3A and 2C forms may be involved in formation of the catechol metabolite, and that multiple catalysts may become modified by reactive metabolites, the relative proportions of adducts differing according to liver P450 expression. Studies with recombinant P450 forms indicated that P450 2C19 was effective at catalyzing both catechol and adduct formation, but that P450 2C9 variants and P450 3A forms may also contribute to phenytoin bioactivation. Adduct formation generally paralleled catechol production. P450 3A and 2C forms are the two most common P450 subfamilies in adult human liver, averaging ∼30 and ∼20% total hepatic P450, respectively, comprising predominantly P450s 3A4 and 2C9 (Shimada et al., 1994). Accordingly, the quantitative contribution of P450s 3A4 and 2C9 to overall catechol formation may be greater than that of 2C19, consistent with the results obtained with sulfaphenazole and troleandomycin.
The activity of the three principal 2C9 variant forms toward HPPH oxidation and adduct formation was found to vary in the same manner as found for other substrates, i.e., the Leu 350 variant (2C9*3) showed markedly diminished activity relative to the wild type (2C9*1) and Cys 144 (2C9*2) forms. Interestingly, the 2C10 form, which is here established to have the wild type sequence except at position 417, showed similar activity toward phenytoin (measured at 150 μM substrate) as wild type, yet the turnover of HPPH (at 150 μM) was significantly reduced. This result and the failure to demonstrate any adduct formation (measured at 5.6 μM [14C]HPPH) with 2C10 may reflect a loss of affinity for HPPH relative to the wild type 2C9 form due to the Asp 417 mutation.
Two specific targets of adduct formation were apparent in human liver microsomes. Adduct formation was reduced in recombinant bacterial preparations compared with human liver microsomes at comparable P450 concentrations (assuming that each form comprised only a fraction of the total P450 concentration in microsomes), an effect that may be due to dilution of the fixed amount of radiolabeled drug in a larger volume of membrane phase. This effect may also explain the failure to see adduct formation in P450 2C9 samples that were supplemented with reductase to 5-fold the P450 concentration. We cannot dismiss the possibility that the N-terminal changes engineered into the recombinant proteins to facilitate expression alter the topology of the channel through which the reactive product exits the P450. This could result in removal or reorientation of nucleophilic target residues in such a way as to allow the reactive metabolite to move farther afield before modifying protein.
Alternatively, the higher reductase-to-P450 ratio in bacterial membrane preparations may have facilitated reduction of the quinone and semiquinone species back to the catechol. This would be expected to reduce the overall adduct formation relative to the same enzyme in human liver microsomes as well as making it more likely that the catechol could escape the microenvironment of the P450 and migrate more widely in the membrane phase before covalently modifying other membrane proteins. Even so, radiolabel appeared to be relatively concentrated within bands in bacterial membrane preparations that comigrated with P450s consistent with preferential modification of the P450s catalyzing catechol formation.
In summary, these results suggest that P450s 2C19, 2C9, 3A4, 3A5, and 3A7 may catalyze the secondary metabolism of phenytoin to a catechol metabolite and thereby bioactivation of the drug to protein-reactive species. It is likely that these forms become modified in the process, and this process may alter antigenic determinants on the P450s. Further studies are required to determine whether and how this process may initiate an autoimmune reaction directed toward related P450s.
Acknowledgments
We thank Professor F. P. Guengerich, Dr. T. Baba, Professor D. J. Birkett, Professor M. E. McManus, Asit Parikh, and Hermann Esselmann for donating plasmids and antibodies used in this work, and Professor F. P. Guengerich, Dr. T. Baba, and H. Esselmann for helpful discussions regarding some of the results obtained.
Footnotes
-
Send reprint requests to: Dr. Elizabeth M.J. Gillam, Department of Physiology and Pharmacology, University of Queensland, St. Lucia, Australia 4072. E-mail: gillam{at}plpk.uq.edu.au
-
↵1 This work was presented in preliminary form at the 5th International ISSX meeting, 25–28 Oct 1998, Cairns, Australia.
- Abbreviations used are::
- P450
- cytochrome P450
- HPPH
- 5-(p-hydroxyphenyl-),5-phenylhydantoin
- hNPR
- human NADPH-cytochrome P450 reductase
- Received March 24, 2000.
- Accepted May 11, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}