


Abstract
The expression and inducibility of four CYP2C genes, including CYP2C8, -2C9, -2C18, and -2C19, was investigated in primary cultures of human hepatocytes. By the use of RNase protection assay and specific antibodies, each CYP2C mRNA and protein were quantified unequivocally. The four CYP2C mRNAs were expressed in human livers and cultured primary hepatocytes, but only the CYP2C18 protein was not detected. Compounds known to activate the pregnane X receptor (PXR) such as rifampicin, or the constitutively activated receptor (CAR) such as phenobarbital, induced CYP2C8, CYP2C9, and to a lesser extent CYP2C19 mRNAs and proteins. CYP2C18 mRNA was expressed but not inducible. The concentration dependence of CYP2C8 and CYP2C9 mRNAs in response to rifampicin and phenobarbital paralleled that of CYP3A4 and CYP2B6, the maximum accumulation being reached with 10 μM rifampicin and 100 μM phenobarbital. In contrast, dexamethasone produced maximum induction of CYP2C8 and CYP2C9 mRNAs at 0.1 μM while in these conditions neither CYP3A4 nor CYP2B6 was significantly induced. Moreover, the concentration dependence of CYP2C8 and CYP2C9 mRNAs in response to dexamethasone paralleled that of tyrosine aminotransferase. Furthermore, dexamethasone, which has been recently shown to up-regulate PXR and CAR expression through the glucocorticoid receptor, potentiated CYP2C8 and CYP2C9 mRNA induction in response to rifampicin and phenobarbital. Collectively, these results suggest the possible implication of at least three receptors in the regulation of CYP2C8 and CYP2C9expression, i.e., glucocorticoid receptor, PXR, and/or CAR.
In humans, the cytochrome P450 (CYP1) 2C subfamily includes at least four functional genes: CYP2C8, -2C9, -2C18, and -2C19 (Goldstein and de Morais, 1994). CYP2C8, -2C9, and -2C19 proteins are primarily located in the liver where they account for approximately 20% of total cytochrome P450 (Shimada et al., 1994). In contrast, CYP2C18 protein seems to be primarily expressed in the skin (Zaphiropoulos, 1997). Low levels of CYP2C mRNAs and proteins have also been detected in small intestine and other extra-hepatic tissues (Klose et al., 1999). The CYP2C proteins play a significant role in metabolizing currently marketed drugs, including tolbutamide, phenytoin, tienilic acid, warfarin (at position 7), diclofenac as substrates of CYP2C9 (Miners and Birkett, 1998); and S-mephenytoin, warfarin (at positions 6 and 8), omeprazole, diazepam, imipramine, pentamidine, and moclobemide as substrates of CYP2C19 (Wrighton et al., 1993; Goldstein and de Morais, 1994; Goldstein et al., 1994). CYP2C8 and CYP2C18 proteins exhibit a substrate specificity that is close to that of -2C9 or -2C19 but with either a lower Vmax or a higher Km (Romkes et al., 1991; Goldstein et al., 1994). CYP2C8 is also specifically involved in the oxidative metabolism of Taxol, benzo[a]pyrene, arachidonic acid, and retinoids (Nadin and Murray, 1999). The polymorphisms of CYP2C19 have been well characterized using the 4-hydroxylation of theS-enantiomer of mephenytoin. Several defective mutants including CYP2C19*2 to CYP2C19*8 have been described and shown to encode either a truncated, inactive, or partially defective protein (De Morais et al., 1994; Ibeanu et al., 1999). Several allelic variants of CYP2C9 includingCYP2C9*2 and 2C9*3 have also been described, the latter being possibly responsible for the rare polymorphism of tolbutamide (Stubbins et al., 1996; Bhasker et al., 1997; Gill et al., 1999; Yasar et al., 1999).
In contrast to the large amount of data on the biochemistry, enzymology, pharmacology, toxicology, and genetics of CYP2Cgenes, little is known on the inducibility of the different members of this subfamily in response to xenobiotics in humans. Yet, significant induction of these genes could further amplify the interindividual variability of CYP2C-related monooxygenase activities observed in human populations. In this respect, a number of clinical studies (reviewed inJang and Maurel, 1999) have shown increased clearance and/or a decreased half-life of drugs known to be CYP2C substrates when given in association with known enzyme inducers, including rifampicin, phenobarbital, and glucocorticoids. On the other hand, in vitro studies in human hepatocyte cultures provided conflicting observations. For example, Runge et al. (2000) reported recently that CYP2C9 to -2C19 were not inducible in response to rifampicin and phenobarbital, whereasMorel et al. (1990) and Chang et al. (1997) reported that the levels of CYP2C immunoreactive proteins and mRNAs were increased in human primary hepatocytes treated with rifampicin, dexamethasone, and phenobarbital. However, because of the high level of similarity in both nucleotide and amino acid sequences, these last studies did not discriminate between the inducibility of the various members of this subfamily at the mRNA and protein levels. Previous papers from different groups have shown that rifampicin- and phenobarbital-mediated induction of CYP3A4 to -3A7 and CYP2B6 is controlled by two nuclear receptors: the pregnane X receptor (PXR) (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998) and the constitutively activated receptor (CAR) (Baes et al., 1994; Honkakoski et al., 1998; Sueyoshi et al., 1999), respectively. Whether these receptors are involved in the control of CYP2C gene regulation is currently unknown.
In the present work, we used a RNase protection assay and specific antibodies to investigate the inducible expression ofCYP2C8, CYP2C9, CYP2C18, andCYP2C19 genes in response to various xenobiotics, including rifampicin, dexamethasone, and phenobarbital, in cultured human hepatocytes. CYP2C8, CYP2C9, and CYP2C19 mRNAs and proteins were induced by previously characterized PXR and CAR activators, the rank order of inducibility being CYP2C8 > CYP2C9 > CYP2C19, while CYP2C18 was not inducible. The data suggest that several nuclear receptors, including the glucocorticoid receptor (GR), PXR, and/or CAR, might be involved in the control of basal and xenobiotic-inducible expression of CYP2C genes.
Experimental Procedures
Human Liver Samples.
Liver samples were lobectomies resected from adult patients for medically required purposes unrelated to our research program. The use of these human hepatic specimens for scientific purposes has been approved by the French National Ethics Committee. Clinical characteristics of liver donors are presented in Table1.
Clinical characteristics of liver donors
Materials.
Ham F-12 and Williams' E culture media, vitamins and hormones, collagenase (type IV), dimethylsulfoxide (DMSO), actinomycin D, dexamethasone, rifampicin, and mifepristone (RU486) were purchased from Sigma (St. Louis, MO). Phenobarbital was purchased from Specia (Paris, France). 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) was obtained from BCP instruments (Lyon, France); 1,4-bis[2-(3,5-dichloropyridyloxy)]-benzene (TCPOBOP) was a gift from P. Lesca (INRA, Toulouse, France). Collagen-coated culture dishes were obtained from Corning (Iwaki Glass, Tokyo, Japan). α-[32P]dCTP, α-[32P]UTP, and enhanced chemiluminescence developing reagent were purchased from Amersham Pharmacia Biotech (Amersham, England).
Primary Cultures of Human Hepatocytes.
Primary cultures of human hepatocytes were prepared and cultured as described in previous papers (Diaz et al., 1990). Xenobiotic inducers were diluted in DMSO and added to the culture medium at the indicated concentrations. In all cases the concentration of DMSO was 0.1%, and control cultures received only DMSO at the same concentration. The treatments lasted for 8 to 96 h and were renewed every 24 h as the culture medium was changed.
Preparation of Total RNA.
Total RNA and protein were isolated using Trizol reagent (Life Technologies, Cergy-Pontoise, France), from 107 cultured hepatocytes according to the manufacturer's instructions. For quality control, 30 μg of total RNA were analyzed by Northern blot using a rat glyceraldehyde phosphate dehydrogenase cDNA probe (J.M. Blanchard, IGMM, Montpellier, France).
Preparation of Plasmids.
The CYP2C cDNA probes were synthesized by reverse transcriptase-PCR from human liver total RNA by using specific oligonucleotides (Kimura et al., 1987; Ged et al., 1988; Romkes et al., 1991; Ged and Beaune, 1992). These probes do not overlap with the regions in which the mutations are found for the known variants of both CYP2C9and CYP2C19 (Fig. 1A).

RNase protection assay.
A, restriction map and length of CYP2C cDNA riboprobes used for the RNase protection assay. B, control experiments with synthetic sense CYP2C9 mRNA fragment. Note that the protected riboprobe migrates slightly ahead of the native probe (NP) because of the presence of a residual fragment from the plasmid polylinker in the native probe.
pGEMT-2C8.
A 311-bp fragment (nucleotides 982-1293) was amplified by PCR with oligonucleotides sense 5′-GATCATGTAATTGGCAGACACA and reverse 5′-CCTGCTGAGAAAGGCATGAAG and cloned in pGEMT vector (Promega, Madison, WI). The Ribonuclease Protection Assay probe was synthesized with SP6 RNA polymerase after linearization of the plasmid withApalI. The native probe contains 365 bp, and the digested probe is 239 bp long.
pGEMT-2C9.
A 437-bp fragment (nucleotides 856-1293) was amplified by PCR with oligonucleotides sense 5′-AGCTTGGAAAACACTGCAGT and reverse 5′-CCTGCTGAGAAAGGCATGAAG and cloned in pGEMT vector (Promega). The Ribonuclease Protection Assay probe was synthesized with T7 RNA polymerase after linearization of the plasmid with ApalI. The native probe contains 284 bp, and the digested probe is 239 bp long.
pGEMT-2C18.
A 431-bp fragment (nucleotides 869-1293) was amplified by PCR with oligonucleotides sense 5′-CTGTAACTGATATGTTTGGG and reverse 5′-CCTGCTGAGAAAGGCATGAAG and cloned in pGEMT vector (Promega). The Ribonuclease Protection Assay probe was synthesized with SP6 RNA polymerase after linearization of the plasmid with ApalI. The native probe contains 365 bp, and the digested probe is 239 bp long.
pGEMT-2C19.
A 431-bp fragment (nucleotides 862-1293) was amplified by PCR with oligonucleotides sense 5′-GTAATCACTGCAGCTGACTTAC and reverse 5′-CCTGCTGAGAAAGGCATGAAG and cloned in pGEMT vector (Promega). The Ribonuclease Protection Assay probe was synthesized with SP6 RNA polymerase after linearization of the plasmid with ApalI. The native probe contains 365 bp, and the digested probe is 239 bp long.
For in vitro sense RNA synthesis, pGEMT-2C plasmids were linearized using NdeI/NcoI/NdeI/NdeI, and synthetic RNA was generated using T7/SP6/T7/T7 RNA polymerase for CYP2C8/9/18/19, respectively.
pBS-IIK-CYP2B6.
A 263-bp fragment of CYP2B6 (nucleotides 25–288) was amplified by PCR with oligonucleotides sense 5′-AGCCTCCCACCAGGGCCCCGCCC and reverse 5′-TGGCAAAGATCACACCATATCCCC, digested by Pst-1 and Sal-1, and cloned in Pst-1/Sal-1 digested pBluescript II-KS+ vector (Stratagene, La Jolla, CA). The Ribonuclease Protection Assay probe was synthesized with T3 RNA polymerase after linearization of the plasmid with Sma-1. The native CYP2B6 probe contains 192 bp, and the digested probe is 180 bp long. CYP3A4 plasmid was prepared as previously described (Greuet et al., 1996). All CYP inserts were verified by nucleotide sequencing.
Ribonuclease Protection Assay.
Total RNA (30 μg) was analyzed by the RNase protection assay using specific riboprobes as previously described (Greuet et al., 1996) with minor modification. Total RNA was hybridized with the radiolabeled antisense RNA probe (100,000–150,000 cpm) overnight at 37°C after incubation for 10 min at 95°C. For Northern blot experiments, 30 μg of total RNA were analyzed using α-[32P]dCTP-labeled human CYP3A4, rat glyceraldehyde phosphate dehydrogenase, or mouse tyrosine aminotransferase (TAT, kindly provided by Dr. T Grange, Institut J. Monod, Paris, France). cDNA probes and autoradiography were carried out by exposing the dried gel to Kodak X-AR film; the signals were quantified by analyzing the radioactivity with a PhosphoImager apparatus and ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
Hepatocyte Microsome Preparation.
Microsomes were prepared from liver samples and hepatocyte cultures by differential centrifugation and stored as described previously (Diaz et al., 1990). Protein concentration was determined by the bicinchoninic acid method, according to the protocol provided by the manufacturer (Pierce Chemical Co., Rockford, IL). Bovine serum albumin (Pierce Chemical Co.) was used as the standard.
Immunoquantitation of CYP Proteins.
Thirty micrograms of microsomal proteins or 80 μg of total proteins were separated by SDS-polyacrylamide gel electrophoresis (10%), then electroblotted onto Immobilon P (Millipore, Bedford, MA). Membranes were incubated with specific antibodies, AK1 (a monoclonal antibody directed against rat CYP2C6 cross reacting with CYP2C9/18/19), or a rabbit polyclonal antibody that recognizes CYP2C8/18 (kindly provided by Drs. P. Beaune and I. de Waziers, INSERM, Paris, France), and the blots were developed with the enhanced chemiluminescence detection system (Amersham Pharmacia Biotech). The relative amount of CYP protein was estimated from densitometric analysis (NIH Image Software, by Dr. W. Rasbond). Authentic standards for immunoblots were microsomes from human lymphoblastoid cells transfected with the human CYP1A1, -1A2, -2A6, -2B6, -2C9, -2C19, -2D6, -2E1, and -3A4 cDNA (GENTEST Corp., Woburn, MA), or microsomes from yeast transfected with the human CYP2C8 or -2C18 cDNA.
Measurement of Tolbutamide 4-Hydroxylation Activity.
The rate of tolbutamide 4-hydroxylation was measured directly in cultured hepatocytes. After 96 h of induction, the culture medium was renewed in the absence of inducers and in the presence of 26.4 μM tolbutamide and [3H]tolbutamide (1 μCi, Amersham Pharmacia Biotech). Extracellular medium samples collected after 8 or 24 h were analyzed for tolbutamide and 4-hydroxytolbutamide by high-performance liquid chromatography using a Zorbax Bond C18 (150- × 4.6-mm; Interchim, Montluçon, France) column, protected with a column of the same phase. Elution was performed at room temperature with a 1.3-ml/min flow of isocratic mobile phase consisting of 40% acetonitrile (Carlo Erba, Val de Rueil, France), 59.96% water, and 0.04% orthophosphoric acid (Carlo Erba). The elution times of tolbutamide and 4-hydroxytolbutamide were 5 min 36 s and 2 min 30 s, respectively. The radioactivity of the effluent from the high-performance liquid chromatography column was analyzed in a Radioactivity Monitor LB 506-CI (Berthold, Wildbad, Germany). Radioactivity peaks were integrated with a computer and converted to molar concentrations of tolbutamide and 4-hydroxytolbutamide.
Statistical Analysis.
Statistical analysis of data was performed using the Macintosh Stat View program (Abacus Concepts, Inc., Berkeley, CA). For mRNA analysis (Table 2), induction ratios (mean ± S.D.) relative to each inducer (mRNA level in inducer-treated cells to mRNA level in untreated cells) were calculated for all different cultures, and the statistical significance of the induction was assessed using the paired t test. For protein analysis (Table 3), relative levels were standardized with respect to levels measured in rifampicin-treated cells (arbitrarily taken as 100 for each culture), because in some cultures no CYP2C protein was detectable in untreated cells. The statistical significance of these relative level variations (mean ± S.D.) were then analyzed using the paired t test.
Average CYP2C mRNA induction ratios in response to dexamethasone, rifampicin, and phenobarbital in human hepatocyte cultures
Relative levels of CYP2C8, CYP2C9, and CYP2C19 proteins and rates of tolbutamide and S-mephenytoin 4′-hydroxylation in human hepatocyte cultures
Results
RNase Protection Assay.
CYP2C mRNAs display a high similarity in their nucleotide sequences (82–94%) (Goldstein and de Morais, 1994). We therefore used RNase protection to quantify these mRNAs individually (Fig. 1A). The specificity of the assay was verified by using synthetic sense mRNA fragments prepared from the CYP2C plasmids. The results presented in Fig. 1B for CYP2C9 show that no interference was detected with CYP2C8, -2C18, or -2C19 mRNAs. The intensity of the band relative to the protected probe increased with the amount of synthetic sense CYP2C9 mRNA within the range 0.1 to 50 ng. This assay enabled us to detect 0.1 ng of the CYP2C9 mRNA riboprobe, i.e., 1 fmol of CYP2C mRNA. Similar results were obtained with the other CYP2C riboprobes in terms of specificity and sensitivity (not shown).
Expression of CYP2C mRNAs and Proteins in Human Livers.
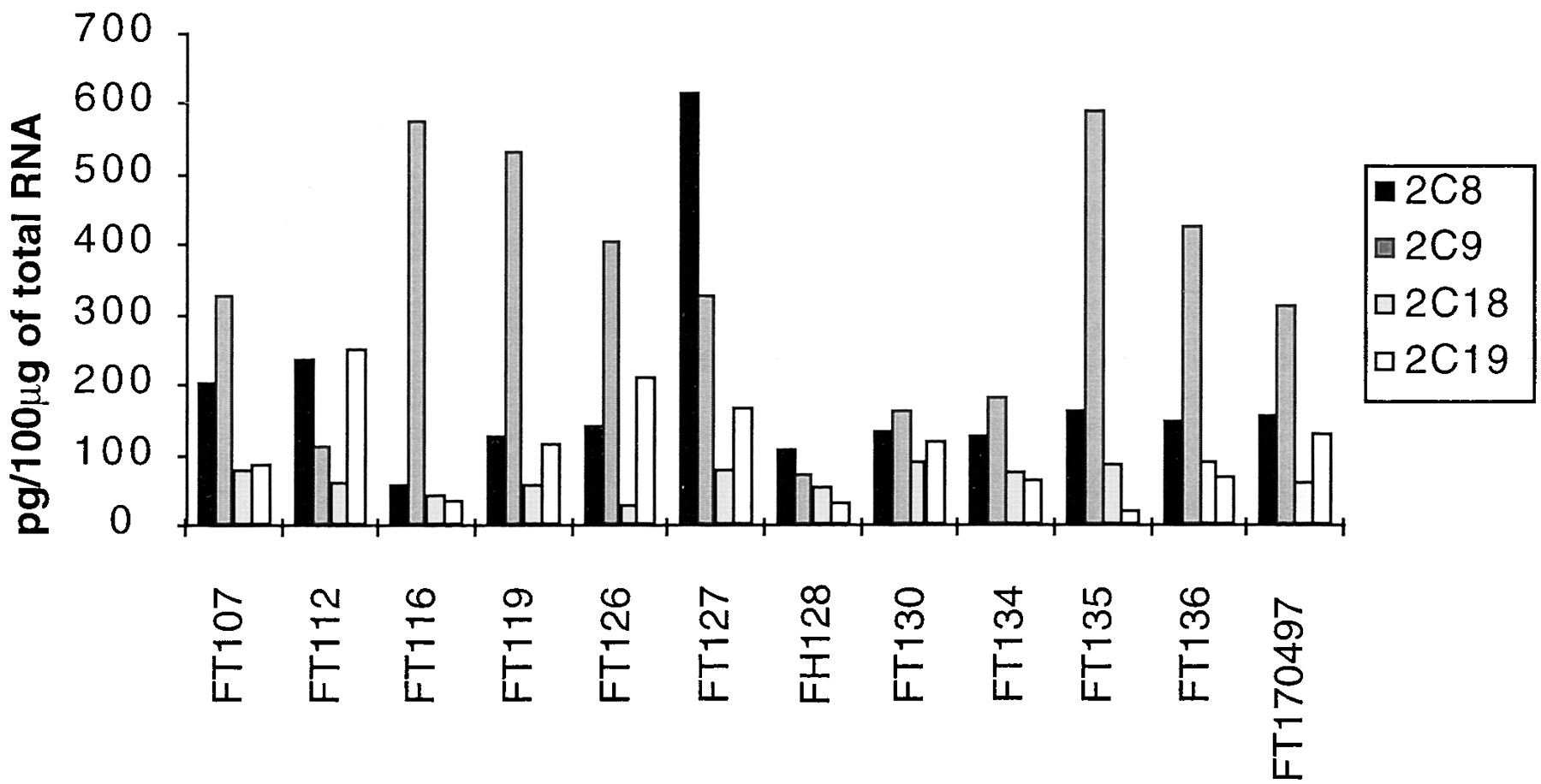
This assay was used to evaluate the levels of CYP2C mRNAs in a bank of human livers (n = 12). The results are shown in Fig.2. First, we verified that this assay was linear on a range of total liver RNA amount between 10 and 100 μg. The four different CYP2C mRNAs were expressed at detectable levels in all the human liver samples analyzed. The levels of CYP2C8, -2C9, and -2C19 mRNA exhibited a large interindividual variability (by factors of 11, 10, and 14, respectively), while CYP2C18 mRNA was more stable (3.4-fold variations). CYP2C9 mRNA accounted for approximately 50% of CYP2C mRNAs, while the relative amount of the other mRNAs was 26% for CYP2C8, 9% for CYP2C18, and 16% for CYP2C19, in reasonable agreement with previous evaluations by other methods (Furuya et al., 1991; Romkes et al., 1991). Statistical analysis of the data indicated that CYP2C mRNA levels are not correlated.

Concentration of CYP2C mRNAs in human livers as assessed by RNase protection assay.
Total RNA was extracted from different human livers and 30 μg were analyzed by RNase protection assay with specific CYP2C cDNA probes. Every CYP2C mRNA was quantified using synthetic specific sense RNA as shown in Fig. 1B.
A monoclonal antibody termed “AK1”, prepared against rat CYP2C6 and shown to cross-react with human CYP2C proteins, and a polyclonal antibody against CYP2C8 were used to quantify CYP2C proteins. As reported in Fig. 3A, AK1 antibody recognized CYP2C9, CYP2C18, and CYP2C19 but did not cross-react with CYP2C8 nor with other CYP forms, including CYP1A1, -1A2, -2A6, -2B6, -2D6, -2E1, and -3A4. On the other hand, the anti-CYP2C8 antibody cross-reacted with CYP2C18 protein but not with other CYPs. In immunoblots of liver microsomes (Fig. 3B), AK1 antibody revealed only two main bands that comigrated with authentic CYP2C9 and CYP2C19, and anti-CYP2C8 revealed only one band comigrating with authentic CYP2C8. CYP2C18, which migrates slightly above CYP2C19, was never detected. The specific content of CYP2C8, CYP2C9, and CYP2C19 was on average 24 ± 11, 120 ± 29, and 28 ± 21 pmol/mg of microsomal protein, respectively, in fair agreement with previous data (Inoue et al., 1997;Lasker et al., 1998). A significant correlation was found between the levels of CYP2C19 mRNA and protein (r = 0.895;P < 0.0001) and the levels of CYP2C8 and CYP2C9 proteins (r = 0.863; P < 0.001). However, neither the levels of CYP2C8 and CYP2C9 mRNA and protein nor the levels of CYP2C19 protein with other CYP2C proteins appeared to be correlated. As expected, the levels of CYP2C9 and the rates of 4-hydroxylation of tolbutamide exhibited a significant correlation (r = 0.7 and P = 0.009, not shown).

Immunoblot analysis of CYP2C proteins.
A, microsomes (1 pmol of protein per lane) from human lymphoblastoid cells transfected with the human CYP1A1, -1A2, -2A6, -2B6, -2C9, -2C19, -2D6, -2E1, and -3A4 cDNA (GENTEST Corp.) or microsomes from yeast transfected with the human CYP2C8 or -2C18 cDNA (P. Beaune) were analyzed by immunoblot with two anti-CYP2C antibodies. The upper and lower blots were developed with AK1 (anti-CYP2C6) and anti-CYP2C8, respectively. B, microsomes (20 or 30 μg of protein per lane analyzed with AK1 or anti-CYP2C8, respectively) prepared from human livers were analyzed by immunoblot with two anti-CYP2C antibodies. The upper and middle blots were developed with AK1 (anti-CYP2C6) and the lower blot with anti-CYP2C8, respectively. Different durations of development were necessary to visualize appropriately CYP2C9 and CYP2C19 (upper and middle blots). No patient was deficient for CYP2C19 protein. Note that in this figure, CYP2C19 is not detectable in FT135; however, the protein could be detected under a longer period of exposure of the film (not shown).
Induction of CYP2C mRNAs and Proteins by Various Compounds in Cultured Primary Human Hepatocytes.
Hepatocytes prepared from different patients were cultured for 96 h in the absence or presence of several compounds previously characterized as CYP inducers. Then, total RNA and microsomes were extracted and analyzed by RNase protection assay and immunoblot, respectively.
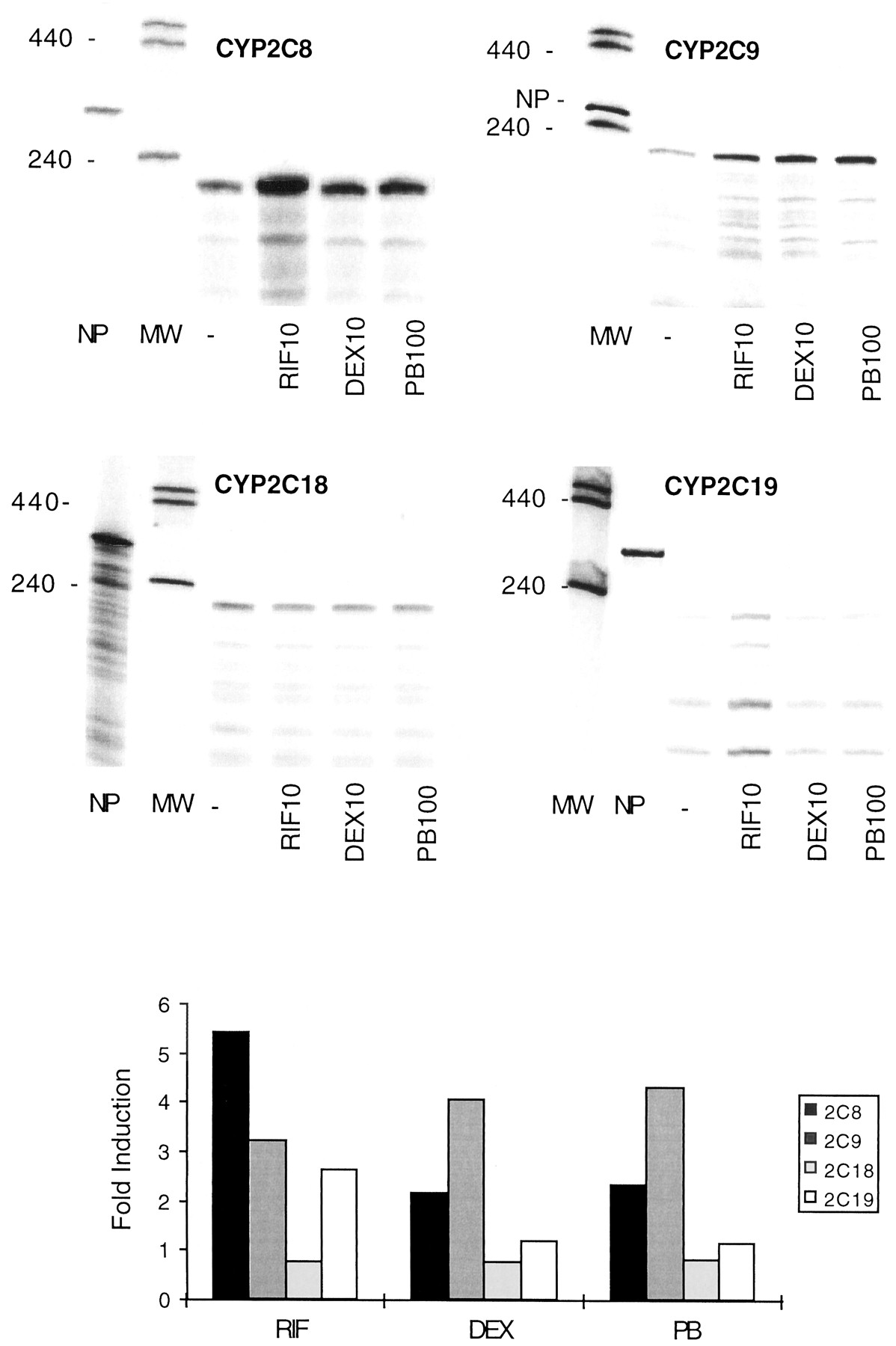
The four CYP2C mRNAs were expressed constitutively in our cultures (Fig. 4). Compounds previously characterized as CYP2B/3A inducers (Maurel, 1996a) including rifampicin, dexamethasone, and phenobarbital were also inducers of CYP2C8 and CYP2C9 mRNAs. However, the fold induction was on average 2 to 6, that is, significantly smaller than that classically observed with CYP2B6 mRNA in response to phenobarbital or with CYP3A4 mRNA in response to rifampicin (see Fig. 6 for example). In one culture (FT137), several other compounds were tested, including TCDD, TCPOBOP, bosentane, omeprazole, prednisone, sulfadimidine, and phenylbutazone. TCDD (a prototypical CYP1A inducer) and TCPOBOP (aCYP2B inducer in rodents) were not significant inducers, whereas the other compounds (which were all CYP3A4 inducers) gave induction ratios close to those found with rifampicin, dexamethasone, and phenobarbital (not shown). Interestingly, the levels of CYP2C8 and CYP2C9 mRNAs exhibited a significant correlation after induction (withr = 0.66; P = 0.03), suggesting that the expression of these genes may be coregulated, at least in part, in response to inducers. CYP2C19 mRNA was also inducible, notably by rifampicin and phenobarbital but to a lesser extent as compared with CYP2C8 and -2C9 (Table 2). In contrast, the levels of CYP2C18 mRNAs were either not affected or decreased in response to the various treatments.

Analysis of CYP2C mRNAs by RNase protection assay in human hepatocytes treated with different inducers.
Human hepatocytes (FT137) were cultured for 96 h in the absence (−) or presence of different inducers. Total RNA was extracted and 30 μg were analyzed by RNase protection assay with specific CYP2C cDNA probes. The signals relative to protected probes were quantified by analyzing the radioactivity with a PhosphoImager apparatus and ImageQuant software and normalized with respect to the levels in untreated cells taken as 1. Inducers: RIF, rifampicin; DEX, dexamethasone; PB, phenobarbital. Concentrations are indicated in micromolar. MW, size markers; NP, native probe.

Time dependence of CYP2C8, CYP2C9, CYP2B6, CYP3A4, and TAT mRNA increase in response to inducers.
A and B, human hepatocytes were cultured for 36 h in the standard culture medium but in the absence of dexamethasone. Cells were then cultured for 8, 16, 24, and 48 h in the presence of 1 μM dexamethasone (DEX). Total RNA was extracted and analyzed by RNase protection assay (20 μg of RNA per lane) with specific CYP2C cDNA probes, or by Northern blot with a specific TAT (30 μg of RNA) cDNA probe. Fold-induction ratios or actual levels (in arbitrary units) are plotted against time. C and D, human hepatocytes were cultured for 48 h in the standard culture medium (0.1 μM dexamethasone), and the culture was continued for 8, 16, 24, and 48 h in the presence of 10 μM rifampicin (RIF). Total RNA was extracted and analyzed by RNase protection assay (20 μg of RNA per lane) with specific CYP2C probes, or by Northern blot with a specific CYP3A4 (20 μg of RNA) cDNA probe. E and F, same experiments as in C and D, except that rifampicin was replaced with 100 μM phenobarbital (PB). Total RNA was extracted and analyzed by RNase protection assay (20 μg of RNA per lane) with specific CYP2C and CYP2B6 cDNA probes. These results represent average data (±S.D.) from three different cultures prepared from three different patients (FT165, FT166, and FT167).
The immunoblot analysis of microsomes from several cultures with AK1 or anti-2C8 antibodies is shown in Fig. 5and Table 3. CYP2C8, -2C9, and -2C19 proteins were clearly induced by rifampicin, dexamethasone, and phenobarbital. Interestingly, the levels of CYP2C9 protein correlated with the rates of 4-hydroxylation of tolbutamide in culture FT137 treated with various inducers (not shown,r = 0.82; P = 0.0005). These results show that CYP2C8, CYP2C9, and CYP2C19genes respond to the same inducers as CYP3A4 (rifampicin, dexamethasone, and phenobarbital).

Analysis of CYP2C proteins in human hepatocytes treated with different inducers.
Human hepatocytes prepared from different patients (FT155, FT156, FT159, and FT160) were cultured for 96 h in the absence (−) or presence of different inducers: rifampicin (RIF, 5 or 10 μM), dexamethasone (DEX, 1 or 5 μM), or phenobarbital (PB, 0.5 mM). Microsomes were extracted and 30 μg were analyzed by immunoblot with AK1 (upper blots) or with anti-CYP2C8 antibodies (lower blot). Note that the same immunoblot (upper right) is presented after different durations of development (10 and 30 s) to allow appropriate detection of CYP2C19. Authentic antigen markers (1 pmol): CYP2C9 and CYP2C19 microsomes from transfected lymphoblastoid cells (GENTEST Corp.), and CYP2C8 and CYP2C18 microsomes from transfected yeast (P. Beaune).
We would like to emphasize here that although induction of CYP2C8 and CYP2C9 mRNA and protein was observed in all cultures tested in this work, the extent of induction in response to inducers was very different from one culture to another. This variability, also observed for CYP1As and CYP3A4 expression as reported previously (Curi-Pedrosa et al., 1994; Pichard-Garcia et al., 2000), was not related to the sex, age, or pathology of the patient from whom hepatocytes were isolated, nor to the quality of the cell monolayer as assessed by phase contrast microscopy.
Comparative Analysis of CYP2C8, CYP2C9, CYP3A4, and CYP2B6 mRNA Induction.
Since these observations suggest that CYP2C8 andCYP2C9 share the same inducers as CYP3A4 andCYP2B6, we sought to determine whether these different CYP families respond identically to inducers in terms of time and concentration dependence. Three compounds were selected for this detailed investigation, i.e., dexamethasone, rifampicin, and phenobarbital.
Effect of dexamethasone.
The effect of this glucocorticoid must be considered separately from the other compounds as it is not only an inducer of CYP genes, but it has also an important effect on the phenotype of hepatocytes (up-regulation of PXR and CAR, see next section). In a first series of experiments, hepatocytes were cultured for 36 h in our standard culture medium but in the absence of dexamethasone (our standard culture medium contains 0.1 μM dexamethasone and no other glucocorticoid). Control experiments (cellular morphology, CYP3A4 inducibility in response to rifampicin; see Fig. 8) demonstrated that no significant loss of differentiation of cells occurred under these conditions. Then, culture medium was supplemented with 1 μM dexamethasone, and cells were harvested 8, 16, 24 and 48 h later. As reported in Fig. 6A, CYP2C8 and CYP2C9 mRNAs were induced with close if not identical time dependence. In contrast, CYP3A4 was only detectable after 48 h of treatment, and CYP2B6 mRNA remained undetectable for the duration of experiments (Pascussi et al., 2000a; data not shown). The same experiments were carried out on TAT mRNA, known to be controlled by the GR. Interestingly, TAT mRNA levels displayed a time dependence identical to that of CYP2C8 and CYP2C9 mRNAs in response to dexamethasone (Fig. 6B). To determine whether induction of CYP2C mRNAs was of transcriptional origin or due to mRNA stabilization, hepatocytes were cultured for 48 h in the standard culture medium (0.1 μM dexamethasone) and exposed to actinomycin D for 32 h, in the absence or presence of 0.1 μM dexamethasone. Data reported in Fig.7 show that CYP2C8 and CYP2C9 mRNAs were rather stable as they exhibited minor decay for the duration of exposure to actinomycin D (i.e., 32 h), irrespective of the presence of dexamethasone. These results suggest that the increased accumulation of CYP2C8 and CYP2C9 mRNAs in response to dexamethasone is of transcriptional origin.

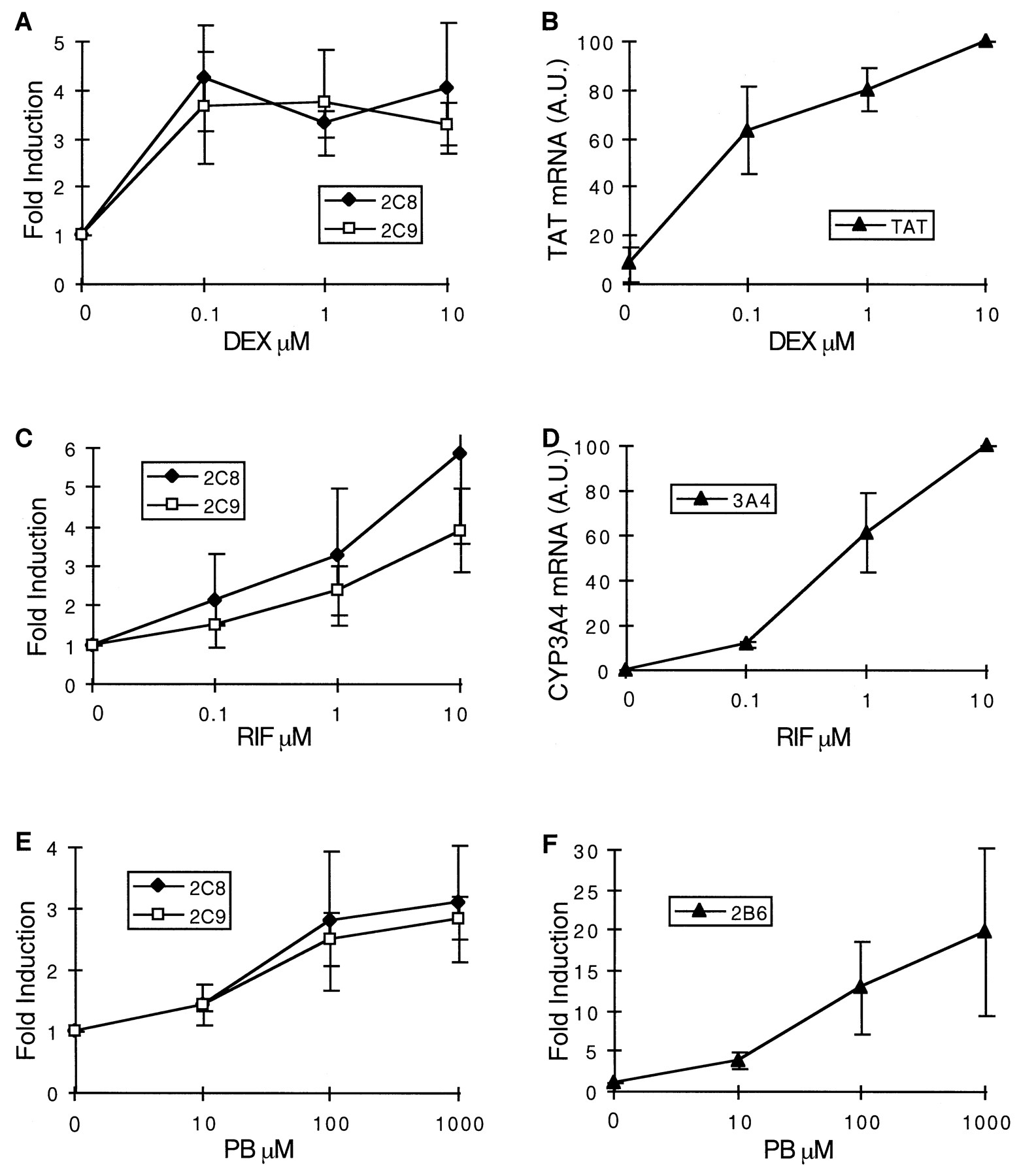
Concentration dependence of CYP2C8, CYP2C9, CYP2B6, CYP3A4, and TAT mRNA increase in response to inducers.
A and B, human hepatocytes were cultured for 36 h in the standard culture medium but in the absence of dexamethasone. Cells were then cultured for 24 h in the presence of 0.1, 1, and 10 μM dexamethasone (DEX). Total RNA was extracted and analyzed by RNase protection assay (20 μg of RNA per lane) with specific CYP2C cDNA probes, or by Northern blot with a specific TAT (30 μg of RNA) cDNA probe. C and D, human hepatocytes were cultured for 48 h in the standard culture medium (0.1 μM dexamethasone), and cells were then exposed to 0.1, 1, and 10 μM rifampicin (RIF) for 24 h. Total RNA was extracted and analyzed by RNase protection assay (20 μg of RNA per lane) with specific CYP2C probes, or by Northern blot with a specific CYP3A4 (20 μg of RNA) cDNA probe. E and F, same experiments as in C and D, except that rifampicin was replaced by 10, 100, and 1000 μM phenobarbital (PB). Total RNA was extracted and analyzed by RNase protection assay (20 μg of RNA per lane) with specific CYP2C and CYP2B6 cDNA probes. Fold-induction ratios or actual levels (in arbitrary units) are plotted against inducer concentration. These results represent average data (±S.D.) from three different cultures prepared from three different patients (FT159, FT166, and FT167).

Effect of actinomycin D on CYP2C8 and CYP2C9 mRNA expression in response to dexamethasone.
Hepatocytes from patient FT167 were cultured for 48 h in the standard culture medium (0.1 μM dexamethasone) and, at time 0 (T0), cells were exposed to 5 μg/ml actinomycin D for 32 h, in the absence (−) or presence (+) of 0.1 μM dexamethasone. At the indicated time, total RNA was extracted and analyzed by RNase protection assay (20 μg of RNA per lane) with specific CYP2C cDNA probes. CYP2C8 and CYP2C9 mRNA levels are presented in arbitrary units. MW, size markers; NP, native probe; DP, digested unprotected probe.
In a further series of experiments, hepatocytes were cultured for 36 h in our standard culture medium but in the absence of dexamethasone. Then, increasing concentration of dexamethasone (0.1, 1, and 10 μM) was added to the culture medium, and cells were harvested 24 h later. The results are presented in Fig.8A. Dexamethasone was a very potent inducer of CYP2C8 and CYP2C9 mRNAs, producing nearly maximum levels at 0.1 μM. In contrast, CYP3A4 and CYP2B6 mRNAs were not detectable in these experimental conditions (Pascussi et al., 2000b; data not shown). Control experiments showed that accumulation of TAT mRNA reached at least 60% of its maximum level at 0.1 μM dexamethasone and only slightly increased at greater concentrations (Fig. 8B). In aggregate, these experiments show that induction of CYP2C8, CYP2C9, and TAT mRNAs in response to dexamethasone display close time and concentration dependence.
Effect of rifampicin and phenobarbital.
Next, we investigated similarly both the time and concentration dependence of CYP2C8 and CYP2C9 mRNAs' increase in response to rifampicin and phenobarbital under the same conditions as those described above for dexamethasone, except that dexamethasone was not withdrawn from the culture medium (concentration: 0.1 μM). Here again, expression of CYP3A4 and CYP2B6 mRNAs in response to rifampicin and phenobarbital, respectively, was analyzed in parallel. The time dependence of CYP2C8 and CYP2C9 mRNAs was not drastically different from that of CYP3A4 or CYP2B6 mRNAs, irrespective of the inducers (Fig.6, C–F). However, in contrast to what was observed with dexamethasone, greater concentrations of rifampicin and phenobarbital were necessary to induce CYP2C8 and CYP2C9 mRNAs, the maximum levels being reached with approximately 10 and 100 μM, respectively (Fig. 8, C–F). Interestingly, the changes in CYP2C8 and CYP2C9 mRNAs roughly paralleled those of CYP3A4 and CYP2B6 mRNAs in response to rifampicin and phenobarbital. Thus, CYP2C8, CYP2C9, CYP3A4, and CYP2B6 mRNAs' inductions display similar time and concentration dependence in response to rifampicin and phenobarbital, but not in response to dexamethasone.
Dexamethasone Enhances Induction of CYP2C8 and CYP2C9 mRNAs in Response to Rifampicin and Phenobarbital.
We observed recently that both PXR (Pascussi et al., 2000a) and CAR (Pascussi et al., 2000b) are up-regulated by dexamethasone in human primary hepatocytes most likely through the glucocorticoid receptor pathway. Consequently, the levels of both PXR and CAR decrease in cells cultured in the absence of dexamethasone (or other glucocorticoid). This accounts for the previous observations that induction of CYP3A4 or CYP2B6 mRNAs in response to compounds known to activate PXR and/or CAR (i.e., rifampicin and/or phenobarbital, for example) is potentiated by pretreatment of hepatocytes with dexamethasone. We report a similar observation here with respect to CYP2C8 and CYP2C9 mRNA induction.
In these experiments, hepatocytes were cultured for 36 h in our standard culture medium (0.1 μM dexamethasone) or in the absence of dexamethasone. Then, the culture was continued under the same conditions, in the absence or presence of rifampicin (0.1, 1, and 10 μM) or phenobarbital (10, 100, and 1000 μM), and the cells were harvested 24 h later. As shown in Fig. 9 and Table 4, induction of both CYP2C8 and CYP2C9 mRNAs in response to rifampicin and phenobarbital was significantly enhanced in cells cultured in the presence of dexamethasone (high levels of PXR and CAR) as compared with the same cells cultured in the absence of dexamethasone (low levels of PXR and CAR). As expected from our previous work (Pascussi et al., 2000a,b), this was also the case for CYP3A4 and CYP2B6 mRNA induction, in the same cultures (Table 4). Control experiments clearly confirmed that, in the presence of dexamethasone, GR, PXR, and CAR were expressed in our cultures at a level similar to that observed in the tissue (not shown). In aggregate, these observations suggest that CYP2C8 and CYP2C9 mRNAs are coregulated in response to rifampicin and phenobarbital and that this response is likely to be mediated, at least in part, by PXR and/or CAR, both receptors being up-regulated by glucocorticoids.

Effect of pretreatment of hepatocytes with dexamethasone on CYP2C8 and CYP2C9 mRNA induction in response to rifampicin and phenobarbital.
Human hepatocytes were cultured for 48 h in our standard culture medium in the presence (0.1 μM DEX) or in the absence of dexamethasone (DMSO). Then, the culture was continued under the same conditions for 24 h, in the absence (−) or presence of increasing concentrations of rifampicin (RIF; 0.1, 1, and 10 μM) or phenobarbital (PB; 10, 100, and 1000 μM). Total RNA was extracted and analyzed by RNase protection assay (20 μg of RNA per lane) with specific CYP2C cDNA probes. A, hepatocytes from patient FT159. B, hepatocytes from patient FT160. MW, size markers; NP, native probe.
Potentiation by dexamethasone of CYP2C8, CYP2C9, CYP3A4, and CYP2B6 mRNA induction in response to rifampicin and phenobarbital
Discussion
Primary cultures of human hepatocytes represent the most reliable in vitro model for evaluating the inducibility of CYP genes in response to xenobiotics in humans. Indeed, we and others have reported on the reasonably good in vivo-in vitro correlation concerning the inducibility of CYP1A1/1A2 and CYP3A4by several drugs (Maurel, 1996b; Li et al., 1997). In this work we show that CYP2C8, CYP2C9, and CYP2C19respond to the same inducers as CYP3A4 andCYP2B6, suggesting that PXR and/or CAR might be involved in this process.
PXR (Bertilsson et al., 1998; Blumberg et al., 1998; Kliewer et al., 1998; Lehmann et al., 1998) and CAR (Baes et al., 1994; Honkakoski et al., 1998; Sueyoshi et al., 1999) are members of the steroid/retinoid/thyroid hormone receptor superfamily (NR1) of ligand-activated transcription factors. These receptors share a common heterodimerization partner, retinoid X receptor (RXR). Heterodimers PXR/RXR and CAR/RXR bind their target CYP genes by common nuclear receptor AGGTCA-based DNA response element motifs, including everted repeats spaced by 6 bp (ER-6) or direct repeats spaced by 4 bp (DR-4), respectively. PXR and CAR have been implicated in the inducible expression of CYP3A4 through -3A7 and CYP2B6, respectively, in response to xenobiotics. The present findings that 1) CYP2C8 and CYP2C9 mRNAs are inducible in response to compounds previously characterized as CYP3A4 inducers and PXR activators (such as rifampicin); 2) the concentration and time dependence of CYP2C8 and CYP2C9 mRNA induction in response to rifampicin is identical to that of CYP3A4 mRNA; and 3) dexamethasone enhances the induction of CYP2C8 and CYP2C9 mRNAs in response to rifampicin, as previously observed with CYP3A4 mRNA (Pascussi et al., 2000a), suggest collectively that PXR is involved at least partly in the control of the inducible expression ofCYP2C8 and CYP2C9 in response to rifampicin. Data obtained on CYP2C19 are more difficult to interpret because of the moderate induction of the mRNA and the large interindividual variability. However, the finding that rifampicin is the strongest inducer of this gene is again in favor of a possible implication of PXR.
Phenobarbital is not a potent inducer of CYP2C8 andCYP2C9 genes. Indeed large concentrations of this compound (at least 100 μM) are required to produce significant induction. This would appear to be consistent with recent data from Lehmann et al. (1998) showing that this barbiturate is able to activate the human PXR at concentrations greater than 100 μM. However, recent work (Honkakoski et al., 1998; Sueyoshi et al., 1999) has shown that induction of CYP2B6 and CYP3A4 by phenobarbital could be mediated through CAR. Although this receptor binds to and activates NR1 cis-element in the CYP2B6 upstream region, it can bind to and activate as well the PXR-responsive ER-6 element present in the CYP3A4 regulatory region. It is therefore not known whether PXR and/or CAR is/are responsible for phenobarbital-mediated induction of CYP2C8 and CYP2C9 mRNAs. To attempt to answer this question, we decided to evaluate the actual contribution of CAR by investigating the effect of androstenol, a potent CAR deactivator, on this process (Moore et al., 2000). Conflicting observations were made: this compound inhibited the induction of CYP2C8 and CYP2C9 mRNAs in response to phenobarbital in some cultures but not in others. A possible reason for this is that androstenol per se is able to activate PXR as revealed by its ability to induce CYP3A4 mRNA in our cultures (not shown). Therefore, although androstenol might inhibit the phenobarbital-mediated induction of CYP2C8 and CYP2C9 mRNA by deactivating CAR, its ability to activate PXR might reverse this effect. The relative amplitude of these two pathways probably varies from one culture to another with the respective levels of CAR and PXR proteins, thus explaining our conflicting observations.
The finding that induction of CYP2C8 and CYP2C9 mRNAs is obtained within 24 h in response to submicromolar concentrations of dexamethasone is interesting (Figs. 6 and 8). Lehmann et al. (1998)have shown that significant activation of PXR by dexamethasone requires concentrations on the order of or greater than 10 μM, consistent with the finding that this compound is not a CYP3A4 inducer below this concentration. Thus, PXR cannot be responsible for the CYP2C8 and CYP2C9 induction observed here at 0.1 and 1 μM dexamethasone, and we therefore suspected GR to be responsible. Our observations that induction of TAT, CYP2C8, and CYP2C9 mRNAs displays similar time and concentration dependence is consistent with this possibility (Figs. 6, A and B and 8, A and B). Especially interesting in this respect is the finding that neither CYP3A4 nor CYP2B6 mRNAs were inducible in parallel to CYP2C8 and CYP2C9 mRNAs in response to dexamethasone. For supramicromolar concentration of dexamethasone (10 μM) and for much longer duration of treatment (96 h), it is possible that PXR becomes partly activated, which would account for the further induction of CYP2C8 and CYP2C9 mRNAs as observed in experiments shown in Fig. 5 and Table 2. As discussed above for androstenol and CAR contribution, we attempted to use RU486 (a prototypical GR antagonist) to evaluate the contribution of GR in dexamethasone-mediated CYP2C8 and CYP2C9 mRNAs' induction. However, these experiments were not conclusive either (presence or absence of inhibition from one culture to another, not shown). Indeed, the inhibitory effect of RU486 on dexamethasone induction mediated by GR might or might not be masked by the activation of PXR by this compound.
Collectively, our results suggest the possible implication of at least three receptors in the xenobiotic-inducible expression of CYP2C8 and CYP2C9, i.e., GR, PXR, and/or CAR. We propose that GR is responsible, at least in part, for the basal expression of these genes under physiological conditions in our standard cultures (presence of 0.1 μM dexamethasone). Note that GR is expressed constitutively in our cultures (Pascussi et al., 2000a,b), as are CYP2C8 and CYP2C9 mRNAs. In the presence of xenobiotics (including glucocorticoids at supramicromolar concentrations) able to activate PXR and/or CAR,CYP2C8 and CYP2C9 gene expression is further enhanced. The finding that the time course of induction of CYP2C8 and CYP2C9 mRNAs is similar, whatever the inducer (see Fig. 6), suggests that this process is only dependent on the activation of latent receptor molecules (GR, PXR, and/or CAR) present in the cells and thatCYP2C gene transactivation by these receptors is not rate limiting. Analysis of cis-elements responsible forCYP2C8 and CYP2C9 gene induction in response to glucocorticoids and xenobiotics is being performed.
In contrast to the other CYP2C messengers, CYP2C18 mRNA was not inducible in any of the cultures tested. Furthermore, we were unable to detect the CYP2C18 protein in microsomes prepared either from human liver tissue or cultured hepatocytes. These data suggest thatCYP2C18 is not significantly expressed at the protein level in the liver and is regulated differently from the other members of this subfamily. Indeed, Zaphiropoulos (1997) reported recently thatCYP2C18 is the member of the CYP2C subfamily most abundantly expressed in the human epidermis.
Our observations on the induction of proteins CYP2C9 and CYP2C19 and related monooxygenase activities (tolbutamide andS-mephenytoin 4-hydroxylations) in our cultures are consistent with previous observations in vivo. Several clinical reports have focused on the changed pharmacokinetic parameters of drugs known as CYP2C substrates, in patients receiving rifampicin, dexamethasone, phenobarbital, or a high concentration of prednisone (Jang and Maurel, 1999). For example, the systemic clearance of phenytoin, tolbutamide, and S-warfarin exhibited a 2- to 3-fold increase in patients receiving rifampicin, suggesting clinically significant CYP2C9 induction. Zhou et al. (1990) observed an increase in the urinary excretion of 4′-hydroxy-S-mephenytoin (CYP2C19) in extensive (but not in poor) metabolizers of this anticonvulsant treated with rifampicin. Lackner (1991) reported an increase in i.v. phenytoin clearance with concomitant administration of dexamethasone, requiring an increased dose of the drug for therapeutic efficacy, again suggesting a clinically significant induction of CYP2C9. Finally, phenobarbital and prednisone were found to decrease the half-life of elimination of cyclophosphamide, a drug recently shown to be a lowKm substrate of CYP2C9 and CYP2C19 (Jao et al., 1972; Faber et al., 1974), whereas dexamethasone produced an increase in the body clearance of this molecule (Yule et al., 1996). The present in vitro data are consistent with these clinical reports and provide clear evidence in favor of a significant influence of xenobiotics, in addition to genetic determinants, to the wide interindividual variability of the CYP2C-related biotransformations in humans.
Acknowledgments
We are grateful to Drs. Philippe Beaune and Isabelle de Waziers (INSERM, Paris, France) for providing authentic CYP2C8 and -2C18 proteins and anti-CYP2C18 antibodies, and to Dr. Colin Young for careful reading of the manuscript.
Footnotes
-
Send reprint requests to: P. Maurel, INSERM U128, IFR24, Campus CNRS, 1919 Route de Mende, 34293 Montpellier, France. E-mail: maurel{at}u128.crbm.cnrs-mop.fr
-
This work was supported in part by Laboratoires Fournier Dijon, France (S.G.-C.) and la Ligue Nationale contre le Cancer (J.M.P.), and by Hoffman-La Roche (Basel, Switzerland).
- Abbreviations used are::
- CYP
- cytochrome P450
- DMSO
- dimethylsulfoxide
- TAT
- tyrosine aminotransferase
- GR
- glucocorticoid receptor
- PXR
- pregnane X receptor
- RXR
- retinoid X receptor
- CAR
- constitutively activated receptor
- TCDD
- 2,3,7,8-tetrachlorodibenzo-p-dioxin
- TCPOBOP
- 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene
- PCR
- polymerase chain reaction
- bp
- base pair
- Received September 13, 2000.
- Accepted November 16, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}