


Abstract
We have identified CYP2C19 and CYP3A4 as the principal cytochrome P450s involved in the metabolism of flunitrazepam to its major metabolites desmethylflunitrazepam and 3-hydroxyflunitrazepam. Human CYP2C19 and CYP3A4 mediated the formation of desmethylflunitrazepam with Km values of 11.1 and 108 μM, respectively, and 3-hydroxyflunitrazepam withKm values of 642 and 34.0 μM, respectively. In human liver microsomes (n = 4) formation of both metabolites followed biphasic kinetics. Desmethylflunitrazepam formation was inhibited 31% byS-mephenytoin and 78% by ketoconazole, suggesting involvement of both CYP2C19 and CYP3A4. Formation of 3-hydroxyflunitrazepam was also significantly inhibited by ketoconazole (94%) and S-mephenytoin (18%). In support of these chemical inhibition data, antibodies directed against CYP2C19 and CYP3A4 selectively inhibited formation of desmethylflunitrazepam by 26 and 45%, respectively, while anti-CYP3A4 antibodies reduced 3-hydroxyflunitrazepam formation by 80%. Our data also suggest that CYP1A2, -2B6, -2C8, -2C9, -2D6, and -2E1 are not involved in either of these metabolic pathways. We estimate that the relative contributions of CYP2C19 and CYP3A4 to the formation of desmethylflunitrazepam in vivo are 63 and 37%, respectively, at therapeutic flunitrazepam concentrations (0.03 μM). We conclude that the polymorphic enzyme CYP2C19 importantly mediates flunitrazepam demethylation, which may alter the efficacy and safety of the drug, while CYP3A4 catalyzes the formation of 3-hydroxyflunitrazepam.
Flunitrazepam (Rohypnol) is a highly lipophilic benzodiazepine derivative used primarily as a sedative and hypnotic (Scharf et al., 1979). It is the most widely prescribed sedative/hypnotic in Europe but is not marketed in North America. However, its abuse in North America and elsewhere continues to gain notoriety (Saum and Inciardi, 1997), with increasing numbers of reports of young women being sexually assaulted after unknowingly ingesting flunitrazepam. This has lead to the media dubbing flunitrazepam the “date rape drug” (Saum and Inciardi, 1997;Schwartz and Weaver, 1998). Flunitrazepam is also used to enhance the effects of alcohol, marijuana, and heroin or to moderate the stimulant effects of cocaine, and its use is reportedly one of the fastest growing drug problems in the southern United States (San et al., 1993;Calhoun et al., 1996; Saum and Inciardi, 1997).
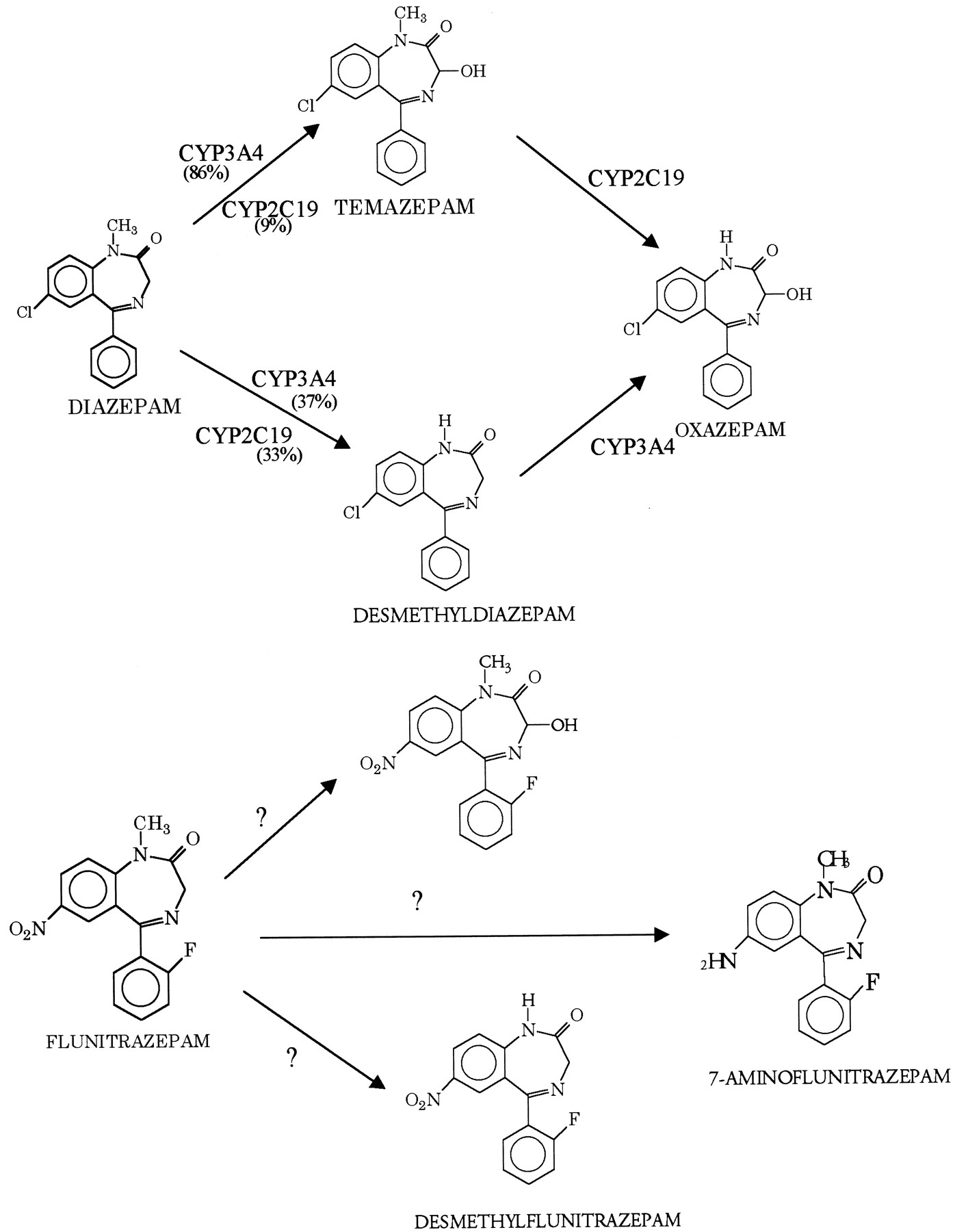
Flunitrazepam is similar in chemical structure and pharmacodynamic and pharmacokinetic properties to other benzodiazepines, e.g., diazepam (Valium) (Wickstrom et al., 1980). However, it is approximately 10-fold more potent than diazepam (Mattila and Larni, 1980). Like diazepam, flunitrazepam is metabolized in the liver and follows a similar catabolic pathway. In humans, flunitrazepam is metabolized to the major metabolites desmethylflunitrazepam, 3-hydroxflunitrazepam, and 7-aminoflunitrazepam (Fig. 1) (Cano and Sumirtapura, 1981). In humans, flunitrazepam is oxidized to the major metabolites desmethylflunitrazepam and 3-hydroxyflunitrazepam and reduced to 7-aminoflunitrazepam. (Fig. 1) (Cano and Sumirtapura, 1981). The relative importance of these metabolic pathways and the enzymes that mediate them has not been determined.

Major enzymatic pathways in the metabolism of diazepam and flunitrazepam.
Once absorbed, flunitrazepam is rapidly distributed to the body tissues from the plasma, resulting in an acute clinical response to the drug that does not correlate with the elimination half-life (approximately 20 h; Mattila and Larni, 1980). The rapid absorption and high lipid solubility of flunitrazepam, which facilitates its speedy entry into the brain and rapid onset of effects, are major contributing factors to its abuse liability. Sedative effects of the drug usually occur within 20 min and last 6 to 8 h with accompanying psychomotor impairment that may last up to 12 h (Mattila and Larni, 1980). The parent drug flunitrazepam is principally responsible for the sleep-inducing effect, although the relative activities of the metabolites are unclear.
CYP2C19 and CYP3A4 have been identified as the major enzymes that mediate the metabolism of diazepam. They contribute approximately 33 and 40% to the formation of desmethyldiazepam, respectively, and 9 and 86% toward the production of 3-hydroxydiazepam (temazepam), respectively (Jung et al., 1997). Both desmethyldiazepam and 3-hydroxydiazepam are pharmacologically active (Baird and Hailey, 1972). CYP2C19 belongs to the human CYP2C subfamily and plays a major role in the metabolism of a number of therapeutically important drugs such as omeprazole, proguanil, and imipramine (Goldstein and de Morais, 1994). The enzyme is genetically polymorphic with large differences in the frequency of the poor metabolizer phenotype among different populations (e.g., approximately 20% of Chinese and 3% of Caucasians are homozygous for deficient CYP2C19 alleles and lack enzyme activity) (Daniel and Edeki, 1996).
Variable clinical responses to flunitrazepam have been reported (Boxenbaum et al., 1978; Wickstrom et al., 1980). If flunitrazepam, like diazepam, is metabolized by CYP2C19, the polymorphic expression of this enzyme may affect the clinical safety and efficacy of this drug. Poor metabolizers lacking enzyme activity might experience the sedative and amnesic effects of flunitrazepam at lower doses and for longer periods of time than those who metabolize the drug more efficiently (extensive metabolizers). The present study investigated, in vitro and in vivo, the role of CYP2C19 and CYP3A4 in the metabolism of flunitrazepam and formation of its major metabolites desmethylflunitrazepam and 3-hydroxyflunitrazepam.
Materials and Methods
Chemicals and Reagents.
Flunitrazepam, desmethylflunitrazepam, 3-hydroxyflunitrazepam, 7-aminoflunitrazepam, and clonazepam were obtained from Hoffmann-La Roche Ltd. (Vaudreuil, QC, Canada). Ketoconazole, sulfaphenazole, α-naphthoflavone, and NADPH were purchased from Sigma Chemical Co. (St. Louis, MO). High-performance liquid chromatography grade solvents were purchased from Fisher Scientific (Fair Lawn, NJ). Omeprazole, 5-hydroxyomeprazole, and H153/52 (internal standard) were generously donated by Astra Hassle (Mölndal, Sweden).S-Mephenytoin and antibodies against CYP1A, CYP2A6, CYP2B1, CYP2C, CYP2D6, and CYP3A4 were purchased from GENTEST Co. (Woburn, MA). Monospecific anti-human CYP2C19 was purchased from Research Diagnostics Inc. (Flanders, NJ). Specific human P4501 enzymes (CYP2B6, -2C9, -2C18, -2C19, -2D6, -2E1, and -3A4) expressed in human lymphoblast cells or the baculovirus expression system were purchased from GENTEST. For initial experiments, CYP2C8, -2C9, -2C18, and -2C19 were expressed in and purified from baculovirus-infected insect cells by previously detailed methods (Haining et al., 1996). The cDNAs for CYP2C8, CYP2C9, CYP2C18, and CYP2C19 were provided by Dr. F. Gonzalez (National Institutes of Health, Bethesda, MD), Dr. M. E. Veronese (Flinders, Australia), and Dr. J. A. Goldstein (National Institute of Environmental Health Sciences, Raleigh, NC), respectively. Cytochrome P450 reductase and cytochrome b5 were also purified as described in Haining et al. (1996). Human liver samples (n = 4) were kindly provided by Dr. T. Inaba (Toronto, ON, Canada). Microsomes were prepared from human livers of kidney donors as previously described by Tyndale et al. (1989). Protein concentrations were determined using a BCA protein assay kit (Pierce Chemical Co., Rockford, IL).
Incubation of Flunitrazepam with Human Liver Microsomes.
All incubations consisted of a 30-min incubation at 37°C; conditions were designed to produce linear formation of the metabolites. Methanol, which was used to dissolve the flunitrazepam (2.5–500 μM final concentration), was evaporated under a gentle stream of N2. NADPH (in a final volume of 200 μl of 25 mM KH2PO4 buffer) was added and reactions were initiated by addition of human liver microsomes (protein = 0.4 mg/ml). Experiments were carried out in duplicate, and the percentage of conversion of all metabolites never exceeded 15% of total substrate added.
The reactions were terminated by removal to ice and addition of the internal standard (clonazepam) and 2.0 ml of dichloromethane. Samples were shaken in a vortex shaker for 10 min followed by centrifugation for 5 min (3000g). The aqueous phase was then aspirated and the organic phase evaporated to dryness under nitrogen. Residues were reconstituted in 200 μl of mobile phase, and a 50-μl aliquot was injected onto the high-performance liquid chromatography column (Spherisorb ODS2, 5 μm, 125 × 4 mm, Agilent, Avondale, PA). Mobile phase of acetonitrile/methanol/water (5:40:55, v/v/v) was delivered with a flow rate of 1.0 ml/min. The analytical system consisted of a Hewlett Packard 1050 liquid chromatographic system with a UV detector set to 234 nm. Under these conditions desmethylflunitrazepam, 3-hydroxyflunitrazepam, internal standard, and flunitrazepam eluted at 11.2, 10.4, 14.3, and 16.1 min, respectively.
Incubation of Flunitrazepam with Expressed P450s.
Incubation conditions were as described for human liver microsomes. Incubations using purified CYP2C8, -2C9, -2C18, and -2C19 microsomes (made by authors) were performed with 200 pmol of enzyme, 200 pmol of reductase, and 200 pmol of cytochrome b5and 100 μM flunitrazepam in a total reaction volume of 1 ml to determine the relative importance of each enzyme in flunitrazepam metabolism. Subsequent experiments used microsomes (CYP2B6, -2C9, -2C18, -2C19, -2D6, -2E1, and -3A4) from baculovirus-transfected insect cells purchased from GENTEST. The amount of enzyme used in initial incubations with flunitrazepam was 40 pmol in a total reaction volume of 200 μl. Once enzymes that metabolized flunitrazepam were identified, the amount of enzyme used was optimized for the linear range of metabolite production. Concentrations of flunitrazepam used in kinetics experiments were between 0 and 500 μM.
For experiments that involved microsomes from human lymphoblast cells (CYP2B6, -2C9, -2C19, -2D6, -2E1, and -3A4), also purchased from GENTEST, the amount of enzyme used in the incubations was adjusted to equal the amount of activity in human liver microsomes as reported by GENTEST.
Within day and between day coefficient of variations in the detection of desmethylflunitrazepam were 4 and 8%, respectively, while coefficient of variations in the detection of 3-hydroxyflunitrazepam were 4% within day and 9% between days.
Chemical Inhibition Studies.
P450-specific inhibitors (α-naphthoflavone (CYP1A1), sulfaphenazole (CYP2C9), ketoconazole (CYP3A4), S-mephenytoin (CYP2C19), and omeprazole (CYP2C19) were used at concentration ranges (0.1–400 μM) that covered the Ki value for each P450 enzyme's specific index reaction in human liver microsomes (Bourrie et al., 1996). The inhibitors, in 50 μl of phosphate buffer, were added to the incubation mixture before addition of the enzyme source. Samples were assayed as described above.
Immunoinhibition Studies.
Experiments were carried out in human liver microsomes with antibodies to CYP2A6 (monoclonal), CYP2B1 (polyclonal), CYP2C (polyclonal), CYP2C19 (monoclonal), CYP2D6 (polyclonal), or CYP3A4 (monoclonal). Antibodies and microsomes were preincubated according to conditions recommended by the manufacturer. Briefly, specific anti-P450 antibodies (0–80 μl) were preincubated on ice with the microsomes for 30 min, after which flunitrazepam was added and incubation carried out as described above. For control experiments, the anti-P450 antibodies were replaced with the addition of appropriate corresponding solutions without the antibody. As in chemical inhibition experiments, inhibition was evaluated at flunitrazepam concentrations equivalent to the apparent Km for the formation of the metabolite of interest in the particular set of microsomes.
Data Analysis.
The metabolism of flunitrazepam to both desmethylflunitrazepam and 3-hydroxyflunitrazepam demonstrated biphasic kinetics. Therefore, a two-enzyme Michaelis-Menten profile was used to obtain the metabolic constants Km andVmax using the GraphPad Prism ver. 2.01 software (San Diego, CA). Relative contributions of CYP3A4 and CYP2C19 to the formation of desmethylflunitrazepam were determined using the following equation:
Results
Kinetic Studies Using cDNA-Expressed P450 Microsomes.
Initially, the data from incubations with P450 microsomes expressed in human lymphoblast cells (CYP2B6, -2C9, -2C19, -2D6, -2E1, and -3A4; GENTEST) suggested that CYP3A4 mediated the formation of desmethylflunitrazepam and 3-hydroxyflunitrazepam with no involvement from CYP2C enzymes. However, incubations with P450 enzymes expressed in the baculovirus system (CYP2B6, -2C9,-2C18, -2C19, -2D6, -2E1, and -3A4; GENTEST) demonstrated that CYP2C19 and CYP3A4 mediated the formation of desmethylflunitrazepam and 3-hydroxyflunitrazepam. CYP2D6 and -2E1 produced no detectable metabolite(s) while CYP2B6, -2C9, and -2C18 produced very small amounts of metabolite(s) at 200 μM flunitrazepam. In a general comparison of flunitrazepam (20 μM) metabolism using purified CYP2C8, -2C9, -2C18, and -2C19, it was demonstrated that CYP2C18 and -2C19 produced significant quantities of desmethylflunitrazepam compared with -2C8 and -2C9 (relative rates: 13.8, 8.3, 1.0, and 1.1, respectively). As CYP2C18 is a minor P450 in the human liver, the detailed kinetic analysis focused on CYP2C19 and CYP3A4 (Shimada et al., 1994).
Baculovirus-expressed CYP2C19 (GENTEST) mediated the metabolism of flunitrazepam to desmethylflunitrazepam with aKm of 11.1 μM andVmax of 0.095 nmol/mg/min, and to 3-hydroxyflunitrazepam with a Km of 642 μM and a Vmax and 0.861 nmol/mg/min (Table 1). CYP3A4 mediated both the demethylation and 3-hydroxylation reactions withKm values of 108 and 34.0 μM andVmax values of 1.27 and 0.420 nmol/min/mg, respectively (Table 1).
Kinetic Studies in Human Liver Microsomes.
Formation of both desmethylflunitrazepam and 3-hydroxyflunitrazepam was characterized by a two-enzyme system. Rates of formation of desmethylflunitrazepam and 3-hydroxyflunitrazepam by human liver microsomes are illustrated in Fig. 2 and summarized in Table 1.
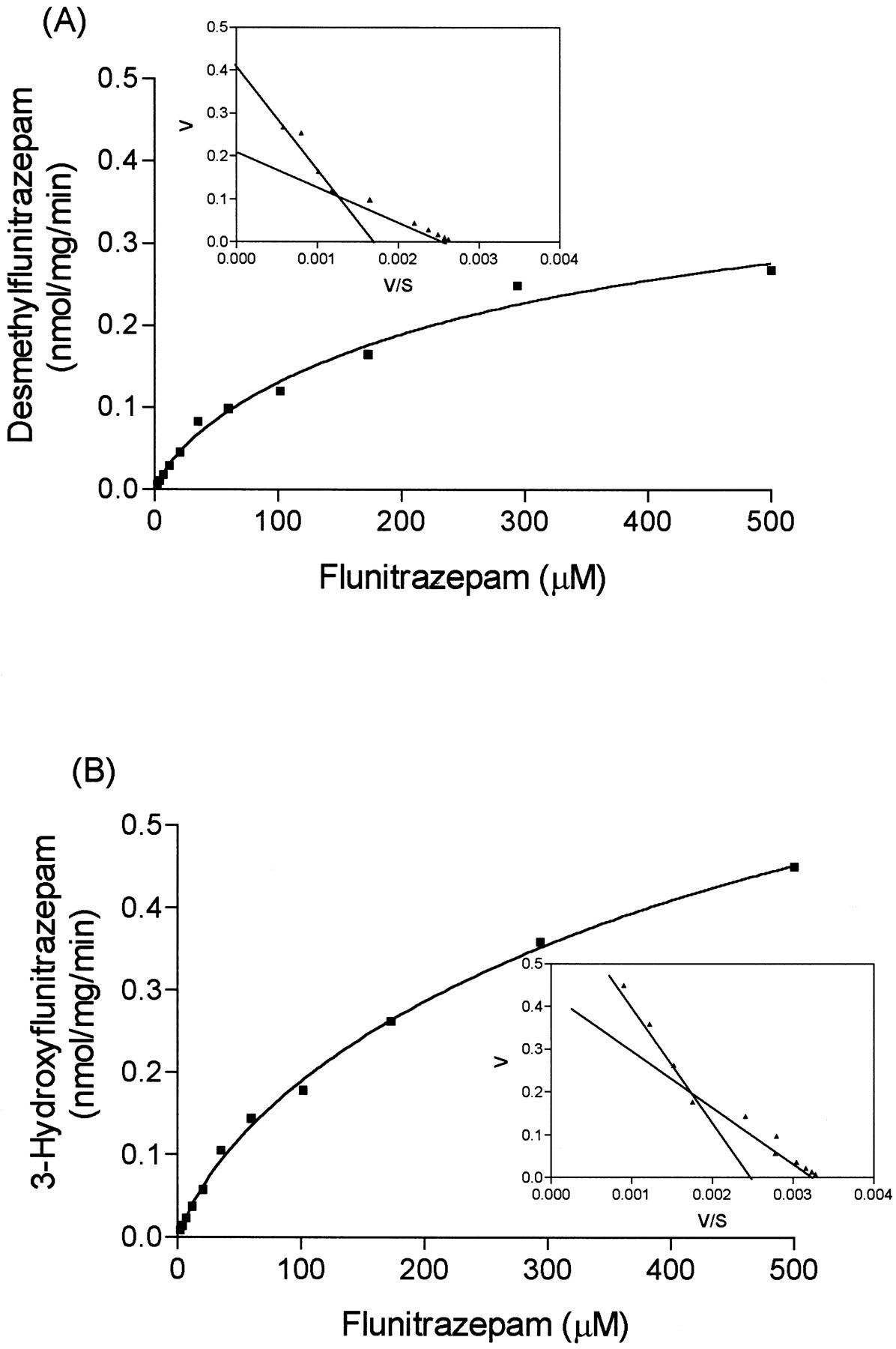
Mean (n = 2) rates of formation of desmethylflunitrazepam (A) and 3-hydroxyflunitrazepam (B) from flunitrazepam by human liver microsomes.
Inset, the data transformed by Eadie-Hofstee plot. Curves were fitted by a nonlinear regression two-site binding equation.
The Km value (11.1 μM) obtained from expressed CYP2C19 was approximately 4-fold lower than the high-affinityKm component of theN-demethylase activity in human liver microsomes (42 ± 17.4 μM; mean ± S.D.; n = 4), while theVmax in human liver microsomes (0.075 ± 0.03 nmol/mg/min) was similar to that obtained with expressed CYP2C19 (0.095 nmol/mg/min). In human liver microsomes the low-affinityKm and Vmaxvalues for the demethylase activity were 165.25 ± 101 μM and 0.171 ± 0.100 nmol/min/mg, respectively, consistent with the idea that metabolism is being mediated by CYP3A4 (Table 1). The kinetics of 3-hydroxyflunitrazepam formation in human liver microsomes was characterized by a high-affinity Km of 86.5 ±20.3 μM and a low-affinity Km of 289.75 ± 84.5 μM. The Vmax values measured for 3-hydroxyflunitrazepam formation in the cDNA-expressed microsomes were higher relative to those measured from human liver microsomes (Table 1).
Chemical Inhibition Studies.
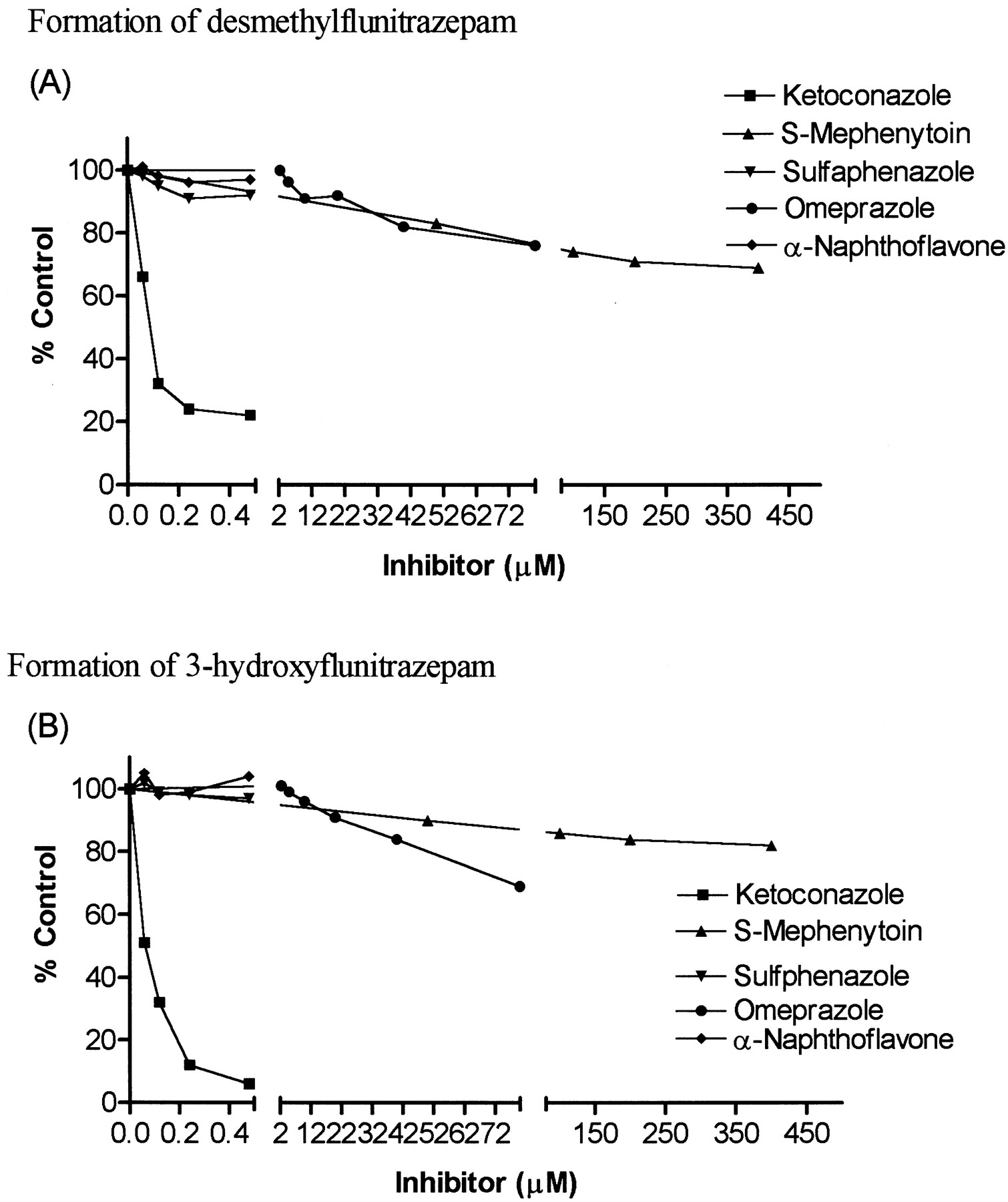
Inhibition of flunitrazepam metabolism in human liver microsomes was performed using the isozyme-specific inhibitors ketoconazole (CYP3A inhibitor), sulfaphenazole (CYP2C9 inhibitor), omeprazole (CYP2C19 substrate), S-mephenytoin (CYP2C19 substrates), and α-naphthoflavone (CYP1A2 inhibitor). Inhibition was evaluated at flunitrazepam concentrations equivalent to the formationKm for the particular set of human liver microsomes being used.
Desmethylflunitrazepam formation was reduced 78% by ketoconazole (0.5 μM), 31% by S-mephenytoin (400 μM), and 24% by omeprazole (80 μM); sulfaphenazole and α-naphthoflavone reduced formation by less than 10%. 3-Hydroxyflunitrazepam formation was reduced 94% by ketoconazole (0.5 μM), 18% byS-mephenytoin (400 μM), and 41% by omeprazole (80 μM), while neither α-naphthoflavone nor sulfaphenazole altered the rate of metabolite formation (Fig. 3).

Effects of different concentrations of inhibitors on the rates of formation of desmethylflunitrazepam (A) and 3-hydroxyflunitrazepam (B) from flunitrazepam.
Reaction rates are expressed as a percentage of the control velocity without inhibitor.
Immunoinhibition Studies.
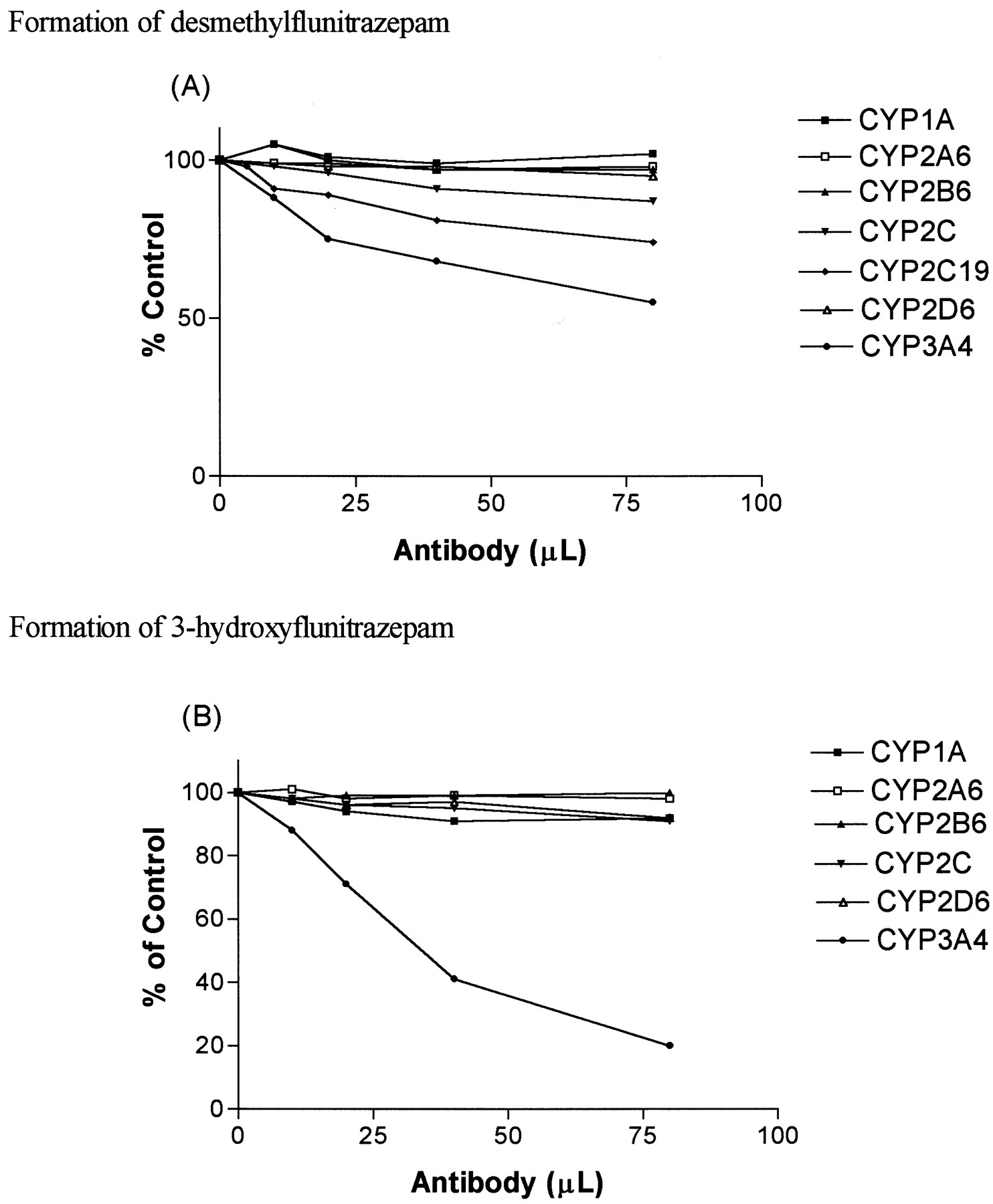
Antibodies raised against CYP1A, CYP2A6, CYP2B1, and CYP2D6 inhibited formation of desmethylflunitrazepam and 3-hydroxyflunitrazepam by less than 10% (Fig. 4). Anti-CYP3A4 antibodies inhibited the formation of desmethylflunitrazepam and 3-hydroxyflunitazepam by 45 and 80%, respectively. Selective anti-CYP2C19 serum inhibited desmethylflunitrazepam formation by 26%, while antibodies to CYP2C13 inhibited formation of desmethylflunitrazepam by 13%. Neither altered the formation of 3-hydroxyflunitrazepam.
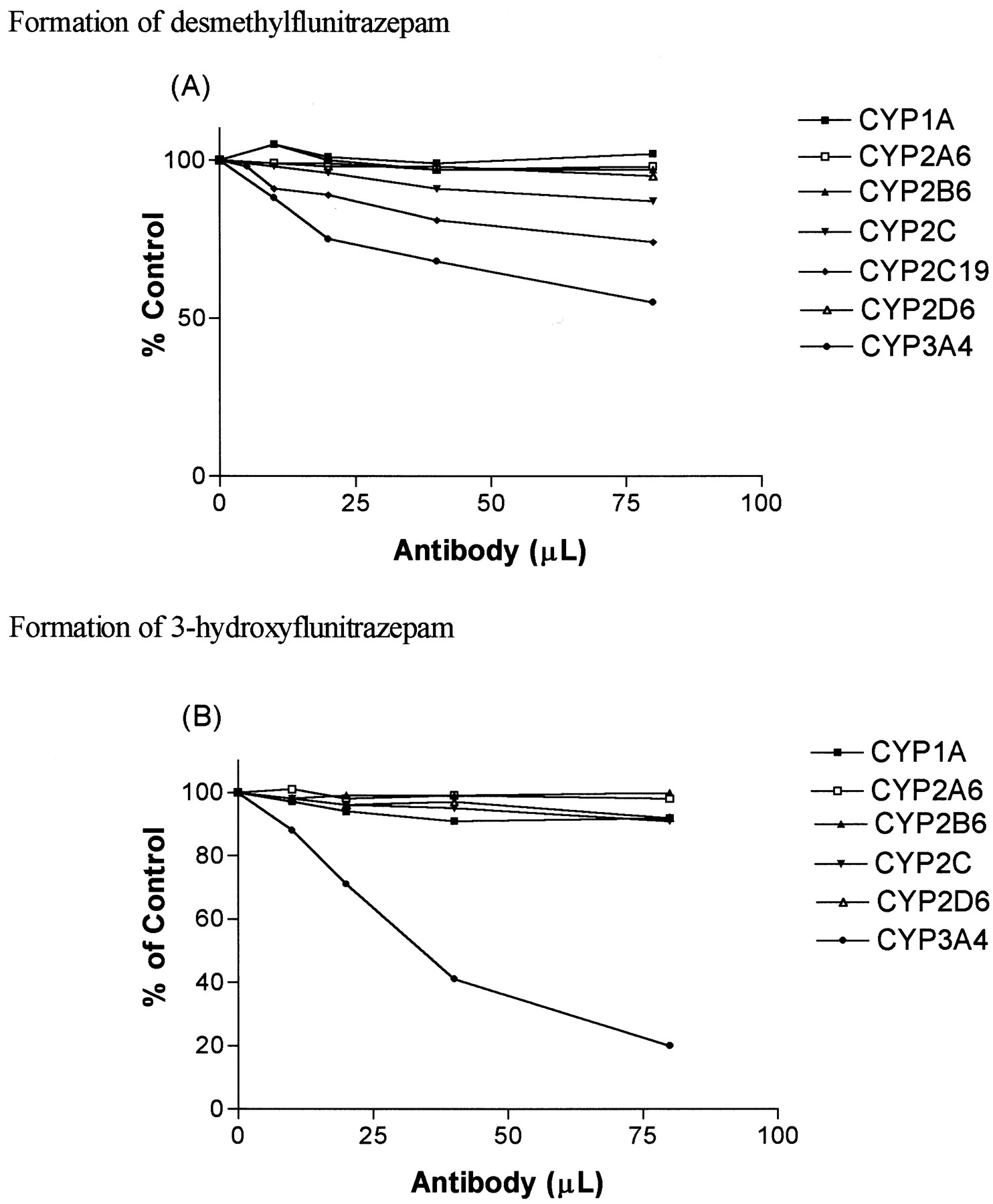
The effect of different P450-selective antibodies on the formation of desmethylflunitrazepam (A) and 3-hydroxyflunitrazepam (B) from flunitrazepam.
Discussion
Despite the extensive use of flunitrazepam in many parts of the world, the characterization of the enzymes involved in its metabolism has not been reported. Expressed CYP2B6, -2C8, -2C9, -2D6, and -2E1 did not metabolize flunitrazepam. In addition, data from our chemical inhibition studies demonstrated a lack of involvement of CYP1A2 (α-naphthoflavone) and CYP2C9 (sulfaphenazole) in the metabolism of flunitrazepam to desmethylflunitrazepam or 3-hydroxyflunitrazepam. Likewise, antibodies raised against CYP1A, -2A6, -2B6, and -2D6 had no effect on flunitrazepam metabolism. Our data using baculovirus-expressed P450s and human liver microsomes strongly suggest that flunitrazepam metabolism to desmethylflunitrazepam and 3-hydroxyflunitrazepam in the human liver is primarily mediated by CYP2C19 and CYP3A4, respectively (Table 1).
Ketoconazole (0.5 μM), at more than 3 times itsKi value (0.015 μM; Bourrie et al., 1996) for CYP3A4 inhibition, decreased the formation of desmethylflunitrazepam by 78%. (S)-Mephenytoin (400 μM), at approximately 2 times its Km as a CYP2C19 substrate (∼200 μM; Goldstein and de Morais, 1994), inhibited desmethylflunitrazepam formation by 31%. Omeprazole (80 μM) inhibited desmethylflunitrazepam formation by 24%. CYP2C19 is responsible for ∼75% of the metabolism of omeprazole to 5-hydroxyomeprazole near therapeutic concentrations (3.0 μM;Km = 5.0 μM) while CYP3A4 contributes to over 80% of omeprazole sulfone formation at the same concentrations (Km = 60.0 μM; Andersson et al., 1990). The chemical inhibition data is supported by the 26% inhibition of desmethylflunitrazepam by anti-CYP2C19 antibodies. At the same substrate concentration, anti-CYP3A4 antibodies inhibited desmethylflunitrazepam formation by 45%. These data together suggest the involvement of both CYP2C19 and CYP3A4 in flunitrazepam metabolism to desmethylflunitrazepam.
Ketoconazole reduced the formation of 3-hydroxyflunitrazepam by over 90%, while omeprazole and (S)-mephenytoin inhibited metabolite formation by 14 and 18%, respectively. 3-Hydroxyflunitrazepam formation was reduced by 80% in the presence of anti-CYP3A4 antibodies, while anti-CYP2C antibodies had no effect on its formation. These data, along with data from expressed P450 kinetic studies (Table 1), strongly suggest that CYP3A4 is the major enzyme involved in the hydroxylation pathway.
Interestingly, flunitrazepam was not metabolized by CYP2C19 microsomes expressed in human lymphoblast microsomes. It is possible that variable coenzyme levels could account for the differences between expression systems. This may also be attributable to the lower levels of activity in this expression system relative to the baculovirus expression system (GENTEST catalog 1996–1997). However, it has been proposed that both the nature of microsomal membranes (phospholipid content) and the environment (such as ionic charge) can be critical in determining the catalytic activity of CYP3A4 (Imaoka et al., 1992; Ingelman-Sundberg et al., 1996; Maenpaa et al., 1998). A similar explanation for the discrepancies between expression systems in flunitrazepam metabolism may be relevant. It is possible that the content of the microsomal membrane of human lymphoblastoid cells and the intracellular environment of the enzyme in which the tertiary structure is formed are different from those found in the baculovirus/insect cells, resulting in altered catalytic activity of CYP2C19 toward specific substrates.
The mean intrinsic clearance, as estimated byVmax/Km, of desmethylflunitrazepam (1.82 ± 0.48) was higher than the intrinsic clearance of 3-hydroxyflunitrazepam (1.52 ± 0.36). This is consistent with observations in vivo indicating that the demethylation pathway may be of greater importance than the hydroxylation route for flunitrazepam metabolism (Cano and Sumirtapura, 1981). After a typical 2.0-mg oral dose of flunitrazepam, plasma concentrations of the metabolites 7-aminoflunitrazepam and desmethylflunitrazepam are 4.6 and 2.8 ng/ml, respectively. 3-Hydroxyflunitrazepam is not detected in the plasma because it undergoes immediate glucuronidation (Singlas, 1979). If active, desmethylflunitrazepam and 3-hydroxyflunitrazepam metabolites may contribute to the hypnotic and amnesic effects of the drug.
Using the kinetic constants derived from the human liver studies (Table1), it is possible to estimate the contribution of CYP2C19 and CYP3A4 to the formation of desmethylflunitrazepam in vivo. At flunitrazepam concentrations equivalent to therapeutic plasma concentrations (0.03 μM) achieved after a typical 2.0-mg oral dose, the relative contributions of CYP2C19 (high-affinity) and CYP3A4 (low-affinity) pathways for desmethylflunitrazepam formation would be 63 and 37%, respectively.
These data together strongly suggested that the desmethylflunitrazepam pathway was quantitatively important and that CYP2C19 plays a major role at therapeutic flunitrazepam concentrations. To test the potential clinical importance of CYP2C19 in vivo, a small pilot study was run in individuals with and without CYP2C19 activity. Individuals who lacked CYP2C19 activity exhibited higher (∼140%) plasma flunitrazepam concentrations, indicating low hepatic first-pass metabolism relative to a subject with full CYP2C19 activity (data not shown). They also demonstrated greater sedation and psychomotor impairment. These data suggest that CYP2C19 is importantly involved in the metabolism of flunitrazepam in vivo and that those individuals with genetically impaired CYP2C19 activity may have elevated and/or prolonged responses to flunitrazepam.
In summary, this research shows that CYP2C19 is an important enzyme involved in the N-demethylation of flunitrazepam and that CYP3A4 is the major contributor to the formation of 3-hydroxyflunitrazepam. The polymorphic expression of CYP2C19 likely influences the clinical safety and abuse liability of flunitrazepam.
Acknowledgments
We thank Dr. T. Inaba for his donation of human livers; Dr. T. Andersson for his donation of omeprazole, omeprazole metabolites, and internal standard used in the omeprazole assay; and Hoffmann-La Roche Ltd. for the generous donation of flunitrazepam, flunitrazepam metabolites, and clonazepam used in the flunitrazepam assay.
Footnotes
-
Send reprint requests to: Dr. E. M. Sellers, Psychopharmacology and Dependence Research Unit, Sunnybrook Women's College Health Sciences Centre, Room 42, 76 Grenville St., Toronto, ON M5S 1B2, Canada. E-mail: e.sellers{at}utoronto.ca
-
Supported in part by National Institute on Drug Abuse Grant DA06889 and the National Institutes of Health Grant GM32165.
-
1 Abbreviation used is: P450, cytochrome P450.
- Received July 31, 2000.
- Accepted December 18, 2000.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}