


Abstract
Most cytochrome P450 (P450 or CYP)-catalyzed reactions are adequately described by classical Michaelis-Menten kinetic parameters (e.g., Km andVmax), which are usually determined by a saturation profile of velocity of product formation versus substrate concentration. In turn, these parameters may be used to predict pharmacokinetics. However, some P450 enzymes exhibit atypical or non-Michaelis-Menten kinetics, due largely to substrate inhibition at higher concentrations of substrate. Although the mechanism of substrate inhibition is unknown, ignoring it and truncating the data can lead to erroneous estimates of kinetic parameters. In the present study, 13 P450 marker substrates were examined with 10 recombinant P450 proteins, and 6 were found, to varying degrees, to exhibit substrate inhibition. To understand the nature of the inhibition, a kinetic model was proposed (assuming that two binding sites exist on the enzyme) and used to fit the experimental data. The derived data indicated that 1) theKI values (substrate inhibition) were approximately 1.2- to 10-fold greater than the respectiveKS values; 2) bothKS and KI values may be affected by the interaction of the two bound substrates within the enzyme, exhibited by a factor α(α = 5.1–23.3); and 3) enzyme activity was inhibited markedly (39–97%) at excess concentrations of the substrates (β = 0.03–0.61). These findings suggest that substrates have access to both the inhibitory site and catalytic site simultaneously (KI >KS). Furthermore, the two sites, in the presence of substrate, can interact with each other. Therefore, the degree of inhibition of the enzyme is dependent on the concentration of the substrate (usually >KI) that sufficiently occupies the inhibitory site.
The cytochrome P450s (CYPs or P450s1) are a superfamily of enzymes that catalyze the metabolism of both endogenous substrates and xenobiotics (Gonzalez, 1988; Nelson et al., 1996). In humans, the CYP1, CYP2, and CYP3 families are involved largely in xenobiotic metabolism (Wrighton and Stevens, 1992; Guengerich, 1995). Substrates for the P450s include a variety of clinically used drugs, environmental pollutants, pesticides, carcinogens, and mutagens. In most cases, the metabolism of these agents can be described using the Michaelis-Menten equation (a saturation profile of velocity of product formation versus a series of concentrations of a given substrate) to yield estimates ofVmax and Km. These parameters can be used to predict the pharmacokinetic or toxicokinetic consequences of exposure to a drug or toxicant (Lin and Lu, 1997; Rodrigues, 1999). However, some P450 enzymes exhibit atypical kinetics and do not adhere to Michaelis-Menten kinetics, due largely to substrate activation (sigmoidal curve) (Ueng et al., 1997; Harlow and Halpert, 1998; Korzekwa et al., 1998; Shou et al., 1999) and substrate inhibition (Vicent et al., 1992; Harris and Davidson, 1993; Musser et al., 1997; Spracklin et al., 1997). Of the P450-mediated reactions, substrate inhibition is commonly observed and has been recognized for decades (Haehner et al., 1996; Spracklin et al., 1997; Fogelman et al., 1999; Tang et al., 2000). A substrate that causes a decrease in the rate of product formation as its concentration increases will lead to a reaction that displays substrate inhibition kinetics. Although the mechanism of P450-mediated substrate inhibition remains unknown, ignoring it and truncating the data can result in substantial errors in the values derived for critical kinetic parameters. In the present study, 13 substrates, used commonly as markers for individual P450 assays, were used to study substrate inhibition. Of all the substrates tested, eight exhibited substrate inhibition. Therefore, a kinetic model was proposed to fit the experimental data. The mechanism of substrate inhibition can be understood in terms of the kinetic constants, resulting from the fitted data, that precisely describe the kinetic features of each equilibrium of the model.
Materials and Methods
Chemicals.
Chemicals were from the following commercial sources. Testosterone, 6β-hydroxytestosterone, progesterone, 6β-hydroxyprogesterone, 16α-hydroxyprogesterone, estrone, 2-OH-estrone, and 17β-estradiol were from Steraloids (Wilton, NH). Phenacetin, acetaminophen, Taxol, flurbiprofen, diazepam, bacctin, temazepam, diclofenac, nordiazepam, dextromethorphan, and NADPH were from Sigma (St. Louis, MO). Dextrorphan and chlorzoxazone were from RBI/Sigma (Natick, MA). Ethoxyresorufin, (S)-mephenytoin, 4′-OH-(S)-mephenytoin, 6-OH-chlorzoxazone, 6α-OH-Taxol (paclitaxel), nirvanol, benzyloxyresorufin, and resorufin were from Ultrafine (Manchester, UK); and benz[a]anthracenetrans-5,6-dihydrodiol (BA t-5,6-diol) was from the National Cancer Institute Chemical Carcinogen Repository (Kansas, MO). Two CYP2C9 mutants, CYP2C9*2 (Arg→Cys144) and CYP2C9*3 (Ile→Leu359) were purchased from GENTEST (Woburn, MA). 4′-OH-Flurbiprofen was kindly provided by Dr. Timothy Tracy at the University of West Virginia (Morgantown, WV). Celecoxib was extracted from the tablets in formulation. Hydroxymethycelecoxib was obtained from the metabolism of celecoxib with CYP2C9 and identified in our laboratory (Tang et al., 2000).
Baculovirus Expression of Human Cytochrome P450s.
Plasmids containing the full-length cDNAs for CYP3A4, CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2E1, CYP2B6, CYP2D6, and P450 oxidoreductase (OR) were provided by Dr. Frank J. Gonzalez of the National Cancer Institute (Bethesda, MD). Each cDNA was constructed in baculovirus pBlueBac 4.5 shuttle vector (Invitrogen, Carlsbad, CA) according to manufacturer's protocols, and the purified recombinant viruses encoding a P450 or OR cDNA were used to infect Spodoptera frugiperda(Sf21) insect cells for P450 protein expression (Mei et al., 1999; Shou et al., 1999). Microsomes of Sf21 cells containing individual P450s were prepared by differential centrifugation of cell homogenates as previously described (Shou et al., 1999), and the resulting P450 content was measured by the CO-difference spectrum. Microsomal membranes were diluted with a buffer containing 0.25 M sucrose, 1 mM EDTA, 0.5 mM dithiothreitol, 1.15% KCl, and 0.1 M potassium phosphate (KPi). Protein concentration in the microsomal suspensions was determined according to the manufacturer's instructions (Pierce Chemical Co., Rockford, IL). The activities of P450 were measured by the following assays: testosterone 6β-hydroxylation (CYP3A4), flurbiprofen 4′-hydroxylation (CYP2C9 and its mutants), bufuralol 1′-hydroxylation (CYP2D6) and phenacetinO-deethylation (CYP1A2), chlorzoxazone 6-hydroxylation (CYP2E1), (S)-mephenytoin 4′-hydroxylation (CYP2C19), paclitaxel 6α-hydroxylation (CYP2C8), and diazepamN-demethylation (CYP2B6) as reported (Mei et al., 1999; Shou et al., 1999).
Substrate Inhibition Kinetics of P450-Mediated Drug Metabolism.
All substrates were dissolved in methanol as stock solution except for benzyloxyresorufin, which was dissolved inN,N-dimethylformamide, and 10 μl of sequential dilutions were added into an equal volume of reaction mixture (usually 1 ml, unless noted otherwise) that gave appropriate concentrations of substrate containing 1% methanol. Microsomal membranes consisting of 10 to 40 nM P450 (varying between different assays) were incubated for analyses of P450-mediated reactions in the presence of 1 mM NADPH and 0.1 M KPi (pH = 7.45). Varying concentrations of substrate, as shown in Table1, were used in experiments for the study of substrate inhibition kinetics. Reactions were terminated (usually after 10–20-min incubation period) with 6 volumes of dichloromethane followed by the addition of their respective internal standard. Samples containing metabolite(s) formed and internal standard were extracted and centrifuged, and organic phase was dried under a nitrogen stream. The residues were dissolved in an appropriate solvent and analyzed by either HPLC or liquid chromatography-mass spectrometry (Table1). For phenacetin O-deethylation assay, incubations (final volume, 0.2 ml) were performed in a 96-well format, initiated with NADPH (1 mM), and allowed to proceed for 30 min (37°C in a shaking water bath). The reaction was terminated with the addition of acetonitrile (0.2 ml), containing 4 nmol of temazepam as an internal standard, and the samples were vortexed. An aliquot of each incubate (0.2 ml) was transferred to a 96-well plate. Samples were centrifuged to precipitate proteins (4000 rpm for 30 min), and 20 μl of the supernatant was analyzed. All the rates were expressed as nanomoles of product formed per minute per nanomole of P450 used. The consumption of the substrate at all concentrations was less than 15% under linear reaction conditions. Concentrations of substrates used for metabolism were maximized by the limitation of their solubility. Each metabolite(s) was identified by comparing the retention time on HPLC with authentic standard and normalized by the ratio of chromatographic peaks between metabolite formed and internal standard added after incubation.
Assays of individual P450-catalyzed metabolism of substrates used for inhibition kinetics
Fluorescent Assay.
Reactions of benzyloxyresorufin O-dealkylation (CYP3A4) and ethoxyresorufin O-deethylation (CYP1A2) were performed in 96-well plates. Each incubation mixture (200 μl) contained 0.1 M KPi buffer, 20 nM CYP3A4 or 10 nM CYP1A2, and varying substrate concentrations as seen in Table 1. In both cases, the reaction mixture was prewarmed in a shaking Jitterbug incubator (Boekel Industries Inc., Philadelphia, PA) at 37°C for 5 min and initiated with the addition of 1 mM NADPH. After a 5-min incubation, the reaction was terminated with 75 μl of acetonitrile containing 0.5 M Tris buffer (20%). The resorufin formed was detected at an excitation wavelength of 530 nm and an emission wavelength of 590 nm.
HPLC Analysis.
HPLC was performed on a Hewlett Packard model 1100 liquid chromatographic system. Assays for estrone 2-hydroxylation, diazepamN-demethylation, paclitaxel 6α-hydroxylation, flurbiprofen 4′-hydroxylation, (S)-mephenytoin 4′-hydroxylation, chlorzoxazone 6-hydroxylation, and testosterone 6β-hydroxylation were performed using published methods (Table 1) with slight modifications. Separation of metabolites, from the dextromethorphanO-demethylation reaction, was carried out using a Zorbax SB-C18 column (5 μ, 4.6 mm × 15 cm, MAC-MOD Analytical, Chadds Ford, PA) eluted with a 20-min linear gradient (20 to 40% acetonitrile in water containing 0.05% acetic acid) at a flow rate of 1 ml/min. Dextromethorphan, dextrorphan, and internal standard (BA t-5,6-diol) were detected by the fluorescence (excitation of 230 nm and emission of 330 nm), and their retention times were 11.5, 4.1, and 22.6 min, respectively. Paclitaxel, 6α-hydroxypaclitaxel, and bacctin (internal standard) were separated on the Zorbax SB-C18 column, eluted with 10% acetonitrile in water for a 5-min and then a 25-min linear gradient from 10 to 65% acetonitrile in water at a flow rate of 1 ml/min, and detected by an UV wavelength of 230 nm. Retention times of bacctin, 6α-OH-paclitaxel, and paclitaxel were 22.8, 27.3, and 29.4 min, respectively. Metabolites of progesterone and internal standard (estradiol) were separated on a ODS 20/20 column (5 μ, 4.6 mm × 15 cm; Thomason Instrument Co., Chantilly, VA) eluted with a mobile phase identical to that of the testosterone assay (Hanioka et al., 1990).
Liquid Chromatography-Mass Spectrometry.
Separation of phenacetin and its metabolite (acetaminophen) was carried out using a PerkinElmer HPLC system, comprising a Series 200 IC pump and autosampler. Separation was achieved using reverse phase chromatography on a SB-C8 column (3.5-μm particle size; 4.6 × 50 mm; MAC-MOD) and a mobile phase consisting of acetonitrile in an aqueous solution of acetic acid (0.05% v/v). The composition of acetonitrile was increased linearly (20 to 80%) over a 10-min period (flow rate of 1.5 ml/min), and the retention times of acetaminophen, phenacetin, and temazepam (internal standard) were 3.3, 5.5, and 6.6 min, respectively. Metabolite and internal standard (temazepam) were identified using an API 150 MCA mass spectrometer in the positive ion mode (m/z 152, acetaminophen; m/z 301.1, temazepam). An acetaminophen calibration curve (versus temazepam) was used to quantitate the concentration of product in the microsomal incubates.
Kinetic Parameter Estimation.
Nonlinear regression was performed using a nonlinear least-squares algorithm. Fits were performed using multiple initial parameter estimates to ensure minimum global variance. To evaluate systematic deviations of experimental data from calculated regression curves, the differences were represented as residual values. Simulations were carried out with AXUM 5.0 software (Mathsoft, Cambridge, MA). All kinetic values were calculated by the models derived from either the Michaelis-Menten kinetics (nonsubstrate inhibition) or the present model (substrate inhibition).
Kinetic Model.
If one assumes a single binding site at the catalytic pocket of P450, the velocity of the substrate conversion to product versus substrate concentrations can be described by single Michaelis-Menten kinetics that gives hyperbolic kinetics in which theKm and Vmax are determined by eq. 1:

Kinetic model for substrate inhibition.
When S binds to the catalytic site of enzyme (ES), the rate andVmax of product formation (ES → P) are determined by kp[ES] andkp [E]total, respectively. When S binds to the inhibitory site (SE) that is nonproductive, the rate of SES → P is reduced by factor β. Dissociation constants for all species are defined byKS =k−1/k1,KI =k−2/k2, αKI =k−3/k3, and αKS =k−4/k4. All parameters are expressed in eq. 2 (see Materials and Methods).
Results
All incubations were performed using a microsomal preparation from insect cells (Sf21) containing cDNA-expressed cytochrome P450s. To coexpress individual P450s and OR, insect cells were infected with the two separate baculoviruses encoding the gene of interest. Activity of P450 in the metabolism of each marker substrate was maximized by appropriate ratios of P450 to OR protein and was accepted for kinetic studies. To accurately evaluate the model and determine kinetic parameters, substrate consumption at all concentrations of substrate was controlled to less than 15%, and production of metabolite(s) used for the quantitation of rate was linear with incubation time (usually 10–20 min). The assay conditions were defined such that metabolism could be treated under the assumptions of both rapid equilibrium and steady state.
Of all reactions, substrate inhibition for flurbiprofen 4′-hydroxylation (CYP2C9), estrone 2-hydroxylation (CYP1A2), phenacetinO-deethylation (CYP1A2), diazepam N-demethylation (CYP2B6), paclitaxel 6α-hydroxylation (CYP2C8), (S)-mephenytoin 4′-hydroxylation (CYP2C19), and chlorzoxazone 6-hydroxylation (CYP2E1), respectively, were not observed at the range of substrate concentrations used (Table 1). Their kinetics conformed fully to the typical hyperbolic saturation profiles (curves not shown), and their Km andVmax values were determined by the Michaelis-Menten equation (eq. 1, Table2).
Kinetic parameters of the Michaelis-Menten and substrate inhibition kinetics
However, the activity of some P450s was inhibited markedly as substrate concentration (usually greater than Km) was increased. The magnitude of the inhibition was dependent upon the structure and concentration of the substrate, the reaction type, and P450 isoform used. The Michaelis-Menten kinetics was used to fit the part of the data for the determination of kinetic values only when the portions of the inhibited rates from the curve were truncated. Therefore, we compared the Km andVmax between the Michaelis-Menten kinetics (dotted line in figures) and the proposed model of the present study (solid line). The two curve fits derived from both models are shown in Figs. 1-6. TheR2 and RSS (Residual Sum of Squares) values are given for the evaluation of the regression equation (eq. 2) and correlation for the independent variables with predicted values. The kinetic parameters from each set of the data in the reaction were calculated by eqs. 1 and 2, respectively, and are listed in Table 2.
Ethoxyresorufin.
As shown in Fig. 1, when substrate concentration was in excess of 7.5 μM the rate of O-deethylation declined. Apparently, our model (curve) fitted the experimental data. Using the proposed model,KS and Vmaxwere 5.3 μM and 733 min−1, respectively, which were different from those calculated using eq. 1(Km = 2.7 μM andVmax = 414 min−1) (Table 1). KI, which defines the binding of the second substrate molecule (inhibitor) to the site of the enzyme (e.g., SE) that results in a reduction of the rate, was 25 μM (4.7-fold greater than its KS). Factors α and β were also determined as 8.1 and 0.03, respectively, suggesting that when the second substrate binds to the enzyme (SES), the apparentKS increased 8.1-fold (αKS = 43 μM) and theVmax for SES was only 3% of theVmax for ES (βVmax = 22 min−1).

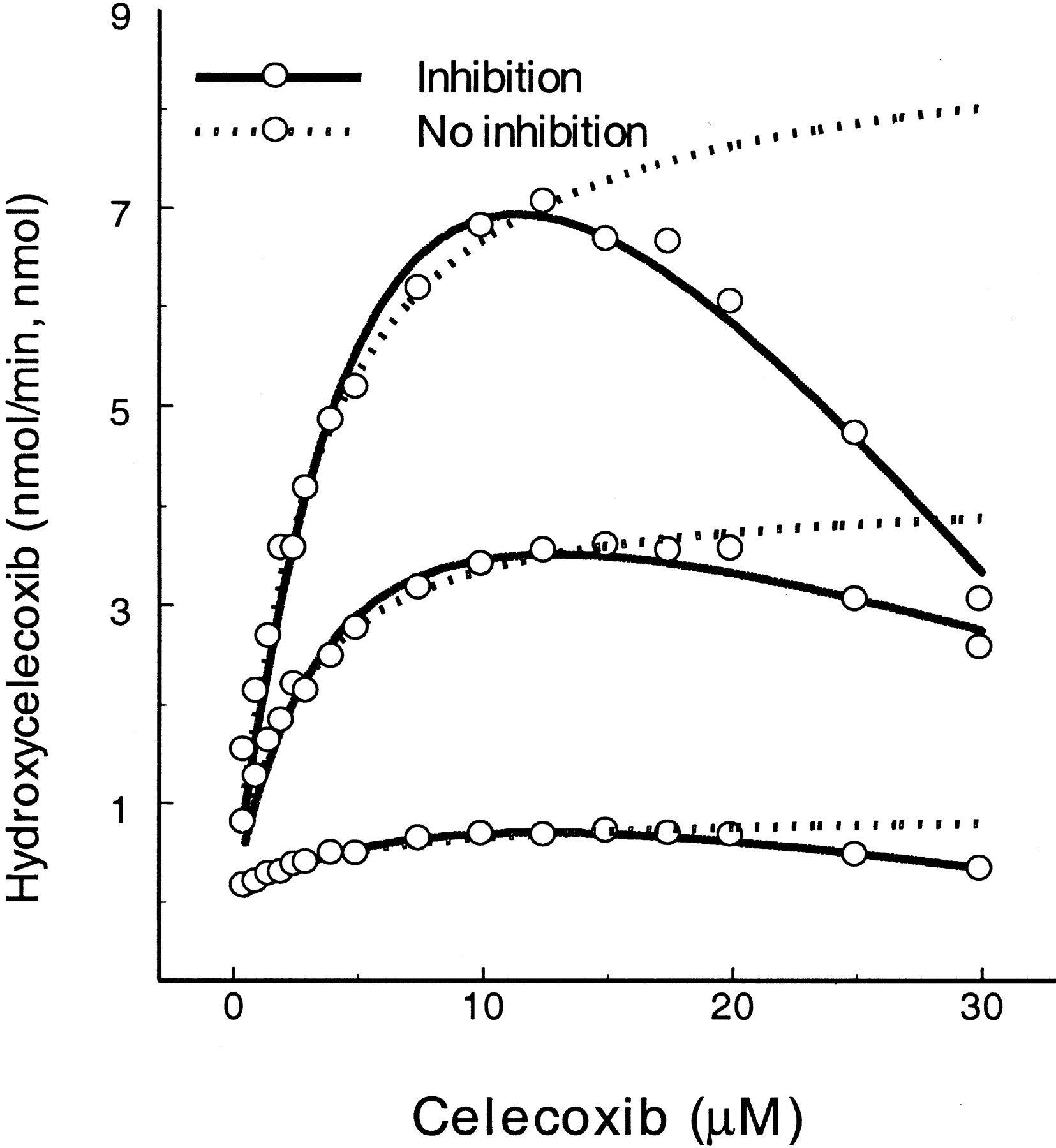
Celecoxib.
Celecoxib is a selective cyclooxygenase II inhibitor that undergoes CYP2C9-catalyzed methyl-hydroxylation (Tang et al., 2000). Celecoxib is also metabolized by the allelic variants CYP2C9*2 and CYP2C9*3, although the activity is reduced by comparison to wild-type CYP2C9 (Fig. 2). However, increasing substrate concentration resulted in substrate inhibition profiles for all three CYP2C9 forms. As a result, kinetic estimates were calculated using the two equations (Table 2). Estimation of kinetic constants using eq. 2 indicates that KS was 2.2- to 3.3-fold (5.8 μM for CYP2C9, 8.2 μM for CYP2C9*2, and 4.5 μM for CYP2C9*3) greater than the Km calculated using eq. 1, and Vmax was 1.2- to 1.7-fold (10.6 min−1 for CYP2C9, 7.2 min−1 for CYP2C9*2, and 1.3 min−1 for CYP2C9*3) greater thanVmax determined using eq. 1.KI values for SE were 1.2- to 5.3-fold greater than KS for ES. In the presence of the second substrate molecule (inhibitor), apparentKS values were increased by factorα up to 23.3-, 12.2-, and 5.1-fold for SE ⇌ SES, respectively, and the activity of the enzyme dropped to 5 to 11% (β = 0.08 for CYP2C9, 0.11 for CYP2C9*2, and 0.05 for CYP2C9*3 of the respective Vmax in comparison to singly bound enzyme complex (ES).

Substrate inhibition of CYP2C9-catalyzed methyl-hydroxylation of celecoxib.
Dotted lines, hyperbolic curves fitted with eq. 1 after truncating the inhibited rates at high substrate concentrations; solid lines, substrate inhibition curves fitted with eq. 2. Curves from top to bottom represent wild-type CYP2C9, CYP2C9*2(Arg→Cys144), and CYP2C9*3 (Ile→Leu359), respectively.
Dextromethorphan.
Dextromethorphan O-demethylation to dextrorphan is catalyzed primarily by the polymorphic CYP2D6 in human with a relatively low Km (<5 μM) (Rodrigues et al., 1994). A wide range of dextromethorphan concentrations (3–2000 μM) was used and substantial substrate inhibition was observed after 200 μM substrate (Fig. 3).KI (48 μM) was about 10-fold greater thanKS (4.8 μM, Table 2), but the inclusion of the second molecule (inhibitor) led to 88% inhibition (12% of theVmax, β = 0.12) and a large increase (23-fold) in apparent KS (α = 23.8, αKS = 114 μM). Noticeably, theKI value for SE was 10-fold greater than the KS value for ES.

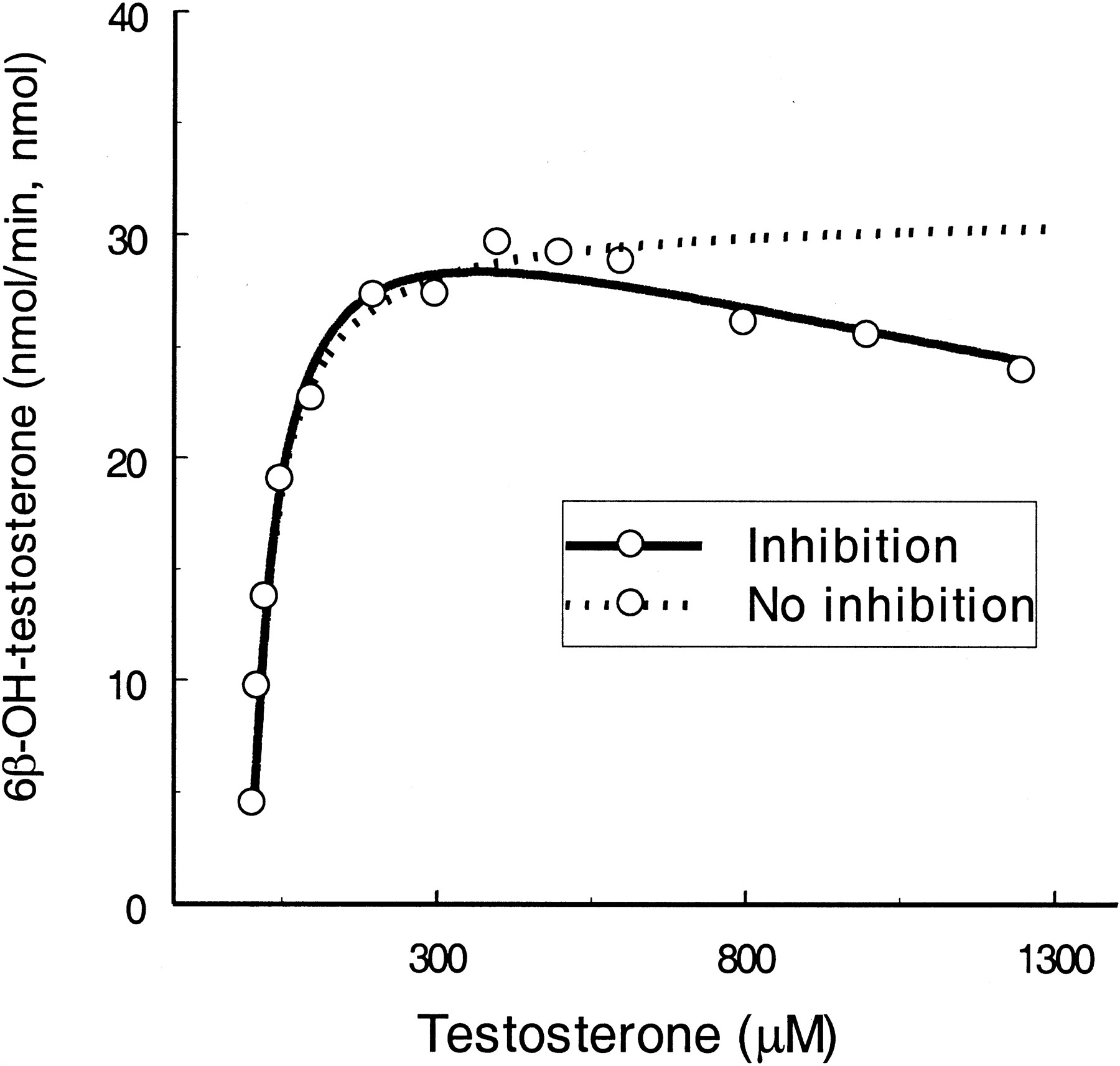
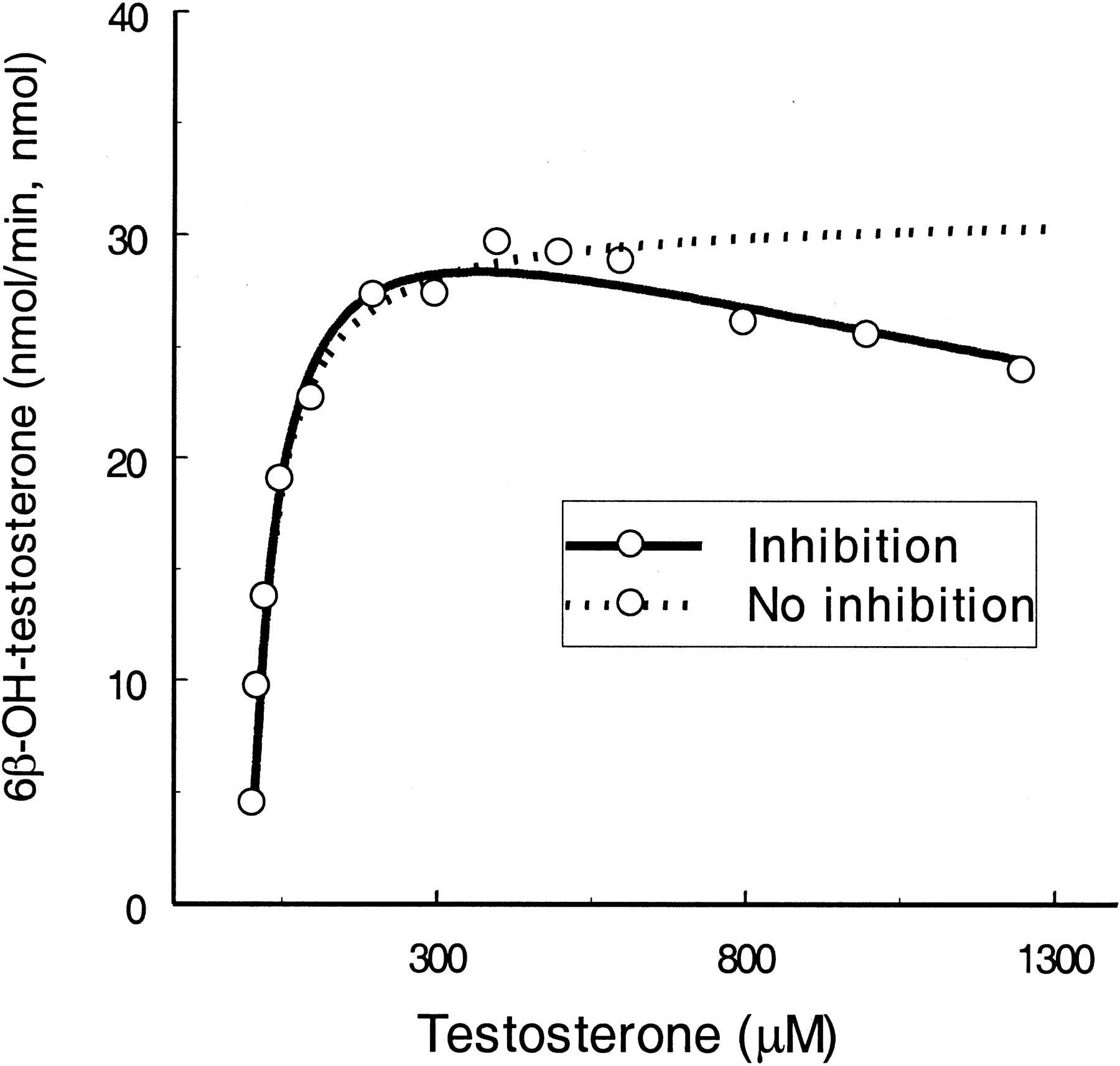
Testosterone.
Testosterone is the most commonly used marker substrate for CYP3A4. Substrate inhibition occurs particularly when testosterone concentration approaches >1000 μM (Fig.4). Using eq. 2,KS and Vmaxvalues were calculated to be slightly higher thanKm and Vmaxvalues derived from eq. 1. KI was 4.7-fold greater than KS. When the two sites on the enzyme were bound simultaneously by the substrate molecules, their binding affinities were changed by 5.7-fold (α = 5.7), and maximal inhibition was achieved by 39% (61% ofVmax for ES, β = 0.61).
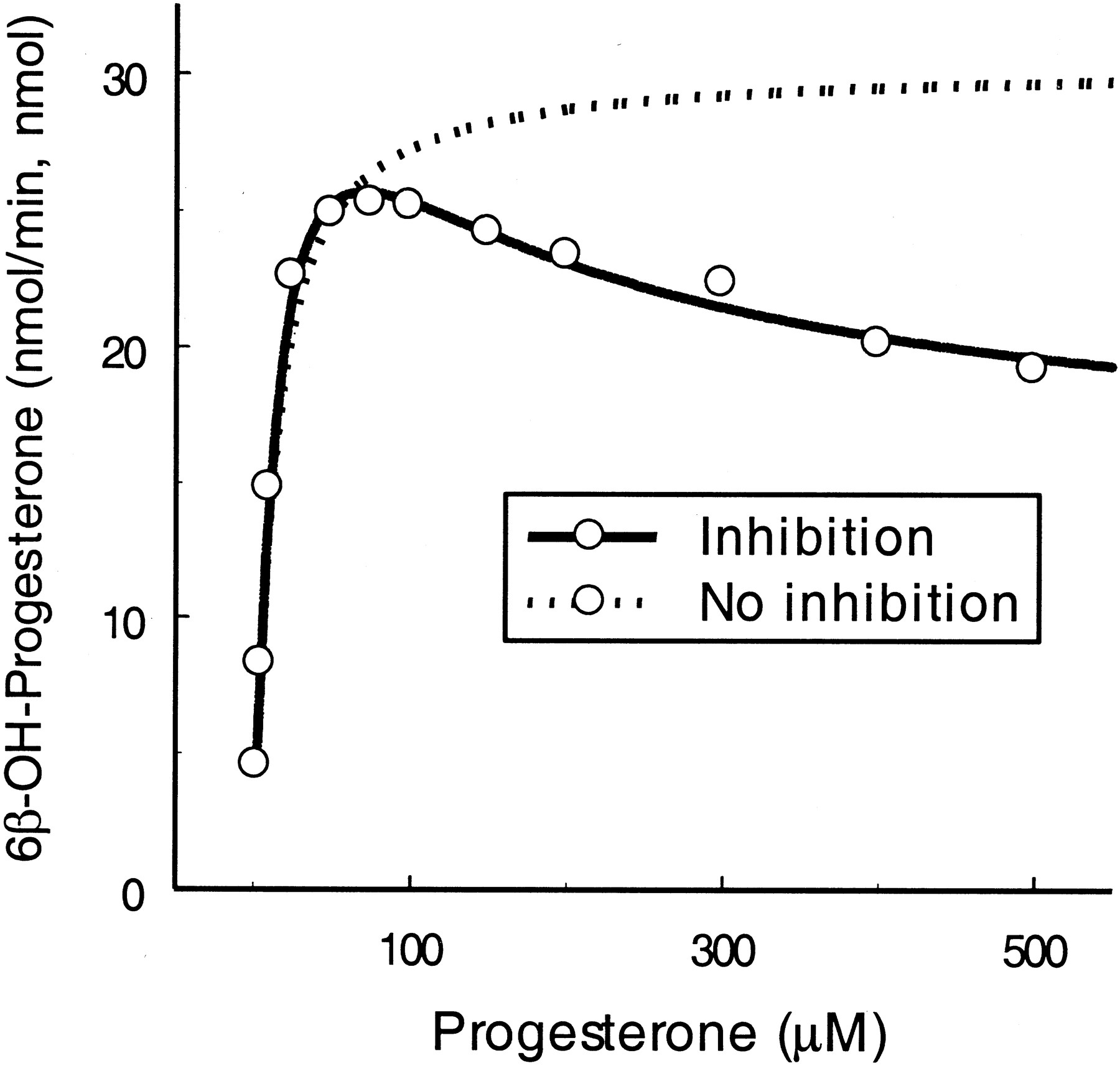
Progesterone.
As with testosterone, the 6β-hydroxylation of progesterone is preferentially catalyzed by CYP3A4 (Yamazaki and Shimada, 1997). TheVmax values from the two equations were shown to be comparable, but the values forKm and KS were less than those observed with testosterone (Table 2). Progesterone substrate inhibition also was noted at higher concentrations (>150 μM and 9-fold greater than KS; Fig.5). The KIvalue (96 μM) for SE was approximately 6-fold higher than theKS value for ES. The α value of 13.2 suggests that presence of the second molecule can alter the binding affinity of the substrate for the catalytic site of the enzyme. The maximal rate for SES ([SES] = [E]total) was 41% (β = 0.41) of Vmax for ES.
Benzyloxyresorufin.
Benzyloxyresorufin is oxidized to fluorescent resorufin and has recently been reported as a CYP3A4 substrate for the high-throughput screening of the enzyme inhibition (Burke et al., 1994). Kinetic studies showed that KS andVmax were 6.1 μM and 231 min−1, respectively. When the substrate concentration exceeded 20 μM, the inhibition appeared to be significant by 92% (β = 0.08; Fig.6).

Discussion
Inhibition of enzyme activity at high substrate concentrations, so called “substrate inhibition”, is one of the most common deviations from kinetics and one of the best documented facts in enzymology. Several models of substrate inhibition in other enzyme systems have been proposed, which include allosteric mechanisms (LiCata and Allewell, 1997), enzymatic chemical oscillations (Shen and Larter, 1994), multiple active sites (Kuhm-Velten et al., 1991), uncompetitive and noncompetitive substrate inhibition (Musser et al., 1997), the Recovery model (Kuhl, 1994), and the King-Altman method (Shin and Kim, 1998). However, these models are not appropriate to describe the kinetics of P450-mediated reactions. Substrate inhibition in most cases behaves as a partial inhibition, since the inhibition of P450 does not approach zero even at very high substrate concentrations.
In the present study, we presume that the substrate that results in an inhibition of P450 activity is due to the access of more than one substrates to the active site. Therefore, the two-site model was proposed for understanding the mechanism of substrate inhibition through the course of kinetic characteristics. The two sites could be either neighboring or at a distant within the active site. In substrate inhibition, one site is favorable for oxidation and the other site is less favorable or nonproductive. Therefore, the metabolism of a substrate can be diminished when the second substrate is present because some interactions in the active site, e.g., steric, electronic or allosteric interaction, may occur, leading to a decrease in activity. When substrate, as an inhibitor binds to the inhibitory site, the inhibited complex (SES) is less capable of converting substrate to product than ES. The β in the model (usually 0 < β < 1) represents the percentage of the maximal inhibition ([E]total = [SES]). Substrate inhibition is more likely due to the direct interaction of the doubly bound sites to which substrate can have access (Shou et al., 1994;Korzekwa et al., 1998). The model presented herein assumes that the active site may contain two distinct binding portions that are neighboring and can be occupied by substrate molecules, one for metabolism (ES) and one for inhibition (SE), which is either nonproductive or poorly productive. When [SES] = [E]total, the maximum inhibition can be observed as expressed by βVmax. The model was tested statistically by curve fitting the observed inhibition data. Consequently, the resulting kinetic constants were used to interpret the nature of the substrate inhibition, and several common features were observed.
First, the substrate inhibition curve could not be fitted to the standard Michaelis-Menten equation (hyperbolic saturation, eq. 1). Therefore, the Km andVmax values obtained using eq. 1 could not be used to truly express the observed kinetics. The results of the present study have shown that the substrate inhibition data were fitted reasonably to a two-site model. There are two dissociation constants in the model, KS for the productive ES andKI for the SE inhibitory to the enzyme (eq.2). However, the two constants for the singly bound complexes can be altered as determined by factor α (usually >1) when both sites are bound. This suggests that the affinity of the substrate for the doubly bound complex is less than that for the singly bound complexes. Accordingly, the maximum velocity (Vmax) of the ES complex can also be changed to βVmax for SES (0 < β < 1). Thus, the magnitude of substrate inhibition at a given concentration of substrate is dependent upon the ratio of [ES] to [SES]. This is probably due to either a steric or allosteric effect of the two-substrate-bound interaction in the active site of the enzyme (SES) (LiCata and Allewell, 1997).
Second, KI values were found to be greater than their respective KS (1.2–10-fold; Table 2). This suggests that the substrate binds with greater affinity to the active site of the enzyme for metabolism. Thus, at the low substrate concentrations, the substrate molecule readily binds to the enzyme to form the ES product-forming complex, and the reaction rate is a function of the ES concentration. The data conform to classical Michaelis-Menten kinetics. However, with increasing substrate concentrations, the proportion of [SE] and [SES] species in [E]total increases, resulting in inhibition even though the inhibitory site has poor affinity for the substrate (KI >KS). Since the two complexes are understood to be noncatalytic (SE) and less productive (β < 1, SES), the degree of inhibition is dependent upon the kinetic characteristics of the bound enzymes (kinetic constants), the ratio of [ES] to the sum of [SE] and [SES], and the type and concentration of the substrate molecule. A decrease in velocity is usually observed at the concentrations of substrate around or greater thanKI. Thus, the potency of substrate inhibition can be defined by parameters KI, α, and β in the model. The α (>1) and β (<1) terms are a result of interaction of doubly bound substrates with the enzyme.
Third, the fitted Vmax values (eq. 2) were shown to be larger than the maximum observed (the highest rates of the substrate inhibition curves) because the observed velocity is the sum of increasing and decreasing components in the substrate inhibition. The earlier the onset of substrate inhibition the higher theKS/KI ratio. The estimated Vmax values, as calculated in eq. 1, were 0.91- to 1.8-fold lower than the fittedVmax data (eq. 2) (Table 2). This implies that truncation of the data cannot aid in assessing trueVmax values for ES, since the contribution of the substrate inhibition to the overall curve shape has occurred and cannot be estimated precisely.
In addition, we hypothesize that at least two sites are located on the enzyme, one productive site (high affinity,KS) and one inhibitory site (low affinity,KI). The two factors (α and β) are used for an adjustment of the curve fitting. The substrate that inhibits the enzyme activity must bind to the site distinct from that substrate binds to for oxidation although it is situated closely. In fact, substrate inhibition appears only at the concentrations of substrate adequate for binding to the inhibitory site (KI > KS). Similarly, the KS for the substrate (SE ⇌ E) also was changed by factor α due to the presence of inhibitory substrate. As a result, increasing SES complex reduces the rate of product formation. The maximum inhibition can be defined by β (0 < β < 1) as the [SES] approaches to [E]total. Our results showed that β values ranged between 0.03 and 0.61, leading to maximum inhibition (39–97%). The α and β values (α > 1 and 0 < β < 1) in each case depend on kinetic characteristics of the enzyme (i.e., allosterism), the steric effects of the two bound substrates, and the physical-chemical properties of the given substrate molecule.
Involvement of product inhibition in substrate inhibition remains unknown because both types of inhibition are not distinguishable. However, in the case of product inhibition, the presence of excess product causes a decrease in rate, and the velocity curve displays a hyperbola and eventually reaches a plateau when maximal inhibition is achieved (a saturation of velocity versus substrate concentration). Product inhibition in nature is similar to competitive inhibition (substrate and product compete for the same site), which basically meets the Michaelis-Menten kinetics. However, note that the kinetic behavior cannot be discriminated between product inhibition and substrate inhibition only if the product formed acts as a noncompetitive inhibitor that binds to the second site of P450 for inhibition. If the substrate exceeds its solubility, increasing substrate concentration should not result in further substrate inhibition because an additional amount of the substrate, which is no longer dissolved in incubation, does not change its concentration. Thus, the velocities versus any substrate concentrations above its solubility remain unchanged.
In conclusion, a two-site model has been used to fit substrate inhibition data for a number of P450-catalyzed reactions. The resulting constants derived from the model were used to interpret the mechanism of substrate inhibition kinetics. KI for the inhibition site was found to be larger thanKS for the site of metabolism.KS and Vmax for ES can be changed significantly by factors α and β (0 < β < 1), respectively, when the second substrate is bound (SES). Thus, increasing substrate concentration leads to a reduction of rate as a result of a decrease in the ratio of [ES] to [SES]. Thus, the doubly bound complex (SES) in the model is believed to be less productive than the singly bound complex (ES), suggesting that binding to the second site on the enzyme inhibits the oxidation of substrate bound to the first site. The interaction between the doubly bound sites that results in substrate inhibition could be steric and allosteric.
Footnotes
-
Send reprint requests to: Magang Shou, Department of Drug Metabolism, WP75A-203, Merck Research Laboratories, West Point, PA. E-mail: magang_shou{at}merck.com
- Abbreviations used are::
- P450
- cytochrome P450
- OR
- oxidoreductase
- Sf21
- Spodoptera frugiperda insect cells
- Kpi
- potassium phosphate
- HPLC
- high-performance liquid chromatography
- ES
- active site of the enzyme
- SE
- inhibitory site of the enzyme
- SES
- two-substrate-bound enzyme
- Received July 10, 2000.
- Accepted September 11, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}