


Abstract
The extent of decreases in apparent hepatic clearance and intrinsic hepatic clearance of midazolam (MDZ) after intravenous administration of MDZ with concomitant oral administration of cimetidine (CIM), itraconazole (ITZ), or erythromycin (EM) was predicted using plasma unbound concentrations and liver unbound concentrations of inhibitors. When MDZ was concomitantly administered with CIM, the observed increase in MDZ concentration was successfully predicted using inhibition constants assessed by human liver microsome and liver-to-plasma unbound concentration ratios in rats. However, the extent of interaction with ITZ or EM was still underestimated even taking into account the concentrative uptake of inhibitors into liver. We could predict the degree of “mechanism-based” inhibition by EM on the hepatic metabolism of MDZ, after repeated administration of EM, by a physiological model incorporating the amount of active enzyme as well as the concentration of inhibitor. The maximum inactivation rate constant and the apparent inactivation constant of EM on MDZ metabolism were 0.0665 min−1 and 81.8 μM, respectively. These kinetic parameters for the inactivation of the enzyme were applied to the physiological model with pharmacokinetic parameters of EM and MDZ obtained from published results. Consequently, we estimated that cytochrome P450 3A4 in the liver after repeated oral administration of EM was inactivated, resulting in 2.6-fold increase in the plasma concentration of MDZ. The estimated extent of increase in MDZ concentration in our study correlated well with the observed value based on metabolic inhibition by EM from published results.
Among drug-drug interactions, there are many reports on increases in drug concentrations caused by inhibition of oxidative metabolism through cytochrome P450 (CYP1). These inhibitions may induce adverse effects and are important problems in clinical cases. Isoforms of metabolic enzymes can be identified using anti-P450 antibodies or specific inhibitors to CYP, which make it possible to predict the possibilities of drug-drug interactions qualitatively. However, to predict the degree of interaction observed in clinical cases quantitatively, it is necessary to investigate the correlation between in vitro inhibitory potency of the inhibitor and in vivo inhibition, taking into account the distribution of the inhibitor into the liver, and extrapolation of data from animal studies. We have estimated drug concentrations in liver from the uptake into rat isolated hepatocytes and have successfully predicted the increasing ratio of plasma concentrations in vivo caused by metabolic inhibition in the liver (Takedomi et al., 1998; Yamano et al., 1999, 2000). Several drugs are eliminated by oxidative biotransformation not only in the liver but also in the small intestine, kidney, lung, and other organs. The plasma concentrations after oral administration of the interacting drug increase due to metabolic inhibitions in the liver and/or the small intestine (Ducharme et al., 1995; Gorski et al., 1998). However, it is difficult to estimate the degree of interaction in the intestine accurately. In this study, we focused on the inhibition on hepatic metabolism and tried to predict the effect of orally administered inhibitors on the plasma concentration of inhibited drugs after intravenous administration to humans. When orally administered, the drug concentration in the portal vein (Cportal) is higher than that in the systemic circulation (Hoffman et al., 1995). Therefore, the concentration of orally administered inhibitor in the liver will be underestimated from plasma concentration in the systemic circulation and liver-to-plasma concentration ratio. In this study, we developed a methodology to predict the degree of the interaction on hepatic metabolism in humans after oral administration of the inhibitor. EM is metabolized by CYP3A and produces a P450 Fe(II)-metabolic intermediate (MI) complex, which is metabolically inactive (Franklin, 1991). As the inhibition of CYP by EM is mechanism-based, the degree of drug-drug interaction is considered to depend on the concentrations of EM and the contact time of CYP3A with EM. Based on a physiological model incorporating the kinetic parameters of inactivated enzyme, pharmacokinetic parameters of EM and MDZ, the concentration of EM, and the amount of active enzyme, we aimed to predict the extent of inhibition on hepatic MDZ metabolism, after repeated oral administration of EM and intravenous administration of MDZ.
Theory
Prediction of Decreasing Ratios of Hepatic Clearance and Increasing Ratios of Drug Concentration in Plasma.
When orally administered, drug concentration in the portal vein (Cportal) is higher than that in the systemic circulation (Hoffman et al., 1995). The maximum Cportal (Cportal,max) after oral administration can be calculated according to eq. 1,
The unbound concentration in the liver (CHf) is expressed as eq. 2,
The intrinsic clearance of the inhibited drug is expressed as a Michaelis-Menten-type equation. Assuming the interaction is based on a competitive inhibition, the intrinsic clearances [CLint(−I), CLint(+I)] of the inhibited drug in the absence or presence of inhibitors are expressed as eqs. 3 and 4, respectively.
The increasing ratio of AUC by concomitant inhibitors is expressed as eq. 10, assuming that the inhibitors do not affect QH or fb.
Prediction of Drug-Drug Interaction by a Physiological Model Based on Mechanism-Based Inhibition.
Ito et al. (1998) simulated the extent of metabolic inhibition in the liver after single oral administration of inhibitor and inhibited drug using a physiological model based on mechanism-based inhibition. In our study, we simulated mechanism-based metabolic inhibition in liver after oral administration of inhibitors and intravenous administration of inhibited drugs using a physiological model (Fig. 1) with the following differential equations, based on a modification of the model ofIto et al. (1998).

Physiological model for mechanism-based inhibition between substrate and inhibitor.
VH, Vportal, Vd, Csys, CH, Cportal, Kp, fb, and CLintrepresent volume of liver, volume of portal vein, volume of distribution at steady state, drug concentration in systemic plasma, drug concentration in liver, drug concentration in portal vein plasma, liver-to-plasma concentration ratio, unbound fraction in plasma, and intrinsic hepatic metabolic clearance, respectively. Solid line, disposition of inhibitor; broken line, disposition of substrate or inhibited drug.
Experimental Procedures
Materials.
ITZ was supplied by Janssen-Kyowa Co. (Tokyo, Japan). EM was supplied by Dainippon Pharmaceutical Co. (Osaka, Japan). CIM was purchased from Sigma Chemical Co. (St. Louis, MO). Human liver microsomes were purchased from GENTEST Corporation (Woburn, MA). MDZ was purchased as MDZ injection (Dormicam Inj.) from Yamanouchi Pharmaceutical Co. (Tokyo, Japan). All other chemicals used were of reagent grade or reagents for high-performance liquid chromatography (HPLC).
Animals.
Sprague-Dawley male rats (7 weeks) weighing 220 to 250 g were purchased from Nippon Bio-Supp Center (Tokyo, Japan). The rats were allowed access to water and food pellets ad libitum.
Pharmacokinetic Data and Inhibition Constants in Humans.
MDZ as an inhibited drug, and CIM, ITZ, and EM as inhibitors were used in this study. When MDZ was administered intravenously after oral administration of those inhibitors, the plasma concentrations of MDZ and those inhibitors were cited from published data (Klotz et al., 1985; Olkkola et al., 1993,1996). The interaction study between CIM and MDZ was performed according to the following schedule (Klotz et al., 1985). At 2 h after oral administration of CIM at a dose of 800 mg, the subjects received an intravenous bolus of 0.05 mg/kg MDZ over 30 sec, followed immediately by a constant infusion of 0.025 mg/kg/h for 10 h. The concentrations of MDZ in plasma at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, and 10 h after the start of infusion were measured.Olkkola et al. (1993) performed an interaction study of EM and MDZ according to the following study design. The subjects were given 500 mg of EM three times a day orally for 1 week. EM was administered at 7 AM, 3 PM, and 11 PM, except on the 6th day when MDZ was administered. On the 6th day, EM was given at 7 AM, 1 PM, and 11 PM, and 0.05 mg/kg MDZ was intravenously administered at 3 PM (2 h after administration of EM). The concentrations of MDZ in plasma at 0.25, 0.5, 0.75, 1, 2, 3, 4, 5, 6, and 18 h after intravenous administration were measured. The interaction study of ITZ and MDZ was performed according to the following design (Olkkola et al., 1996). The subjects were given 200 mg of ITZ once a day orally for 4 days. On the 4th day, 0.05 mg/kg MDZ was intravenously administered at 2 h after administration of ITZ. The concentrations of MDZ in plasma at 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, and 17 h after intravenous administration were measured.
The in vitro inhibition activities of those inhibitors against MDZ metabolism on human liver microsomes were cited from published data (Gascon and Dayer, 1991; Wrighton and Ring, 1994; Von Moltke et al., 1996). The metabolic parameters (Km and Vmax) of MDZ and EM on human microsomes were quoted from published data (Wang et al., 1997; Riley and Howbrook, 1998; Wandel et al., 1998).
Absorption Rate Constants of CIM, ITZ, and EM.
We calculated absorption rate constants of CIM, ITZ and EM according to the methods described below. The plasma concentrations after oral administration of CIM, ITZ, or EM to humans obtained from published data were fitted to a two-compartment model with first-order absorption (eq. 20) using the nonlinear least squares method (Yamaoka et al., 1981) to calculate the absorption rate constants.
Liver-to-Plasma Concentration Ratios of CIM, ITZ, EM, and MDZ.
Because the drug concentrations in human liver could not be measured directly, we estimated the concentrations in human liver by using the liver-to-plasma concentration ratios in rats determined previously. CIM was administered through the femoral vein at doses of 3, 5, or 9 mg/body and then infused at constant rates of 1.4, 2.8, or 5.7 mg/h/body, respectively. ITZ, EM, or MDZ was administered through the femoral vein to rats by bolus injection at doses of 5, 50, or 10 mg/kg, respectively. At 6 h after starting the infusion of CIM at 0.25, 1, 4, 8, and 24 h after administration of ITZ, or at 1, 2, and 3 h after administration of EM or MDZ, the blood was collected from the abdominal aorta, and the liver was removed. The blood was centrifuged at 12,000 rpm for 2 min to obtain plasma. The liver was homogenized with 4 volumes of ice-cold distilled water. We determined the concentrations of each drug in the plasma and liver by the methods described below and calculated the liver-to-plasma concentration ratios.
Unbound Fractions of Inhibitors in the Plasma and Liver Tissue.
Unbound fractions of CIM and ITZ in the plasma and liver tissue evaluated by the equilibrium dialysis method were cited from our previous reports (Takedomi et al., 1998; Yamano et al., 1999). Unbound fractions of EM in the plasma and liver tissue were evaluated by the equilibrium dialysis method.
Dialysis was performed using an apparatus made of clear acrylic resin and consisting of two 1.5-ml chambers separated by a cellulose dialysis membrane (SC-101-M10H, DIACHEMA). EM was added to rat plasma at concentrations of 5 and 20 μg/ml and applied to one chamber, and isotonic phosphate buffer (pH 7.4) was applied to the other chamber. After incubation at 37°C for 6 h, 0.1 ml of sample was collected from both chambers for assay.
For determination of the liver tissue binding of EM, liver tissues were homogenized with 0.1 M phosphate buffer (pH 7.4) to prepare 10, 20, and 30% tissue homogenates. Tissue homogenates were dialyzed two times with 100 volumes of 0.1 M phosphate buffer (pH 7.4) for 12 h to remove coenzymes. EM was added to the tissue homogenates at concentrations of 2, 10, or 50 μg/ml. The mixture and isotonic phosphate buffer (pH 7.4) were added to the diaylsis chamber and incubated at 25°C for 6 h. After the incubation, 0.5 ml of sample was collected from both sides for assay.
The liver tissue unbound fraction was calculated according to the following equation:
Blood-to-Plasma Concentration Ratio of MDZ in Humans.
The blood-to-plasma concentration ratio (CB/Cp) of MDZ was measured as follows. MDZ was added to fresh human blood at a concentration of 0.5, 2, or 10 μg/ml, and 1 ml of blood sample was incubated at 37°C for 15 min. Subsequently, 0.2 ml of sample was taken, plasma was obtained by centrifugation, and CB/CP ratios were calculated. From the preliminary experiment, we confirmed that CB/CP ratios were substantially constant after incubation at 37°C for 15 min, and MDZ was stable during incubation.
Inactivation Kinetics by EM, ITZ, and CIM in Human Hepatic Microsomes.
To investigate whether EM, ITZ, and CIM are mechanism-based inhibitors, we calculated inactivation kinetic parameters of EM, ITZ, and CIM in human hepatic microsomes. From the preliminary experiment, we confirmed that MDZ depletion increased proportionally up to 15 min and in the range of 0.1 to 2 mg/ml of protein concentration on the condition of metabolic inhibition experiment. Incubation mixture (180 ml) containing human liver microsomes (protein concentration, 5 mg/ml) and a NADPH regenerating system (100 mM, Na2HPO
Prediction of the Plasma Concentrations in Portal Vein and the Liver Concentrations in Humans Using Absorption Rate Constants andKp Values.
We estimated the plasma concentrations in portal vein after administration of inhibitors to humans using absorption rate constants according to eq. 1 and calculated the concentrations in the liver using Kp values in rat liver according to eq. 2.
Prediction of Increasing Ratios of Plasma Concentrations of MDZ by Concomitant Administration of CIM, ITZ, and EM in Humans.
We predicted the decreasing ratios of hepatic intrinsic clearance and hepatic metabolic clearance after intravenous administration of MDZ to humans in the presence of CIM, ITZ, or EM according to eqs. 7 and 10. The maximum unbound plasma concentrations in the portal vein, the average unbound plasma concentrations in the systemic circulation, and the unbound concentrations in liver calculated from those plasma concentrations were used as the inhibitor concentrations to predict the increasing ratios of plasma concentrations of MDZ. The inhibition constants of CIM, ITZ, and EM on the metabolism of MDZ were taken from published data and were 268 μM for CIM, 0.275 (Ki1) and 2.59 μM (Ki2) for ITZ, and 148 μM for EM (Gascon et al., 1991;Wrighton and Ring, 1994; Von Moltke et al., 1996).
Prediction of Drug-Drug Interaction by a Physiological Model Based on Mechanism-Based Inhibition.
We predicted the extent of inhibition on hepatic MDZ metabolism, after repeated oral administration of EM and intravenous administration of MDZ, using kinetic parameters of enzyme inactivation, pharmacokinetic parameters of EM and MDZ, the concentration of EM, and the amount of active enzyme by the physiological model described previously. The concentrations of MDZ after intravenous administration in the absence of EM (Olkkola et al., 1993) were fitted to the physiological model (Fig. 1) using the pharmacokinetic parameters of MDZ shown in Table 6 to calculate Vd,S and Vmax,S. In the present study, the plasma concentration of MDZ and the concentration of active enzyme were simulated according to the above-mentioned study design ofOlkkola et al. (1993) using WinNonlin (version 3.1, Pharsight Corp., Mountain View, CA). It was reported that the total amount of P450 in human liver is 25,000 nmol (Thummel et al., 1997) and that CYP3A4, which is involved in metabolism of MDZ, accounts for 30% of total P450 in the liver (Shimada et al., 1994). The CYP3A4 content in human liver was calculated to be 7500 nmol. To convert Vmax of EM and MDZ into the value per human body, we used 52.5 mg of protein/g of liver as the amount of microsomal protein in human liver (Iwatsubo et al., 1997), and 1800 g as human liver weight (Davies and Morris, 1993). Because there have been no reports on turnover rate of CYP3A4 in human liver, the plasma concentration of MDZ after intravenous administration was simulated using turnover rate constant of CYP3A2 in rat liver (Correia, 1991).
Pharmacokinetic parameters of midazolam and erythromycin
Measurement of the Concentrations of Various Drugs.
The concentrations of CIM, ITZ, EM, and MDZ were measured using HPLC. The concentrations of CIM in plasma and liver were measured by a modification of the method of Takedomi et al. (1998). In brief, 0.1 ml of plasma or 0.5 ml of 20% liver homogenate, 0.1 ml of methanol, 0.5 ml of 1 N NaOH, and 5 ml of dichloromethane were mixed and shaken for 5 min and then centrifuged at 3000 rpm for 5 min. Four milliliters of the organic phase was then transferred to another tube and evaporated under nitrogen gas. The residue was dissolved in 0.2 ml of the mobile phase and 40 μl was injected into HPLC. For detection, a wavelength of 228 nm was used. The column was a reversed-phase YMC-Pack Pro C18, 3.0-mm × 150-mm (YMC, Kyoto, Japan) and was maintained at 40°C. The mobile phase was acetonitrile/10 mM phosphate buffer (pH 6.5) (15:85, v/v), and was pumped isocratically at a flow-rate of 0.4 ml/min. The limit of quantification was 100 ng/ml for plasma and 200 ng/ml for liver.
The concentrations of ITZ in plasma and liver were measured by the method of Yamano et al. (1999). For the determination of EM concentrations in plasma and liver, 0.1 ml of plasma or 0.5 ml of 20% liver homogenate, 0.1 ml of 0.5 N NaOH, and 3 ml oftert-butylmethylether were mixed and shaken for 5 min, then centrifuged at 3000 rpm for 5 min. Two milliliters of the organic phase was transferred to another tube and evaporated under nitrogen gas. The residue was dissolved with 50 μl of the mobile phase and 20 μl was injected into HPLC. The chromatographic system consisted of a pump LC-10AD, and an electron chemical detector ECD-10A (Shimadzu, Kyoto, Japan). The detector cell potential for the oxidation was set at 1000 mV. The column was a reversed-phase TSKgel 80TM ODS, 4.6-mm × 150-mm (Toso, Tokyo, Japan) and was maintained at 30°C. The mobile phase was acetonitrile/100 mM phosphate buffer (pH 6.4) (50:50, v/v), and was pumped isocratically at a flow-rate of 1 ml/min. The limit of quantitation was 200 ng/ml for plasma and 1000 ng/g for liver.
The concentrations of MDZ in plasma were measured by the method ofYamano et al. (1999). For the determination of MDZ concentration in liver, 0.5 ml of 20% liver homogenate, 0.5 ml of 1 N NaOH and 3 ml of n -hexane were mixed and shaken for 5 min, then centrifuged at 3000 rpm for 5 min. Two milliliters of the organic phase was transferred to another tube and evaporated under nitrogen gas. The residue was dissolved with 0.2 ml of the mobile phase and 75 μl was injected into HPLC. The HPLC conditions were the same as those for determining the plasma concentration of ITZ (Yamano et al., 1999). MDZ was detected by measuring the absorption at 245 nm. The limit of quantitation was 100 ng/g for liver.
For the determination of MDZ in blood, 0.1 ml of blood, 0.5 ml of 1 N NaOH, and 5 ml of n -hexane were mixed and shaken for 5 min, and then centrifuged at 3000 rpm for 5 min. Four milliliters of the organic phase was then transferred to another tube and back-extracted with 3 ml of 0.1 N HCl. To 2 ml of the aqueous phase, 0.5 ml of 1 N NaOH was added and extracted with 2.5 ml of n -hexane. Two milliliters of the organic phase was transferred to another tube and evaporated under nitrogen gas. The residue was dissolved in 0.2 ml of the mobile phase and 75 μl was injected into HPLC. The HPLC conditions were the same as those for determining the plasma concentration of MDZ. The limit of quantitation was 100 ng/ml for blood.
In all measurements, coefficients of variation were less than 10%, and within-run accuracies were less than ±10%. When the concentrations in the samples were below the limit of quantitation, levels were determined by increasing the amount of samples.
Results
Absorption Constant of CIM, ITZ, and EM.
Absorption rate constants of CIM, ITZ, and EM were calculated by fitting the plasma concentrations after oral administration of CIM, ITZ, or EM to humans or according to eq. 21 and are shown in Table1.
Pharmacokinetic parameters of cimetidine, itraconazole, and erythromycin in humans
Liver-to-Plasma Concentration Ratios of CIM, ITZ, EM, and MDZ in Rats.
As shown in Fig. 2, liver-to-plasma concentration ratios after administration of EM and MDZ to rats were constant, regardless of plasma concentrations. The ratios of CIM in rats were 3.81 ± 1.03 (mean ± S.D.), which were identical to those ratios in dogs and humans (Berg et al., 1984;Ziemniak et al., 1984). Table2 shows liver-to-plasma concentration ratios of CIM, ITZ, EM, and MDZ (Takedomi et al., 1998;Yamano et al., 1999).
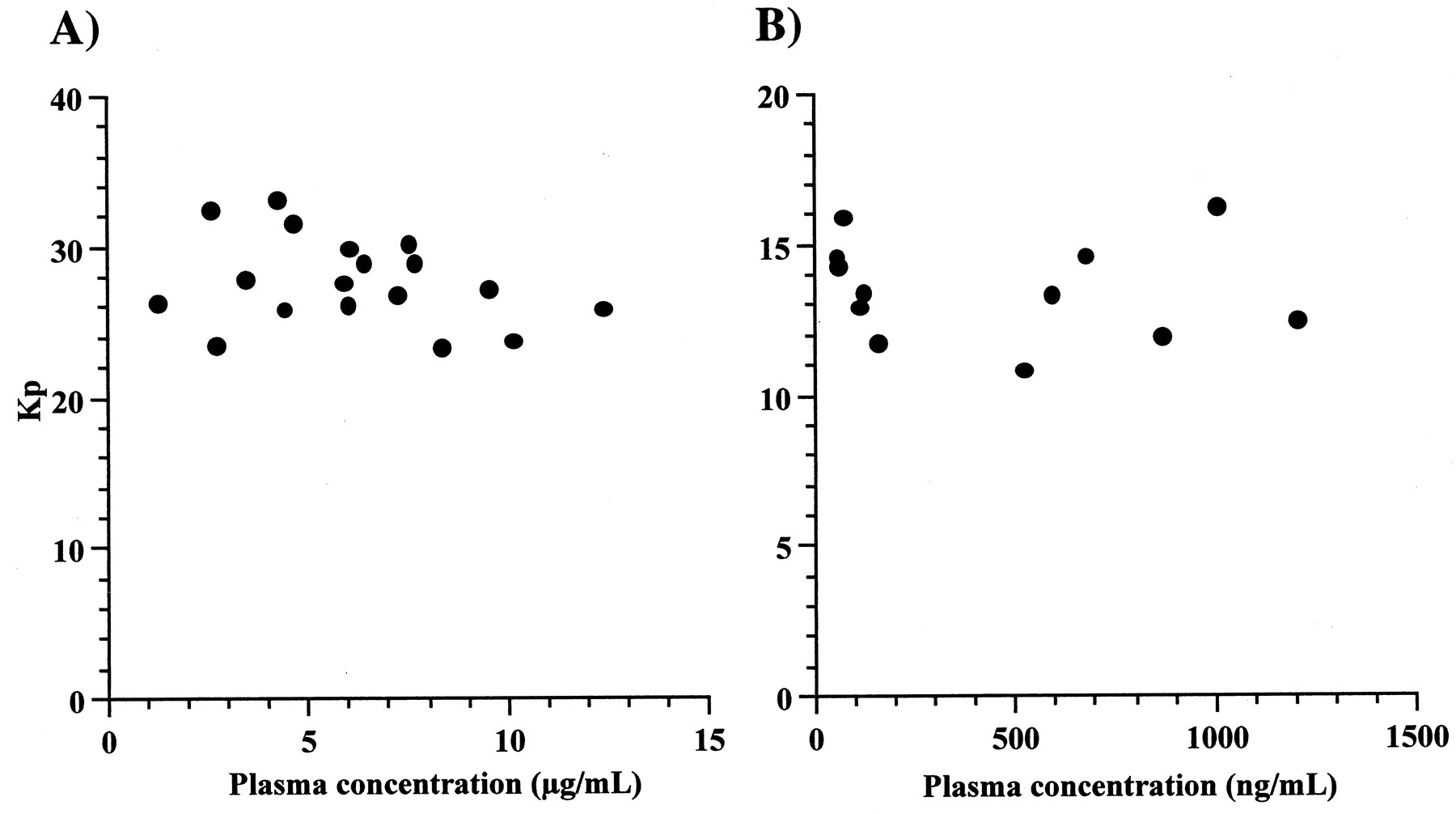
Plasma concentration dependence of the Kp values for EM (A) and MDZ (B).
EM or MDZ were administered through the femoral vein to rats by bolus injection at doses of 50 or 10 mg/kg, respectively. Kp values were liver-to-plasma concentration ratios at 1, 2, and 3 h after administration of EM or MDZ.
Pharmacokinetic parameters of cimetidine, itraconazole, erythromycin, and midazolam in rats
CB/CP Ratios of MDZ.
CB/CP ratios of MDZ were within the range of 0.68 to 0.70 and were constant within the concentration range of 0.5 to 10 μg/ml.
Plasma Protein and Liver Tissue Binding of EM.
The plasma unbound fraction (fp) of EM was 0.78 ± 0.08 (mean ± S.D., n = 3). The liver tissue unbound fractions (fH) of EM calculated from eq. 22 were 0.56 ± 0.02, 0.52 ± 0.03, and 0.53 ± 0.03 (mean ± S.D., n = 3) for 10, 20, and 30% homogenate of liver tissue. Table 2 summarizes the unbound fractions of CIM, ITZ, and EM in plasma and liver (Takedomi et al., 1998; Yamano et al., 1999).
Prediction of the Plasma Concentrations in Portal Vein and the Liver Concentrations in Humans.
Table 3 shows the concentrations in human liver after oral administration of inhibitors, which were predicted using Kp values of rats and the plasma concentrations in portal vein calculated from absorption rate constants. The plasma concentrations in portal vein of ITZ and EM were almost the same as the systemic plasma concentrations. However, for CIM, the plasma concentration in portal vein was higher than the systemic plasma concentration.
Predicted plasma and liver concentrations of cimetidine, itraconazole, and erythromycin in humans
Inactivation Kinetic Analysis by CIM, ITZ, and EM on Human Liver Microsomes.
Figure 3 shows the time and concentration dependency of inactivation on MDZ metabolism by EM. The maximum inactivation rate constant and the apparent inactivation constant of EM on MDZ metabolism were calculated to be 0.0665 min−1 and 81.8 μM, respectively, according to eq. 23 (Fig.4, Table6). However, because inactivation of enzyme was not dependent on preincubation with ITZ and CIM, inactivation rate constants of ITZ and CIM could not be calculated.

Time- and concentration-dependent inactivation of enzyme by EM (A), ITZ (B), and CIM (C).
The concentrations of inhibitors were EM, 20, 40, 100, 250, and 400 μM; ITZ, 0.1, 0.5, and 2.5 μM; CIM, 100, 250, and 500 μM. Panel A: ●, with 20 μM EM; ○, with 40 μM EM; ▴, with 100 μM EM; ▵, with 250 μM EM; ■, with 400 μM EM. Panel B: ●, with 0.1 μM ITZ; ○, with 0.5 μM ITZ; ■, with 2.5 μM ITZ. Panel C: ●, with 100 μM CIM; ○, with 250 μM CIM; ■, with 500 μM CIM.

Determination of kinetic parameters for mechanism-based inhibition.
Apparent inactivation rate constants (kobs) were fitted to eq. 23 using the nonlinear iterative least squares method to estimate Ki,app and kinact.
Prediction of Increasing Ratios of Plasma Concentrations of MDZ with Concomitant Administration of CIM, ITZ, and EM in Humans.
Tables 4 and5 represent the observed decreasing ratios of hepatic intrinsic clearance and hepatic metabolic clearance after intravenous administration of MDZ to humans in the presence of CIM, ITZ, or EM, and the predicted values using unbound plasma concentrations or unbound liver concentrations of inhibitors, respectively. In the case of CIM, by using the unbound concentrations in liver calculated from maximum plasma concentrations in the portal vein, the predicted values were very close to the observed values, whereas the degree of interaction with ITZ or EM were still underestimated even when taking into account the concentrative uptake of inhibitors.
Effects of concomitant administration of inhibitors on hepatic metabolic clearance of midazolam in humans
Effects of concomitant administration of inhibitors on hepatic intrinsic clearance of midazolam in humans
Prediction of Drug-Drug Interaction by a Physiological Model Based on Mechanism-Based Inhibition.
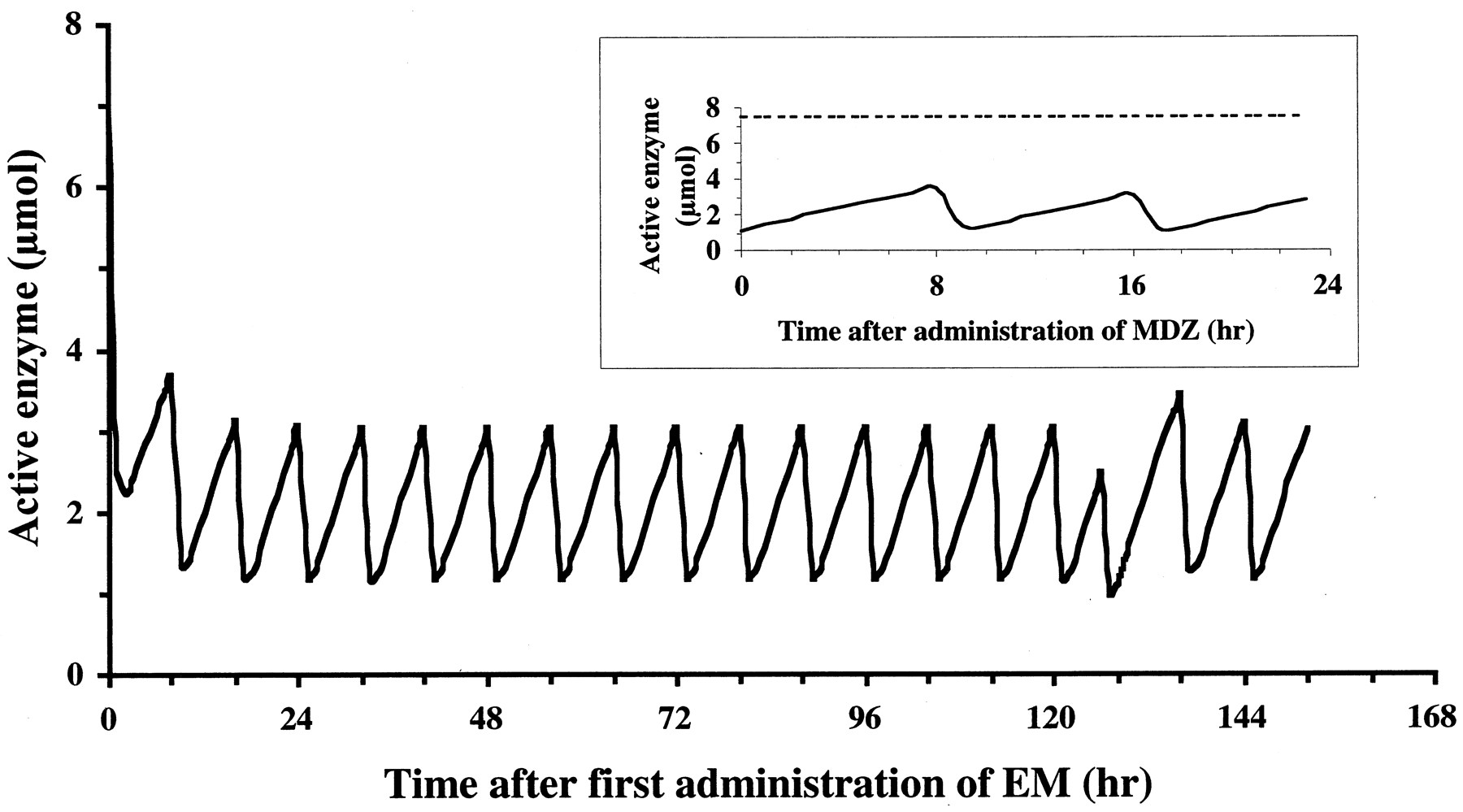
We predicted the degree of inhibition on hepatic MDZ metabolism after repeated oral administration of EM and intravenous administration of MDZ using kinetic parameters of inactivated enzyme, pharmacokinetic parameters of EM and MDZ, the concentration of EM, and the amount of active enzyme using a previously described physiological model of drug-drug interaction based on mechanism-based inhibition. We also fitted the concentrations of MDZ after intravenous administration in the absence of EM (Olkkola et al., 1993) to the physiological model (Fig. 1) using pharmacokinetic parameters of MDZ to estimate Vd,s and Vmax,sto be 20.2 liter and 2410 μmol/h, respectively. Estimated kinetic parameters for the inactivation of enzyme by EM are listed in Table 6, along with pharmacokinetic parameters of EM and MDZ obtained from published data. Simulation of CYP3A4 activity in the liver during repeated oral administration is shown in Fig. 5. EM was administered at intervals of 8 h except on the 6th day. On the 6th day, when MDZ was administered, EM was given at intervals of 6 or 10 h. Therefore, active CYP3A4 content profile on the 6th day was different from that on other days. Figure 6 shows the plasma concentrations of MDZ in the absence or presence of EM from published data (Olkkola et al., 1993) and the simulated plasma concentration of MDZ, incorporating the physiological model of drug-drug interaction based on mechanism-based inhibition. The AUC of MDZ was predicted to increase up to 2.6-fold compared with that in the absence of EM (Table 4). The estimated extent of increase in MDZ concentration in our study correlated well with the observed values based on metabolic inhibition by EM from published data.

Simulated effect of the administration of EM on the content of active enzyme.
For the simulation exercise, subjects are given 500 mg of EM three times a day orally for 1 week. EM is administered at 7 AM, 3 PM, and 11 PM, except on the 6th day when MDZ is administered. On the 6th day, EM is given at 7 AM, 1 PM, and 11 PM, and 0.05 mg/kg MDZ is intravenously administered at 3 PM (2 h after administration of EM). Inset, active enzyme content on the 6th day during repeated oral administration of EM; solid line, the content of active enzyme in EM-treated; broken line, the content of active enzyme in EM-untreated. Simulation study was performed using eqs. 11 to 19. Pharmacokinetic parameters and enzymatic parameters are shown in Table 6.

Effect of the administration of EM on the concentration of MDZ in plasma.
○, the concentration of MDZ observed in the presence of EM; ●, the concentration of MDZ observed in the absence of EM obtained from published data (Olkkola et al., 1993); line, the concentration of MDZ simulated in the absence or the presence of EM by a physiological model based on mechanism-based inhibition.
Discussion
To estimate the degree of drug-drug interaction based on metabolic inhibition quantitatively, inhibitory potencies of drugs to the enzymes, and the concentrations in the metabolic organs after administration of inhibitors need to be determined. The liver, small intestine, kidney, lung, and others have been characterized as metabolic organs. When the interacting drugs are administered orally, their concentrations in the plasma increase due to the metabolic inhibitions in the liver and/or the small intestine. However, it is difficult to estimate the degree of interaction in the intestine accurately. In this study, we focused on the metabolic inhibition in the liver and tried to predict the decreasing ratio of hepatic clearance after intravenous administration of the interacting drugs and oral administration of inhibitors from in vitro data and animal experiments.
To estimate the decreasing ratios of hepatic clearance by concomitant administration of inhibitors in humans, unbound concentrations of inhibitors in the liver, which inhibit the metabolism, need to be determined. When the inhibitors are orally administered, the concentrations of the inhibitors in the portal vein are higher than those in the systemic circulation (Hoffman et al., 1995). However, it is difficult to measure drug concentrations in the portal vein directly. Absorption rate constants from the gastrointestinal tract are necessary to estimate the concentrations in the portal vein from those in the systemic circulation. In this study, absorption rate constants of CIM, ITZ, and EM were calculated to estimate the concentrations in the portal vein from those in the systemic circulation. We tried to predict the decreasing ratios of hepatic clearance of MDZ by using unbound concentrations in plasma and liver calculated from average plasma concentrations in the systemic circulation and maximal portal plasma concentrations after oral administration. It is considered that the prediction by maximal portal plasma concentrations is appropriate to avoid the interaction based on metabolic inhibition in liver from a safety perspective. However, in this case the interaction may be overestimated. On the other hand, the interaction may be underestimated by using average plasma concentrations in the systemic circulation as the concentrations in the liver of inhibitor calculated from plasma concentrations in the systemic circulation are lower than that calculated from the concentrations in the portal vein. Therefore we consider that the concentrations in liver of inhibitor calculated from average plasma concentrations in the portal vein should be used to predict the degree of metabolic inhibition in liver accurately. As it is impossible to measure plasma concentrations in the portal vein in humans directly, to estimate those concentrations during the period of MDZ elimination the analysis based on physiological model by using parameters, such as absorption rate constants, elimination rate constants, and hepatic blood flow is necessary. Further detailed studies must be made to establish the methodology to predict the degree of metabolic inhibition in liver by taking into account that plasma concentrations in the portal vein change higher than plasma concentrations in the systemic circulation.
There was no interspecies difference in the liver-to-plasma concentration ratios on the elimination phase after administration of CIM (Berg et al., 1984; Ziemniak et al., 1984). As the concentrations in the systemic circulation are almost equivalent to those in the portal vein on the elimination phase, it is conceivable that there is no interspecies difference on liver-to-portal vein concentration ratios of CIM. We assumed that there is little interspecies difference on liver-to-portal vein concentration ratios of EM and ITZ and substituted Kp values of rats for those of humans. It is generally considered that this assumption holds good on scale-up from animals to human using the physiological model. However, as for drugs that are actively transported into the liver (Yamazaki et al., 1996), an experiment examining the uptake into human hepatocytes would be necessary to estimate the concentration in the human liver because of interspecies difference on the disposition and character of the transporters. Among the three inhibitors we examined, oral CIM-induced decrease in hepatic clearance of intravenously administered MDZ was successfully predicted by using the concentrations in liver of inhibitor calculated from maximal plasma concentrations in the portal vein, suggesting the validity of our methodology. Levine and Bellward (1995) showed that CIM selectively inhibits CYP2C11 by formation of MI complex in rats. Since the inactivation of MDZ metabolism was not evoked by CIM in this study (Fig.3), the increase of plasma concentration of MDZ by CIM was not due to a decrease in active P450 content by the formation of MI complex. Therefore, this increase can be conceivably attributed to competitive inhibition by CIM. However, the degrees of interaction with ITZ or EM were still underestimated, even when taking into account the concentrative uptake of inhibitors, suggesting the presence of other factors which influence the degree of the drug-drug interaction. Absorption rate constants after oral administration do not exceed gastric emptying time generally. Although Ito et al., (1998) recommend using the theoretical maximum value (0.1 min−1) as ka, if it is unknown, for the estimation of the maximum concentrations of inhibitors to avoid false negative prediction, substitution of 0.1 min−1 for ka of EM and ITZ still caused an underestimation of the decrease of hepatic intrinsic clearance and hepatic metabolic clearance (data not shown).
ITZ inhibits P450 activities by coordination of an imidazole group to P450 in the same way as CIM. Because ITZ is water-insoluble and lipophilic (log P, 5.66) (Heykants et al., 1987), it seems to be conceivable that ITZ, which dissolves into lipid bilayer, affects P450 activities. Repeated administration of ITZ for 4 days potentiated an increase in the concentration of intravenously administered MDZ. Therefore, it is necessary to investigate the change in pharmacokinetics and metabolic inhibitory properties of ITZ after its repeated administration. To investigate whether ITZ is a mechanism-based inhibitor, inactivation kinetics by ITZ in human hepatic microsomes were estimated. However, as inactivation of enzyme was not dependent on preincubation with ITZ, the inactivation rate constant of ITZ could not be calculated (Fig. 3).
Because EM is metabolized by CYP3A and the metabolite of EM produces a MI complex that is a metabolically inactive P450 species (Delaforge et al., 1983), there was a possibility that the recovery of active enzyme was delayed by repeated administration of EM. Because the inhibition by EM is a mechanism-based inhibition, the degree of inhibition is dependent on the concentration of the inhibitor and the contact time of the enzyme with the inhibitor. Indeed, the degree of inhibition on hepatic MDZ metabolism after repeated oral administration of EM and intravenous administration of MDZ was successfully predicted by a physiological model using kinetic parameters of inactivated enzyme, pharmacokinetic parameters of EM and MDZ, the concentration of EM, and the amount of active enzyme. After repeated oral administration of EM, CYP3A4 in the liver was inactivated (Fig. 5) and the AUC of MDZ was estimated to increase up to 2.6-fold, compared with that in the absence of EM (Fig.6). Although the prediction overestimated the observed value based on metabolic inhibition by EM from published data (2.2-fold), it was considered that the degree of drug-drug interaction based on mechanism-based inhibition was successfully predicted from in vitro data. In this study, we assumed that the amount of active enzyme is only decreased by mechanism-based inhibition and increased by turnover in the model. However, EM is also an inducer of CYP3A (Maurel, 1995), and the induction by EM may antagonize the inhibitory effect. This induction may explain why the predicted increase in AUC was somewhat greater than the observed one. A more accurate prediction may be attained by incorporating competitive inhibition and the induction of enzyme in this model.
In conclusion, we predicted the increasing ratio of AUC of MDZ due to metabolic inhibition by CIM in the liver quantitatively using the unbound concentrations in human liver, which we estimated, and the inhibition constants on human hepatic microsomes from published data. When the inhibitors, such as CIM, were concentratively transported into the liver, the inhibitory effects were underestimated using unbound concentration in the plasma. However, the predicted values in the presence of ITZ or EM calculated by taking into account the concentrative uptake of inhibitors were still underestimated. Because the inhibition by EM was due to mechanism-based inhibition, the increase ratio of the plasma concentration of MDZ, which we predicted by fitting kinetic parameters for inactivation of the enzyme to a physiological model, was very close to the observed ratio value due to metabolic inhibition by EM from published data (Fig. 6). To quantitatively evaluate the extent of the drug interaction by hepatic metabolic inhibition, it is important to investigate the contribution of the competitive inhibition and mechanism-based inhibition. In the case of mechanism-based inhibition, such as with EM, the interaction is considered to remain even after the inhibitor completely disappears from the metabolic organ. However, ITZ-induced metabolic inhibition still remains unpredictable even taking into account the concentrative uptake of ITZ. In the future, it will be necessary to examine the time-dependent change in the amount of inactive enzyme by ITZ and the effect of ITZ, which dissolves into lipid bilayer around P450, on enzyme activity.
Footnotes
-
Send reprint requests to: Katsuhiro Yamano, Biopharmaceutical and Pharmacokinetic Research Laboratories, Fujisawa Pharmaceutical Co., Ltd., 1-6, Kashima 2-chome, Yodogawa-ku, Osaka 532-8514, Japan. E-mail:katsuhiro_yamano{at}po.fujisawa.co.jp
- Abbreviations used are::
- CYP
- cytochrome P450
- EM
- erythromycin
- MI
- metabolic intermediate
- MDZ
- midazolam
- AUC
- area under the plasma concentration curve
- ITZ
- itraconazole
- CIM
- cimetidine
- HPLC
- high-performance liquid chromatography
- Received April 19, 2000.
- Accepted December 6, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}